
Abstract
Genome-wide association studies (GWAS) have identified hundreds of genetic variants associated with complex traits and diseases. However, elucidating the causal genes underlying GWAS hits remains challenging. We applied the summary data-based Mendelian randomization (SMR) method to 28 GWAS summary datasets to identify genes whose expression levels were associated with traits and diseases due to pleiotropy or causality (the expression level of a gene and the trait are affected by the same causal variant at a locus). We identified 71 genes, of which 17 are novel associations (no GWAS hit within 1 Mb distance of the genes). We integrated all the results in an online database (http://www.cnsgenomics/shiny/SMRdb/), providing important resources to prioritize genes for further follow-up, for example in functional studies.
Similar content being viewed by others

Background
Genome-wide association studies (GWAS) have identified thousands of genetic loci associated with various complex traits, disorders, and diseases [1, 2]. The GWAS paradigm exploits the linkage disequilibrium (LD) correlation structure of the genome, which means that the majority of the variation in the genome can be captured in a cost-effective way by genotyping only a few hundred thousand variants, followed by imputation of non-genotyped variants using a densely genotyped reference panel [3]. However, the LD structure also means that identified associations frequently point to genomic regions that harbor many genes, and it is extremely difficult to prioritize among these genes to identify the most functionally relevant genes using GWAS data alone. Laboratory-based follow-up of the associated regions is costly and prohibitive given the number of putatively causal variants in a typical genome-wide significant locus. GWAS of gene expression levels has allowed identification of expression quantitative trait loci (eQTL) [4–6]. Several recent methods [7–11] have used analytical approaches to integrate eQTL and complex trait associations as strategies to prioritize genes for further studies. In this study, we apply the recently developed summary data-based Mendelian randomization (SMR) method to 28 complex traits (including diseases), which have GWAS summary statistics available in the public domain, to obtain a list of genes to prioritize for further follow-up such as functional studies, and develop a database to query all the data and results. We use the SMR method because: it implements a transcriptome-wide association analysis in a formal statistical framework using summary data so that the statistical power is increased by using the latest GWAS and eQTL data of very large sample size; it provides a test to distinguish pleiotropy (or causality) from linkage (see below for more details) [11]; and it is implemented in a user-friendly software tool [12, 13].
Construction and content
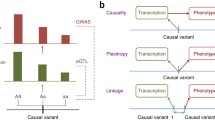
Details of the SMR method can be found in the Zhu et al. paper [11]. In brief, SMR applies the principles of Mendelian randomization (MR) to jointly analyze GWAS and eQTL summary statistics in order to test for association between gene expression and trait due to a shared causal variant at a locus. Mendelian randomization is an instrumental variable analysis approach that uses genetic variant(s) as instrumental variable(s) (Z) to test whether an exposure (X) has a causal effect on an outcome (Y) [14, 15]. Equivalently, it is an analysis to test whether the effect of Z on Y is mediated by X (a model of Z - > X - > Y). The instrumental variable estimate of the effect of X on Y (b XY) can be expressed as b XY = b ZY/b ZX, where b ZY is the effect size of Z on Y and b ZX is the effect size of Z on X [16]. This approach is usually used to test for the causative effect of a modifiable risk factor on health outcomes but the same principle can be used to test whether the effect size of a SNP (Z) on a trait (Y) identified from GWAS is mediated by the expression level of a gene (X). The SMR test [11] is a two-sample MR approach [17, 18]. It allows us to estimate and test b XY using summary data from independent studies [11]. For the purpose of testing for the association between gene expression and trait, it uses the estimate of SNP effect on the trait (b ZY) from GWAS summary data and the estimate of SNP effect on gene expression (b ZX) from summary data of an independent eQTL study. In this case, trait is the outcome (Y), gene expression is the exposure (X), and the top cis-eQTL that is strongly associated with gene expression is used as the instrument (Z) (we used cis-eQTL with P eQTL <5e-8 in this study). Here we use “association” rather than “causal association” because previous results [11] suggest that there are at least three models consistent with a significant association from the SMR test using only a single genetic variant. These models are causality (Z - > X - > Y), pleiotropy (Z - > X and Z - > Y), and linkage (Z1 - > X, Z2 - > Y, and Z1 and Z2 are in LD). We provide details below of a test to distinguish pleiotropy (or causality) from linkage that is of less biological interest. The purpose of this study is to identify genes whose expression levels are associated with complex traits due to a shared causal variant. We therefore do not further distinguish between causality and pleiotropy (which is also impossible to achieve using only the cis-eQTLs).
As mentioned above, significant SMR results could also reflect linkage (i.e. the top associated cis-eQTL being in LD with two distinct causal variants, one affecting gene expression and the other affecting trait variation), which may be of less interest, at least in the first round of gene prioritization. To exclude SMR results that may reflect linkage, Zhu et al. [11] proposed the heterogeneity in dependent instruments (HEIDI) test, which considers the pattern of associations using all the SNPs that are significantly associated with gene expression (eQTLs) in the cis-region. The null hypothesis is that there is a single causal variant affecting trait and gene expression (pleiotropy or causality), which is of biological interest and should be prioritized for follow-up studies. The alternative hypothesis is that gene expression and trait are affected by two distinct causal variants, which is of less biological interest. Under the null hypothesis that there is a single causal variant, b XY estimated at any of the cis-SNPs that are associated with gene expression (e.g. SNPs with P eQTL <1.6 × 10−3, equivalent to χ 2 > 10) is expected to be equal to that estimated at the top associated cis-eQTL (see Equation 7 of Zhu et al. [11] for more details). Therefore, it is equivalent to test whether there is heterogeneity in b XY estimated at the significant cis-eQTLs (null hypothesis: no heterogeneity, causality or pleiotropy model; alternative hypothesis: heterogeneity, linkage model). Note that the HEIDI test takes into account non-independence of cis-eQTLs due to LD (using individual-level data from a reference sample to estimate LD between the cis-SNPs). Probes that show evidence of heterogeneity (e.g. P HEIDI <0.05) are rejected.
The previous SMR study analyzed three traits (body mass index (BMI), height, and waist-to-hip ratio adjusted by BMI) and two diseases (rheumatoid arthritis and schizophrenia) and identified 21 novel genes (genes that passed the SMR and HEIDI tests and that are located >1 Mb from the nearest GWAS hit) [11]. In this study, the SMR analysis is extended to an additional 28 complex traits and diseases (Table 1) which have summary data available in the public domain from large-scale GWAS. The results from the SMR analyses are made available in an online query database (http://www.cnsgenomics.com/shiny/SMRdb/) [13], which is implemented in R Shiny.
Utility and discussion
After quality control (QC) steps [11], associations between 5967 probes and 757,479 SNPs from the blood gene expression study by Westra et al. [5] were used in the analysis. The Westra eQTL summary data are available in the public domain and on the SMR website [12]. It should be noted that all the probes included in the analysis have at least a cis-eQTL at P eQTL <5 × 10–8. For each probe, the top associated cis-eQTL was used as the instrument for the SMR test. The SMR test was performed for each of the 5967 probes on 28 traits and disorders/diseases (Additional file 1: Table S1). The genome-wide significance level for the SMR test, corrected for multiple testing, is defined as 0.05/5967 = 8.4 × 10–6. For probes with P SMR <8.4 × 10–6, we conducted the HEIDI test and retained for further investigation only those probes with little evidence of heterogeneity P HEIDI ≥0.05. All the analyses were performed using the SMR software tool [11, 12]. We particularly emphasized results that are considered to be novel, i.e. no previously identified SNP, reported as genome-wide significant in the primary GWAS paper, within a 1 Mb window of the probes. We identified 247 gene-trait associations (271 probes) with P SMR <8.4 × 10–6 (Additional file 1: Table S2). After application of the HEIDI test (P HEIDI ≥0.05), this was reduced to 71 gene-trait associations (77 probes) (Additional file 1: Table S3). Of these, 17 gene-trait associations were considered novel (Table 2 and Additional file 1: Table S4).
There were 15 genes associated with more than one trait or disease (Additional file 1: Table S5). Where a gene was associated across more than one trait, there was a strong correlation between the traits, with only two cross trait associations being between disparate traits or diseases. Crohn’s disease (CD) and ulcerative colitis (UC) are chronic gastrointestinal disorders that represent as intestinal inflammation; collectively they are known as inflammatory bowel disease (IBD). GWAS to date have identified 200 loci associated with IBD [19], 71 with CD [20], and 47 with UC [21], as well as evidence for trans-ancestry shared genetic risk for IBD [19]. The SMR analyses predicted ten gene targets for a combination of IBD, CD, and UC (Additional file 1: Table S6), of which four were novel gene associations (in total there were two novel gene associations for CD and three each for IBD and UC). The other traits that shared gene associations were the lipids, i.e. high-density lipoprotein (HDL), low-density lipoprotein (LDL), and total cholesterol (TC) (Additional file 1: Table S7).
The results from this analysis can be queried and viewed in the online application [13]. Results from the initial Zhu et al. study are also included in this database. We intend that as more GWAS summary data becomes available, SMR analysis will be conducted using the summary data and the results database will be updated accordingly. This application enables users to query the database by trait, gene, or both and apply thresholds based on the p value from the SMR method and the HEIDI test. In addition, Manhattan plots are given based on the p value from the SMR analysis and regional association plots are provided for those probe-trait associations that pass both the SMR and HEIDI tests.
Conclusion
SMR, as indicated by the results, provides a means of using summary statistics from GWAS and eQTL data to prioritize likely functionally relevant genes within previously identified regions of association and in some cases identify novel gene associations.
Abbreviations
CD, Crohn’s disease; eQTL, Expression quantitative trait loci; GWAS, Genome-wide association study; HDL, High-density lipoprotein; HEIDI, Heterogeneity in dependent instruments; IBD, Inflammatory bowel disease; LD, Linkage disequilibrium; LDL, Low-density lipoprotein; MR, Mendelian randomization; QC, Quality control; SMR, Summary data-based Mendelian randomization; TC, Total cholesterol; UC, Ulcerative colitis
References
Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–7.
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–1006.
Visscher Peter M, Brown Matthew A, McCarthy Mark I, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24.
Westra HJ, Franke L. From genome to function by studying eQTLs. Biochim Biophys Acta. 2014;1842(10):1896–902.
Westra H-J, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–43.
Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16(4):197–212.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, Jansen R, de Geus EJC, Boomsma DI, Wright FA et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–52.
Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Consortium GT, Nicolae DL et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–8.
He X, Fuller CK, Song Y, Meng Q, Zhang B, Yang X, Li H. Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am J Hum Genet. 2013;92(5):667–80.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, Montgomery GW, Goddard ME, Wray NR, Visscher PM et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
SMR software tool. http://www.cnsgenomics.com/software/smr/. Accessed 22 July 2016.
SMR Results Database. http://www.cnsgenomics.com/shiny/SMRdb/. Accessed 22 July 2016.
VanderWeele TJ, Tchetgen Tchetgen EJ, Cornelis M, Kraft P. Methodological challenges in mendelian randomization. Epidemiology. 2014;25(3):427–35.
Boef AG, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. 2015;44(2):496–511.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178(7):1177–84.
Inoue A, Solon G. Two-sample instrumental variables estimators. Rev Econ Stat. 2010;92:557–61.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–86.
Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42(12):1118–25.
Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43(3):246–52.
Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381(9875):1371–9.
Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Robinson EB, St Pourcain B, Anttila V, Kosmicki JA, Bulik-Sullivan B, Grove J, Maller J, Samocha KE, Sanders SJ, Ripke S et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet. 2016;48:552–5.
Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJ, Nurnberger JI, Rietschel M, Blackwood D et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–83.
Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18(4):497–511.
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30.
The International Consortium for Blood Pressure Genome-Wide Association Studies. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9.
Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–83.
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–90.
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–16.
The Tobacco Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–7.
Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW, Westra HJ, Shakhbazov K, Abdellaoui A, Agrawal A et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science. 2013;340(6139):1467–71.
Benyamin B, Pourcain B, Davis OS, Davies G, Hansell NK, Brion MJ, Kirkpatrick RM, Cents RA, Franic S, Miller MB et al. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol Psychiatry. 2014;19(2):253–8.
de Moor MH, Costa PT, Terracciano A, Krueger RF, de Geus EJ, Toshiko T, Penninx BW, Esko T, Madden PA, Derringer J et al. Meta-analysis of genome-wide association studies for personality. Mol Psychiatry. 2012;17(3):337–49.
de Moor MH, van den Berg SM, Verweij KJ, Krueger RF, Luciano M, Arias Vasquez A, Matteson LK, Derringer J, Esko T, Amin N et al. Meta-analysis of genome-wide association studies for neuroticism, and the polygenic association with major depressive disorder. JAMA Psychiatry. 2015;72(7):642–50.
van den Berg SM, de Moor MH, McGue M, Pettersson E, Terracciano A, Verweij KJ, Amin N, Derringer J, Esko T, van Grootheest G et al. Harmonization of neuroticism and extraversion phenotypes across inventories and cohorts in the Genetics of Personality Consortium: an application of Item Response Theory. Behav Genet. 2014;44(4):295–313.
Acknowledgements
This work has only been made possible by the generous sharing of summary statistics data who each request recognition in different ways. We thank all the consortia who make their summary statistics data available for download (a full list of acknowledgements can be found in Additional file 2: Text S1).
Funding
This research was supported by the Australian Research Council (130102666, 160101343), the Australian National Health and Medical Research Council (1107258, 1078901, 1087889, 1083656), and the Sylvia and Charles Viertel Charitable Foundation.
Availability of data and materials
The summary statistics used in this analysis are available in the public domain. Links to these websites are provided in Additional file 1: Table S1. This information is also made available in the online database (http://www.cnsgenomics.com/shiny/SMRdb/) under the GWAS information tab. The Westra eQTL data can be downloaded from the SMR website (http://www.cnsgenomics.com/software/smr/).
Authors’ contributions
JY and NRW conceived and designed the study. JMWP and ZZ conducted the analysis. JMWP developed the database with contributions from JG and AFM. JMWP, NRW, and JY wrote the manuscript. All authors reviewed and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
GWAS information. Table S2. SMR results (P SMR <8.4 × 10–6). Table S3. SMR and HEIDI results (P HEIDI ≥0.05). Table S4. Novel genes. Table S5. Genes across more than one trait. Table S6. IBD, CD, and UC gene associations. Table S7. HDL, LDL, and TC gene associations. (XLSX 141 kb)
Additional file 2:
Full list of acknowledgements. (DOCX 109 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pavlides, J.M.W., Zhu, Z., Gratten, J. et al. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med 8, 84 (2016). https://doi.org/10.1186/s13073-016-0338-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-016-0338-4