
Abstract
Alzheimer’s disease (AD) is caused by synaptic and neuronal loss in the brain. One of the characteristic hallmarks of AD is senile plaques containing amyloid β-peptide (Aβ). Aβ is produced from amyloid precursor protein (APP) by sequential proteolytic cleavages by β-secretase and γ-secretase, and the polymerization of Aβ into amyloid plaques is thought to be a key pathogenic event in AD. Since γ-secretase mediates the final cleavage that liberates Aβ, γ-secretase has been widely studied as a potential drug target for the treatment of AD. γ-Secretase is a transmembrane protein complex containing presenilin, nicastrin, Aph-1, and Pen-2, which are sufficient for γ-secretase activity. γ-Secretase cleaves >140 substrates, including APP and Notch. Previously, γ-secretase inhibitors (GSIs) were shown to cause side effects in clinical trials due to the inhibition of Notch signaling. Therefore, more specific regulation or modulation of γ-secretase is needed. In recent years, γ-secretase modulators (GSMs) have been developed. To modulate γ-secretase and to understand its complex biology, finding the binding sites of GSIs and GSMs on γ-secretase as well as identifying transiently binding γ-secretase modulatory proteins have been of great interest. In this review, decades of findings on γ-secretase in AD are discussed.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common form of dementia1. Two major pathological hallmarks of AD are senile plaques, which result from extracellular accumulation and deposition of amyloid β-peptide (Aβ), and neurofibrillary tangles containing the hyperphosphorylated tau protein in neurons2,3. AD progresses slowly, and the progression is estimated to occur 25 years prior to the onset of symptoms4. The current treatment for AD is to use acetylcholinesterase inhibitors and the N-methyl-d-aspartate receptor antagonist memantine for the symptomatic improvement of AD5, and there is no cure available. Recently, aducanumab targeting Aβ aggregates in the brain6 was approved with some controversy.
According to the amyloid cascade hypothesis, the accumulation of Aβ in the brain is the primary cause of AD7. The chronic imbalance between the production and clearance rate of Aβ may lead to increased Aβ42 levels, followed by Aβ oligomerization, fibril formation, and accumulation in plaques7. Both Aβ oligomers and plaques damage neurons by astrocytic activation, oxidative injury, and altered kinase/phosphatase activities, followed by the formation of neurofibrillary tangles7. Therefore, therapeutics aimed at lowering Aβ levels could be clinically useful for the treatment of AD7. Genetically inherited familial Alzheimer’s disease (FAD) genes also support Aβ as the key driver in the amyloid cascade hypothesis. In most cases, APP mutations increase the ratio of Aβ42/Aβ40 or total Aβ production8. Missense mutations, insertions, or deletions in PSEN are mostly located in the transmembrane regions or hydrophilic loops in the cytosol, and they result in an increased ratio of Aβ42/Aβ408.
APP processing and Aβ
The amyloid plaques in the brains of AD patients consist of fibrils formed by Aβ. Aβ is produced from amyloid precursor protein (APP) by sequential proteolytic cleavages of β-secretase (β-site APP-cleaving enzyme, BACE) and γ-secretase (Fig. 1)9. In the amyloidogenic pathway of APP processing, APP is initially cleaved by BACE, resulting in soluble APPβ (sAPPβ) and membrane-bound APP-CTF (C99)9. C99 is further cleaved by γ-secretase to release Aβ extracellularly and the APP intracellular domain (AICD) for nuclear translocation9. Alternatively, APP is cleaved by α-secretase to produce sAPPα and APP-CTF (C83) (Fig. 1)9. C83 is further cleaved by γ-secretase to produce p3 and AICD9.

In the amyloidogenic pathway, β-secretase cleaves APP extracellularly to release sAPPβ and a membrane-bound APP-CTF (C99). C99 is subsequently cleaved by γ-secretase to release Aβ and the APP intracellular domain (AICD). In the non-amyloidogenic pathway, APP is cleaved by α-secretase to release sAPPα and a membrane-bound APP-CTF (C83). C83 is cleaved further by γ-secretase to release p3 and AICD.
It has been commonly referred to as the γ-secretase cleavage to release Aβ40 or Aβ42, and AICD. The γ-secretase cleavage site can be further separated into γ-, ζ-, and ε-cleavage sites (Fig. 2)10. The γ-site ends at Aβ40 or Aβ42, and AICD starts at Aβ49 or Aβ50. This discrepancy with missing amino acid residues led to the new identification of the ε-cleavage site at Aβ4911,12,13,14. The question of whether the γ- and ε-cleavages occur sequentially or independently from each other was answered by a new identification of the ζ-cleavage site at Aβ4615,16. Aβ peptides are cleaved mainly by tripeptide trimming via the Aβ40 product line (Aβ49→46→43→40→37) or the Aβ42 product line with the last cleavage step by tetrapeptide trimming (Aβ48→45→42→38) (Fig. 2)17. In addition, other Aβ peptides found in varying lengths support the link between two major Aβ40 and Aβ42 product lines and multiple interactive pathways releasing tri-, tetra-, penta-, and hexapeptides18,19.

After APP is cleaved by β-secretase, APP-CTFs are processed by ε-cleavage, resulting in Aβ49 and AICD50-99 or Aβ48 and AICD49-99. Aβ49 is further cleaved at the ζ-site to Aβ46, and the Aβ40 product line follows (Aβ49→46→43→40→37). The Aβ42 product line is Aβ48→45→42→38. The β-, α-, γ-, ζ-, and ε-cleavage sites are indicated by arrows. Membranes are indicated in pink. Aβ sequence numbering starts from 1 (after β-secretase cleavage) to 49 (after ε-cleavage).
The physiological role of Aβ is not yet clear. The length of Aβ found in CSF or brain varies from 37 to 43 amino acids20,21,22. Aβ42 is more prone to aggregate and more toxic than Aβ40, even though the ratio of production for Aβ42 and Aβ40 is approximately one to nine23. Aβ42 is the major component of amyloid plaques23,24,25, and Aβ43 was reported to exist in amyloid deposition of the human AD brain20.
γ-Secretase
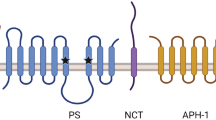
γ-Secretase carries out a sequential cleavage of the substrate C99 to generate Aβ peptides9. As such, γ-secretase has been an attractive target for the potential treatment of AD. However, it has been found to be challenging, and more studies are needed to fully understand γ-secretase. γ-Secretase is a transmembrane protein complex containing presenilin (PS), nicastrin, anterior pharynx defective-1 (Aph-1), and presenilin enhancer-2 (Pen-2) (Fig. 3a, b). γ-Secretase belongs to a new class of proteases, intramembrane-cleaving proteases (I-CliPs), and its unusual cleavage processes substrates in the lipid bilayer of membranes26.

a γ-Secretase complexes require at least four essential components: presenilin (PS), nicastrin (Nct), Aph-1, and Pen-2. The two catalytic aspartyl residues in PS are indicated by ‘D’ (Asp257 in TM6 and Asp385 in TM7). PS undergoes endoproteolysis (indicated by arrow) and becomes a PS-NTF/PS-CTF heterodimer. b The γ-secretase complex structure is shown in the surface view. Presenilin (blue), nicastrin (magenta), Aph-1 (green), and Pen-2 (yellow). Rendered from Protein Data Bank entry 7D8X. The structural figure was prepared with UCSF ChimeraX 1.2.5.
PS, nicastrin, Aph-1, and Pen-2
Compared to BACE, γ-secretase is not strictly site-specific and yields Aβ peptides that are 37–43 amino acids long20,21,22. The topology of PS has nine transmembrane spanning domains27. PS has two mammalian homologs, PS1 and PS2, which share 67% sequence similarity28. γ-Secretase is an aspartyl protease with essential aspartyl residues at positions 257 and 385 within transmembrane domains 6 and 7 of PS (both PS1 and PS2) that constitute the active site of the protease (Fig. 3a)29,30. The mature form of PS is cleaved endoproteolytically between the sixth and the seventh transmembrane domains into an N-terminal and a C-terminal fragment (NTF and CTF), and the PS1-NTF/PS1-CTF heterodimer forms the catalytic site of γ-secretase30,31,32,33. Transition state analog (TSA) γ-secretase inhibitors (GSIs) that bind to PS1-NTF and PS1-CTF also support this finding34,35. In addition, >300 PSEN-harboring FAD mutations increased the Aβ42/40 ratio, and knockout (KO) of PSEN1 decreased γ-secretase cleavage of APP and reduced Aβ production31.
Nicastrin was discovered by its association with PS after immunoaffinity purification using an anti-PS antibody36. Two additional cofactors, Aph-1 and Pen-2, were discovered by genetic screening in Caenorhabditis elegans37,38. Nicastrin is a single-pass transmembrane protein with a large extracellular domain. Immature nicastrin is ~110 kDa, and the apparent molecular weight is increased to ~130 kDa after N-glycosylation in the Golgi/TGN compartments39. This mature form of nicastrin is associated with the active γ-secretase complex39,40,41,42. Aph-1 is required for the cell-surface localization of nicastrin38, and Pen-2 is required for both the expression of PS and the maturation of nicastrin43.
The mRNA and protein expression of the γ-secretase complex subunits is ubiquitously expressed in the body44. The physiological functions of γ-secretase complex subunits were studied by using KO mice. PS1 KO mice are lethal, resulting Notch signaling deficiency, while the phenotype of PS2 KO mice is normal, and double KO of PS1 and PS2 is embryonic lethal, showing a severe Notch deficiency45,46. Nicastrin KO mice showed a Notch phenotype with embryonic lethality47. Aph-1a KO mice showed embryonic lethality, and Aph-1b/c KO mice (equivalent to human Aph-1b loss) showed reduced APP processing in several regions in the adult brain48. A KO study in zebrafish showed that Pen-2 is important for neuronal cell survival and protects cells from apoptosis49.
Whether these four subunits of γ-secretase are essential for its activity was investigated. The γ-secretase activity was reconstituted in Saccharomyces cerevisiae, which lacks endogenous γ-secretase activity, by co-expressing PS, nicastrin, Aph-1, and Pen-250. Thus, these four proteins appear to be necessary and sufficient for γ-secretase activity50. This was also shown in Drosophila and mammalian cells50,51,52,53,54. Co-expression of all four components also increased the PS heterodimeric form, fully glycosylated nicastrin, and γ-secretase activity in mammalian cells52. In a postmortem human brain study, it was shown that human brain-derived γ-secretase is present as a high molecular weight protein complex containing PS, nicastrin, Aph-1, and Pen-2 and that these are associated with γ-secretase activity55. The activity of the γ-secretase complex was inhibited by the specific GSI L-685,458, suggesting that this γ-secretase complex isolated from the human brain is functional55.
The assembly of the γ-secretase complex is initiated in the endoplasmatic reticulum (ER)56, where Aph-1 and nicastrin interact, followed by the binding of PS53. Thereafter, Pen-2 binds to the complex and facilitates the endoproteolysis of PS to PS-NTF and PS-CTF, resulting in an active γ-secretase complex53. In a γ-secretase activity study using a biotinylated affinity ligand, it was confirmed that PS heterodimers and mature nicastrin exist in the active enzyme complex57. It was also reported that bacterially synthesized recombinant proteins in liposomes such as PS1-ΔE9 (FAD mutation with PS1 exon 9 deletion) alone or PS1-full-length (FL)/Pen-2 have active γ-secretase activity58.
Stoichiometry of γ-secretase
PS has two homologs, PS1 and PS2. Aph-1 has two homologs, Aph-1a and Aph-1b, in humans and one additional homolog, Aph-1c, in rodents. Aph-1a has two alternatively spliced forms, Aph-1aL (long form) and Aph-1aS (short form). In total, γ-secretase can form six different complexes in humans44.
The molecular weight of the four components is PS1-NTF (~30 kDa), PS1-CTF (~20 kDa), fully glycosylated nicastrin (~130 kDa), Aph-1 (~30 kDa), and Pen-2 (~12 kDa). The molecular weight of the γ-secretase complex is calculated to be ~220 kDa at a stoichiometry of 1:1:1:1 (PS:glycosylated nicastrin:Aph-1:Pen-2). Different methods have been used for the preparation and analysis of the complex, resulting in observed molecular weights in the range of 200–2000 kDa50,52,55,59,60. The lowest reported molecular weight of the complex is 200–250 kDa, corresponding to a monomeric complex52. The complex at ~440 kDa suggests a possible stoichiometry of 2:2:2:250. Super-resolution imaging showed that a stoichiometry of 1:1 (PS1:NCT) at the cell surface and a BN-PAGE gel showed γ-secretase complexes at ~440 kDa60. In membranes from the postmortem human brain, the γ-secretase components were eluted in a fraction of > 1000 kDa55. Sato et al.61 reported the stoichiometry of active γ-secretase complexes as 1:1:1:1. Differences in the molecular weight of the γ-secretase complex might indicate the possibilities of additional proteins, either novel core components or proteins binding transiently (γ-secretase modulatory protein, GSMP). The molecular weight of the γ-secretase complex with TMP21 was reported to be approximately 660 kDa62. Another binding protein, GSAP co-eluted with γ-secretase complex components at ~670 kDa63. Active γ-secretase complexes captured by Compound 3 showed a GSMP, Hif-1α, with γ-secretase complexes in high molecular weight fractions64.
Trafficking and localization of γ-secretase
How APP, BACE1, and γ-secretase are trafficked and processed through subcellular compartments has been studied to identify the sites for Aβ production in cells. Aβ is found in the TGN65 and endosomes66. The subcellular localization of Aβ in brain tissue is mainly endosomal as well67,68. APP is cleaved by α-secretase at the cell surface69, while BACE1 cleavage occurs mostly in the late Golgi/TGN and endosomes70. γ-Secretase components have been found in many subcellular compartments, such as the ER, ER-Golgi intermediate compartment, Golgi, TGN, endosomes, and plasma membrane71,72,73,74. Interestingly, PS was also found in synaptic compartments75,76,77,78. In addition, all four γ-secretase components were found in phagosomes79. PS1, nicastrin, and APP are localized in the outer membranes of lysosomes80.
Importantly, the sites for γ-secretase activity have been investigated. A biotinylated active site probe labeled γ-secretase in the plasma membrane of cells73,81. Additionally, a small fraction of active γ-secretase was found in mitochondria82. γ-Secretase enriched in endosomes, in the plasma membranes, and at synapses is active to produce Aβ or AICD, and active γ-secretase was labeled by a GSI in the brain or primary cortical neurons74,83.
The lipid membrane environment can also affect the activity of proteins. Since γ-secretase is a transmembrane-bound protein, different detergents have been used to extract proteins from membranes and study the complex. However, γ-secretase can also be studied in a membrane environment, preserving some of its natural interactions with lipids. Cholesterol and sphingolipids are the major lipid constituents of ordered microdomains called lipid rafts in cell membranes84. Lipid rafts are considered to be dynamic platforms for cell signaling, membrane protein sorting, and transport84. Several findings suggest that the trafficking and processing of APP are regulated in lipid rafts85,86,87,88,89. APP, BACE, and γ-secretase have been shown to localize to lipid rafts. APP and BACE residing in separate lipid rafts can merge in endosomes, where amyloidogenic processing occurs85. Active γ-secretase was found in lipid rafts86 and brain lipid rafts90, and γ-secretase was active in lipid rafts from post-Golgi compartments and endosomes87. The reconstitution study of γ-secretase with different lipid mixtures showed that a lipid raft-like condition gave the highest γ-secretase activity91.
A recent high-throughput functional genomics screen using the FLeXSelect human FL cDNA library identified orphan G protein-coupled receptor 3 (GPR3)92. GPR3 appears to promote complex assembly of γ-secretase, resulting in increased trafficking of the γ-secretase components and the mature γ-secretase complex to the cell surface and increased localization in lipid rafts, which eventually leads to an increase in Aβ generation92. Therefore, specific inhibition of γ-secretase in certain organelles or microdomains could be an attractive approach91,93,94, and a membrane-anchored version of a β-secretase transition state inhibitor reduced enzyme activity95.
γ-Secretase structure
The catalytic residues of I-CliPs are located within transmembrane regions, and they hydrolyze the peptide bonds of their substrates in the transmembrane regions96. The I-CliP family can be categorized into aspartyl proteases (including γ-secretase and signal peptide peptidase), metalloproteases (site-2 protease, Eep), and serine proteases (Rhomboid, AarA)97. It had been challenging for γ-secretase structure studies due to its many subunits and transmembrane domains.
An electron microscopy study on the 3D structure of γ-secretase revealed that there is a low-density interior chamber and two pores (apical and basal pores), which allow for water molecule entry into the structure98. These pores for water molecules could explain this unusual intramembrane cleavage (peptide bond hydrolysis) by γ-secretase98. Aβ and AICD could be released through two pores into outer spaces (extracellular and cytosolic spaces, respectively)98. In 2015, single-particle cryoelectron microscopy (cryo-EM) revealed an atomic structure of γ-secretase in a substrate-free state with a 3.4 Å resolution99. In recent years, cryo-EM structures of the γ-secretase complex either bound to APP (C83) at a 2.6 Å resolution or Notch (Notch-100) at a 2.7 Å resolution have shown that PS1 undergoes conformational changes upon substrate binding100,101,102. Substrate-bound γ-secretase showed that the β-strand from the C-terminal of APP together with two APP-induced β-strands of PS1 form a hybrid β sheet, which guides γ-secretase cleavage for substrates101.
Nicastrin acts as a gatekeeper for the entry of γ-secretase substrates to block substrates with long extracellular domains103. APP can enter, either in whole or in part, at the substrate docking site between PS-NTF and PS-CTF to access the internal active site104. In other words, after a substrate binds to the docking site on PS, the substrate is moved into the S1′, S2′, and S3′ sites (three substrate binding pockets) in the active site of PS by lateral gating, and long Aβ peptides are produced105. Then, long Aβ peptides were cleaved by tripeptide trimming (Aβ49→46→43→40→37 or Aβ48→45→42→38) to release Aβ peptides (Fig. 2)17.
γ-Secretase substrates
γ-Secretase has more than 140 substrates and substrate candidates in addition to APP, which are type 1 transmembrane proteins106. These substrates include APLP1 and APLP2 (together with APP, regulating synaptic plasticity and neuronal excitability), proteins involved in cell adhesion (N-cadherin, E-cadherin, CD44), the CSF1 receptor (protein tyrosine kinase), deleted in colorectal cancer (DCC, Netrin-1 receptor), ErbB4 (growth factor-dependent receptor tyrosine kinase), low-density lipoprotein receptor-related protein (endocytic receptor), Nectin-1α (adherens junction formation), Notch 1–4 (signaling receptors), Delta and Jagged (Notch ligands), p75 (neurotrophin co-receptor), and syndecan-3 (cell-surface proteoglycan co-receptor)97,107. γ-Secretase cleavage does not depend on the specific sequence of the substrate. Rather, it depends on ectodomain shedding108. In many cases, the intracellular domains (ICDs) released upon γ-secretase cleavage are involved in the regulation of gene transcription97.
A well-known γ-secretase substrate, Notch, undergoes ectodomain shedding by metalloprotease at the S2 site, which is further cleaved by γ-secretase at the S3 site and releases the Notch intracellular domain (NICD) (Fig. 4)97. Rare genetic variants of TREM2 (ex. R47H) are associated with AD109. The microglial surface receptor TREM2 and its adaptor protein DAP12 (TYROBP) cascade TREM2 signaling, which promotes phagocytosis109. After TREM2 undergoes ectodomain shedding by ADAM10, it was reported that TREM2-CTF can be cleaved by γ-secretase in cells110. The processing of several substrates by γ-secretase was investigated by ICD formation and the accumulation of substrate-C-terminal fragments (CTFs) by western blotting111. Ideally, an in vitro assay can confirm substrate cleavages106. Structurally, the β-strand region of several substrates (CD43, CD44, N-cadherin, ErbB4, and CD33) was aligned with the β-strand sequences of APP and Notch112. CD43 and CD44 closely resemble Notch 1, whereas N-cadherin, ErbB4, and CD33 share similar features with APP (C99)112. There is still much to be investigated for these substrate cleavages by γ-secretase and their physiological functions.

Notch ligands (ex. Delta, Jagged) from signal sending cells bind to Notch receptors (Notch 1–4) at signal receiving cells. Notch undergoes ectodomain shedding by ADAM metalloproteases (ex. ADAM10, TACE) at the extracellular S2 site (S2 cleavage). A membrane-bound truncated form of Notch, NotchΔE substrate, is further cleaved by γ-secretase at the S3 site (S3 cleavage) and releases Nβ and the Notch intracellular domain (NICD). NICD is translocated to the nucleus to regulate transcription genes such as Hes and Hey.
Gain or loss of function of PS
The common feature of PSEN1 or PSEN2 FAD mutations is the increased Aβ42/40 ratio. However, it has been debated whether it is due to a gain or loss of PS function that results in an increased Aβ42/40 ratio113. An Aβ42/40 ratio increase could be due to increased Aβ42 production, decreased Aβ40 production, or a combination of both111. Analysis of the formation of substrate CTFs, ICDs, and Aβ species as the effect of FAD mutations of PSEN1 or PSEN2 on the cleavage of various γ-secretase substrates, such as APP, Notch, syndecan-3, N-cadherin, and β1-integrin, showed that different mutations had a varying effect on substrate processing, indicating “variable” or “partial” loss of PS protein function, and PS2 was less efficient than PS1111. Reconstitution of the PS protein from 138 PS1 FAD mutations with Aph-1aL containing γ-secretase mostly decreased the production of Aβ42 and Aβ40, increased the Aβ42/40 ratio, and suggested the loss of PS1 function114. However, these 138 PS1 FAD mutations also showed that different mutations displayed variations in Aβ42 or Aβ40 production (increase or decrease)114. In addition, further studies addressing the effect of PS FAD mutations on the structure of γ-secretase and how those conformational changes could affect the cleavage of different substrates by γ-secretase remain to be investigated. For instance, E280 in PS1 forms hydrogen bonds with Y159 and Y154102. PS1 E280A (the Columbian mutation) disrupts hydrogen bonds and causes a local conformational change102.
Small molecules targeting γ-secretase
Over the years, small molecule inhibitors and modulators targeting γ-secretase have been developed as potential disease-modifying agents in AD. The main goal is to target γ-secretase and reduce toxic Aβ42 species while sparing other substrate cleavage processing by γ-secretase.
γ-Secretase inhibitors
GSIs bind to the active site of PS and inhibit γ-secretase cleavage, thereby reducing total Aβ production. GSIs such as L-685,45835,115, BrA-1-Bt34, III-31C116, DAPT117, and Merck C57 as well as GSI-based chemical probes have been widely used to study γ-secretase. A GSI-based photoaffinity probe showed that <14% of PS1 is incorporated into active γ-secretase complexes and catalytically active while leaving the rest of PS1 in inactive γ-secretase complexes118. Thus, GSI-based chemical probes are critical to differentiating enzymatically active γ-secretase complexes from inactive complexes119. On the other hand, a co-immunoprecipitation study against γ-secretase complex components pulled down both active and inactive γ-secretase complexes.
In animal studies, GSIs successfully reduced Aβ production. DAPT decreased Aβ levels in the plasma, CSF, or brain of AD transgenic mice117,120. Chronic treatment with LY-411,575 in AD transgenic mice reduced Aβ but also inhibited Notch signaling, leading to side effects121. Semagacestat (LY-450,139) and avagacestat (BMS-708,163) in Tg2576 mice reduced Aβ production while increasing APP-CTF122. However, those GSIs impaired normal cognition in wild-type mice122. Begacestat (GSI-953) reduced Aβ levels in Tg2576 mice123.
In clinical trials, GSIs such as semagacestat (LY-450,139, Eli Lilly) and avagacestat (BMS-708,163, Bristol-Myers Squibb) reduced Aβ production in AD patients124,125. However, the multitude of γ-secretase substrates has made the development of clinically useful inhibitors difficult. Due to the decreased Notch signaling and the accumulation of APP-CTFs122, side effects such as the risk of skin cancer and infection, gastrointestinal bleeding, and worsening cognition led to the pause of clinical trials5,124,125. Therefore, these GSIs are nonselective and inhibit both APP and Notch121,124,126. Avagacestat was reported as a “Notch-sparing” GSI and was shown to have a higher selectivity for APP over Notch cleavage127. However, avagacestat was suggested to be nonselective later based on poor Notch-sparing activity122,128 and its binding site as PS1-NTF128. Another “Notch-sparing” GSI, begacestat (GSI-953, Wyeth/Pfizer), was also discontinued in phase I clinical trial, and the reasons are unclear129. Another concern regarding GSI treatment is the Aβ rebound effect. GSIs at lower doses increased Aβ levels, and discontinuation of GSI treatment was observed with a rebound of Aβ levels130,131. These GSIs target PS1-NTF128. GSIs have been repurposed in the cancer field for Notch signaling inhibition and are currently in clinical trials.
γ-Secretase modulators
Instead of inhibiting the whole γ-secretase activity, modulating γ-secretase activity by γ-secretase modulators (GSMs) has been tested. GSMs are more attractive disease-modifying agents than GSIs because GSMs (1) inhibit selectively aggregation-prone Aβ42 production, (2) increase shorter Aβ37 or Aβ38 species, (3) do not affect the total Aβ production and the accumulation of APP-CTF, and (4) spare Notch processing132.
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen, indomethacin, and sulindac sulfide, were found to modulate γ-secretase and represent first-generation GSMs (NSAID-derived carboxylic acid GSMs)132. These NSAIDs lowered Aβ42 and increased Aβ38 without affecting Notch cleavage133. This Aβ modulation was not due to the inhibition of cyclooxygenase activity, the pharmacological target of NSAIDs133. Sulindac sulfide treatment showed a varying degree of Aβ42 reduction levels while increasing high Aβ38 levels in cells overexpressing PS1 FAD mutants134.
Second-generation GSMs were developed to improve in vivo potency and blood-brain penetrance, including NSAID-derived carboxylic acid GSMs, non-NSAID-derived imidazole GSMs, and natural product-derived GSMs132. Acid GSMs decrease Aβ42, increase Aβ38, and have little effect on Aβ40 levels, total Aβ levels, and NICD production132. GSM-1 (acid GSM, GSM-2, and GSM-10 h as close analogs) reduced Aβ42 in many PS mutants but did not reduce Aβ42 levels in cells overexpressing PS1 L166P or PS2 N141I mutants134,135. GSM-2 improved memory in Tg2576 mice and did not affect cognition in wild-type mice122. Acute and subchronic administration of GSM-10 h decreased Aβ42 with no effect on Notch signaling, and there was no Aβ rebound effect and no accumulation of APP-CTFs (C83 and C99)136,137. E2012 (imidazole GSM) decreased Aβ42, Aβ40, and Aβ39 and increased Aβ37 and slightly Aβ38 without affecting Notch processing138.
Several GSMs have entered AD clinical trials. Tarenflurbil ((R)-flurbiprofen, NSAID GSM, Myriad Genetics & Laboratories) failed in phase III clinical trial due to lack of efficacy139. However, there were no Notch inhibition-related adverse effects140. A safety study in rats indicated that E2012 (Eisai) induced cataracts by inhibiting the final step in cholesterol biosynthesis141. Therefore, E2012 was withdrawn from the phase I clinical trial, and Eisai pursued E2212119. E2212 has a better safety profile than E2012 and was evaluated for safety, tolerability, pharmacokinetics, and pharmacodynamics in healthy subjects in phase I clinical trial140. The most common adverse effect was diarrhea140. PF-06648671 (Pfizer) was well tolerated at single doses in healthy subjects, lowered plasma Aβ40 and Aβ42, and increased Aβ37 and Aβ38142. However, this small molecule was discontinued due to Pfizer’s discontinuation of R&D in neurology in 2018. EVP-0962 (NSAID GSM, Forum Pharmaceuticals, Inc.) was discontinued after the phase II clinical trial, and the results were not reported129. CHF5074 (acid GSM, CereSpir Incorporated, Chiesi Pharmaceuticals, Inc.) was first thought of as a GSM and improved memory and reduced microglial activation in Tg2576 mice143. CHF5074 lowered soluble CD40 ligand levels (microglia activation marker)144 and is considered a microglia modulator145. The natural product GSM NIC5-15 (Humanetics Pharmaceuticals Corporation), which is found in soy, plants, and fruits, reduced Aβ production without affecting Notch processing (ALZFORUM, http://www.alzforum.org)129. Amyloid PET in APP-Swe transgenic mice revealed that chronic treatment with RO5506284 reduced de novo amyloid plaque formation146. The GSM-based brain imaging agent [11C]SGSM-15606 was also developed and showed γ-secretase imaging in the brains of mice and macaques147. Recently, chronic treatment with GSM UCSD-776890 in PSAPP mice reduced amyloid deposition and microgliosis148.
GSI and GSM-binding sites
GSIs bind to the active site of PS and inhibit the γ-secretase cleavage of APP and Notch. The mechanism of how GSMs shift Aβ cleavages from longer Aβ peptides to shorter Aβ peptides and where GSMs bind in γ-secretase complexes are still poorly understood129. Moreover, the physiological role of shorter Aβ peptides (Aβ38 or Aβ37) is not well understood. To understand the mechanism of action of GSMs in modifying γ-secretase activity, researchers have investigated GSM target proteins. Studies have shown that NSAID GSMs bind to APP or γ-secretase132. GSM-1-based photoaffinity probes label PS1 or PS1-NTF, and E2012 targets PS1-NTF in γ-secretase complexes149,150,151. Further identification of potential GSM-binding proteins is important to understand the modulation of γ-secretase and to avoid possible side effects of GSMs in clinical trials.
Various chemical probes have been developed to identify the multiple binding sites within the active γ-secretase complex. The “photophore walking” approach to modifying GSI-based photoaffinity probes with a benzophenone group on P2, P1, P1′, and P3′ of L-685,458 (GSI compounds: L646, GY4, JC8, and L505, respectively) can label subsites such as S2, S1, S1′, and S3′ in the active site of the γ-secretase complex149,152. GSI GY4 photolabeling in the presence of GSM-1 altered the S1 subsite of PS1 and increased GY4 labeling149. These results indicated that GSM-1 (acid GSM) caused an active site shape change (conformational change) in the γ-secretase complex and that there is a GSM-1 allosteric binding site on PS1 apart from the GSI L-685,458 binding site (Fig. 5)149. Photolabeling by E2012-Bpyne (imidazole GSM E2012-based photoaffinity probe) also revealed PS1-NTF as the imidazole GSM-binding site within γ-secretase complexes (Fig. 5)150.

Based on cryo-EM structure studies by Yang et al., there are different binding sites for the active binding site for the transition state analog GSI (TSA GSI) (ex. L-685,458) and for the allosteric binding site for imidazole GSM (ex. E2012) (indicated by asterisks). Based on biochemical studies, there might be an additional allosteric binding site for acid GSM (ex. GSM-1) in PS. Note that the structure of acid GSM-bound γ-secretase has not yet been resolved by cryo-EM. Presenilin (blue), nicastrin (magenta), Aph-1 (green), and Pen-2 (yellow). Rendered from Protein Data Bank entry 7D8X. Structural figures were prepared with UCSF ChimeraX 1.2.5.
In summary, biochemical studies suggest that distinct GSI and GSM modulation sites exist in PS of the γ-secretase complex: binding sites for TSA GSI and allosteric GSMs (acid GSM and imidazole GSM, respectively) (Fig. 5)132. These different classes of small molecules occupy different distinctive sites within the γ-secretase complex. Therefore, they interact and induce conformational changes in γ-secretase complexes, which lead to different Aβ cleavages132. For example, E2012-BPyne shows enhanced labeling of PS1-NTF in the presence of GSI L-685,458150. Recently, cryo-EM structure studies confirmed the different binding sites for TSA GSI (L-685,458) and imidazole GSM (E2012) found in the γ-secretase complex (Fig. 5)112. L-685,458 binds the active site of PS1, while the imidazole GSM E2012 binds to the allosteric binding site in PS1112. For a non-TSA GSI, semagacestat occupies the same location as APP (C99) and Notch (N100)112. It suggests that semagacestat could block hybrid β sheet formation between substrates and PS1, therefore inhibiting substrate cleavages112. Another non-TSA GSI (avagacestat) also occupies a similar binding site as semagacestat except with some variations112. L-685,458, semagacestat, and avagacestat share the same binding pocket in PS1, whereas L-685,458 has an additional unique binding pocket112. Co-incubation with L-685,458 and E2012 also showed that E2012 binds to the interface between Nct and PS1 (Fig. 5)112. Yang et al.112 suggested that GSIs and GSMs could be used in combination for a synergistic effect, and this structural information could also improve the design of substrate-selective small molecules for AD.
γ-Secretase modulatory proteins (GSMPs)
Bateman et al.153 reported that Aβ production and clearance rates per hour in human CSF are 7.6% and 8.3%, respectively. Sporadic AD (SAD) human brain gray matter has 4.8 mg more total Aβ than healthy controls154. This gives the estimated Aβ accumulation rate in the brain ~28 ng/hour, and a 2–5% slight increase in Aβ deposition could lead to AD in ~20 years154. For FAD, PS FAD mutations increase the ratio of Aβ42/40, which leads to AD over several decades155. Therefore, reducing Aβ production by a few percent154 by modulating γ-secretase activity with GSMs and/or other means could be therapeutic for AD patients.
Although PS, nicastrin, Aph-1, and Pen-2 are essential subunits for γ-secretase activity50, it is plausible that other transiently binding proteins could regulate γ-secretase activity and/or substrate specificity in different types of tissues, cells, or subcellular organelles. In addition, the reported size of the γ-secretase complex varies between 200 and 2000 kDa50,52,55, indicating that there is a possibility of unknown components being present in the γ-secretase complex. Moreover, most of the γ-secretase complexes are inactive, while <14% are enzymatically active118. How the activation of inactive γ-secretase complexes occurs is still unclear119. It is possible that the binding of GSMPs could shift γ-secretase complexes from inactive to active. Thus, these GSMPs could be new targets to modulate γ-secretase activity in AD. Over the years, several GSMPs have been identified by using different methods: multiple purification columns followed by amino acid sequencing156, IP followed by mass spectrometry (MS)62, and tandem affinity purification157. To capture GSMPs in enzymatically active γ-secretase complexes, researchers used GSI pulldown or GSM photolabeling followed by MS158,159. The following are some of the GSMPs reported.
CD147
CD147 (also known as basigin, extracellular matrix metalloproteinase inducer (EMMPRIN)) is a transmembrane glycoprotein with two Ig-like domains, and CD147 was initially identified as a regulatory subunit of γ-secretase156. CD147 is ubiquitously expressed in various cells and tissues160 and is suggested to be involved in many biological functions, such as neural-glial cell interactions, reproduction, neural function, inflammation, protein trafficking, and tumor invasion156. The deletion of CD147 in mice has resulted in severe defects in nervous system development, spatial learning deficits, and working memory deficits161. Co-immunoprecipitation with anti-PS1-CTF and nicastrin antibodies indicated that CD147 is present in the γ-secretase complex156. Suppression of CD147 expression by siRNA resulted in dosage-dependent increases in the Aβ40 and Aβ42 levels without changes in the expression levels of the other γ-secretase components or APP substrates156. However, it was later questioned whether CD147 is associated with the γ-secretase complex. GSI pulldown using Merck C (biotinylated GSI L-685,458) did not co-purify CD147, indicating that CD147 is not engaged in active γ-secretase complexes162. Vetrivel et al.163 also reported that CD147 degrades Aβ and that it is independent of γ-secretase activity.
TMP21
TMP21 (also known as p23) was identified by immunoprecipitation with an anti-PS antibody followed by MS62. TMP21 is a type I transmembrane protein164, is a member of the p24 cargo-protein family164 and is involved in protein transport and quality control in the ER and Golgi165. When TMP21 was suppressed by siRNA, Aβ production (Aβ40 and Aβ42) was increased62,166. TMP21 might have two pools. The major pool joins the p23 cargo family without affecting Aβ production, and the minor makes a complex with PS162. Since TMP21 siRNA affects Aβ production (γ-cleavage site) but not AICD or NICD production (ε-cleavage site), TMP21 in the minor pool regulates only γ-cleavage62. The transmembrane domain of TMP21 interacts with γ-secretase to alter γ-cleavage167. Later, it was questioned whether TMP21 is truly a GSMP. It was reported that TMP21 also plays a role in APP trafficking, which affects Aβ production168. In addition, GSI Merck C (L-685,458 derivative) using detergent-solubilized human cell line membranes did not pulldown TMP21, suggesting that TMP21 might not be a part of active γ-secretase complexes162,168. However, TMP21 was found to be associated with endogenous active γ-secretase complexes using GSI GCB (L-685,458 derivative) in the brain159 and brain lipid rafts169. This discrepancy between cell lines and brain studies might be because TMP21 in the minor pool is transiently associated with γ-secretase, and different materials and methods were used62,159,162,169. Interestingly, it was easier to detect TMP21 in lipid raft-associated γ-secretase than in detergent-solubilized membranes from the brain159,169. For AD, TMP21 protein expression levels (normalized to nicastrin protein levels) were decreased in the brains of SAD and FAD patients170, and the TMP21 SNP rs12435391 was associated with SAD171.
GSMPs in membrane microdomains
Previously, it was reported that γ-secretase is localized at membrane microdomains called lipid rafts86,89,90,172. Several GSMPs in lipid raft-like microdomains or lipid rafts were identified. Proteins in the tetraspanin web were identified as GSMPs: EWI-F, CD81, CD98hc, and CD9157. Members of this family form lipid raft-like microdomains in cellular membranes157 and have a role in intracellular and intercellular processes, cell fusion, cell proliferation, adhesion, and migration173. EWI proteins (EWI-F) form a primary interaction with tetraspanins (CD81), followed by a secondary interaction with integrins (β1-integrin), and the tetraspanin web makes a tertiary interaction with the γ-secretase complex157. Suppressing CD81, EWI-F, or CD98hc by siRNA decreased Aβ production157. CD81, CD9, and EWI-F are in γ-secretase complexes, as shown by Aβ production in co-immunoprecipitates157. The α-secretase ADAM10 was also associated with tetraspanins for the cleavage of TNF-α and epidermal growth factor (EGF)174, and a new APP processing model by α- and γ-secretases in the tetraspanin web was proposed175,176.
GSI pulldown using lipid rafts from the brain identified several endogenous GSMPs that regulate active γ-secretase. Voltage-dependent anion channel 1 (VDAC1, also known as porin) and contactin-associated protein 1 (CNTNAP1, also known as Caspr) are associated with active γ-secretase complexes in brain lipid rafts, and silencing those genes in HEK293-APP695 cells decreased Aβ production169. VDAC is a major protein at the outer mitochondrial membrane177. A new member of the VDAC family, B-36 VDAC at 36 kDa, was found during the purification of the GABAA receptor and was localized at the membrane of nonpyramidal neurons in the human prefrontal cortex178. VDAC at the plasma membrane of neurons was also reported179. VDAC accumulates around amyloid plaques in APP/PS1 Tg mice177. Nitrated VDAC1 protein was increased in the hippocampus of AD brains180, and VDAC and estrogen receptor alpha in caveolae are highly expressed in AD human brains179. CNTNAP1 and contactin are essential for forming the paranodal junction in myelinated axons181. Contactin interacts with APP in neurons and brains182,183. CNTNAP1 interacted with APP, and overexpression of CNTNAP1 decreased Aβ production in HEK293 cells overexpressing APP V717F (Indiana mutant)184. Erlin-2, which is known to be located at lipid rafts from the ER, is also engaged in active γ-secretase from lipid rafts to regulate Aβ levels185. VDAC1, Erlin-1, and Erlin-2 were previously shown to interact with PS157. Flotillin-2, syntaxin-binding protein 1, solute carrier family 2 member 3, and growth-associated protein 43 were also found as γ-secretase-associated proteins in lipid rafts186.
Endogenous GSMPs in the brain
Using a biotinylated GSI, GCB (GSI coupled to biotin via a cleavable linker, L-685,458 derivative), in the brain instead of using overexpressed cell lines identified several endogenous GSMPs. Previously, a yeast two-hybrid screening identified a synaptic plasma membrane protein, syntaxin 1A, which binds to PS1187. Less than 1% of syntaxin 1 associates with endogenous active γ-secretase complexes in brain membranes159 and brain lipid rafts169. Proton myoinositol cotransporter (SLC2A13) is another endogenous brain GSMP that regulates Aβ production without affecting Notch processing188.
GSI GCB also pulled down several endogenous GSMPs from synapses in the brain, such as NADH dehydrogenase iron-sulfur protein 7 (NDUFS7) from synaptic vesicles and tubulin polymerization promoting protein (TPPP) from synaptic membranes189. Silencing NDUFS7 decreased Aβ levels, while TPPP increased Aβ production, and both proteins were co-immunoprecipitated with Nct and PS1-CTF in the human brain189. Previously, it was shown that monoamine oxidase B (MAO-B) is increased in AD brains and platelets190. Overexpression of MAO-B increased Aβ production, and MAO-B was associated with active γ-secretase191. MAO-B levels were increased in neurons of AD human brains191.
γ-Secretase activating protein
The treatment for chronic myeloid leukemia, Gleevec (an anticancer drug, imatinib mesylate, STI571), was shown to reduce Aβ production but spare Notch cleavage192. A biotinylated derivative of imatinib identified GSAP (γ-secretase activating proteins)-16 kDa, and GSAP is the C-terminal region of an uncharacterized protein, pigeon homologue protein (PION)63. GSAP-16 kDa, γ-secretase, and APP-CTF form a tertiary complex63. Knockdown (KD) of GSAP by siRNA reduced Aβ production and did not change NICD production, and recombinant GSAP-16 kDa increased Aβ production63. KD of GSAP by crossing AD X 2 mice with doxycycline-inducible GSAP RNAi mice resulted in the reduction of Aβ and amyloid plaques in the brain63. Knockout (KO) of GSAP also decreased Aβ production while sparing Notch cleavage193. Overexpression of FL GSAP in GSAP KO cells increased Aβ generation193. Treatment with imatinib in 3XTg mice (mutant APP, mutant PS1, and mutant MAPT) decreased GSAP-16 kDa protein, Aβ production, brain Aβ deposits, and phosphorylated tau194.
However, the relationship between GSAP and γ-secretase for Aβ generation was later questioned by several groups195. Hussain et al.195 reported that KD of GSAP decreased Aβ levels, but overexpression of GSAP-16 kDa did not increase Aβ production, and APP-CTF/PS1-CTF complexes were immunoprecipitated without GSAP. Hussain et al.195 suggested that Aβ reduction by KD of GSAP might be due to some effects on the trafficking or assembly of γ-secretase but not a direct effect of GSAP on γ-secretase. In addition, imatinib did not decrease Aβ generation in cell lines and in vivo while sparing Notch processing in cell lines. Another study also showed that, unlike GSI L-685,458, imatinib treatment did not inhibit Aβ production in cell lines, mouse primary neurons, and differentiated human embryonic stem cells196. In humans, imatinib treatment in chronic myeloid leukemia patients for up to 12 months also did not result in an Aβ decrease in plasma196. In contrast, overexpression of GSAP-FL in GSAP KO cells rescued γ-secretase activity, and the dual GSI photoprobe L631 for PS1-NTF and PS1-CTF labeled PS1-NTF, PS1-CTF, and FL PS1 when GSAP-FL was overexpressed in GSAP KO cells compared to KO cells193. This result suggested that the presence of GSAP aligned PS1-NTF and PS1-CTF in a specific confirmation with higher γ-secretase activity for Aβ cleavage193. In human brains, an immunohistochemistry study showed that GSAP-positive deposits are present both in control and AD brains, while the quantification of GSAP-positive deposits is higher in AD brains, and these GSAP-positive deposits are closely localized to PS1 and Aβ deposits in AD brains197. It was also reported that the GSAP SNP rs4727380 was associated with APOE4 noncarriers of AD patients from Han Chinese in a small sample size198.
GSMPs induced by other factors
Hif-1α was identified as a GSMP for Notch processing. Hif-1 expression is upregulated by aging in the frontal cortex of the human brain199, and stroke increases the risk for dementia200. Brain ischemia/hypoxia-induced Aβ deposits in the human brain201. The BACE1 gene contains a hypoxia response element (HRE) in the promoter region, and hypoxia increases BACE1 protein expression as well as β-secretase cleavage for APP202. A transcription factor, Hif-1α (hypoxia-inducible factor-1α), works as an oxygen sensor, and Hif-1α is degraded by the ubiquitin-proteasome system under normoxia203. Under hypoxia, the canonical hypoxic response leads to the binding of Hif-1α/Hif-1β to HRE elements in the promoter regions of several genes, such as vascular endothelial growth factor, erythropoietin (Epo), and glucose transporters 1, for angiogenesis, erythropoiesis, and energy metabolism203. Gustafsson et al.204 discovered crosstalk between the noncanonical pathway of Hif-1α and Notch signaling. Under hypoxia, Hif-1α binds to the NICD and induces Notch downstream genes such as Hes and Hey for the undifferentiated cell state in the stem cell population204. Villa et al.64 found that hypoxia also increases active γ-secretase complex formation and upregulates γ-secretase activity to cleave Notch. Nontranscriptional Hif-1α converts the pool of inactive γ-secretase to active γ-secretase, and GSI-34 decreases hypoxia-induced cell invasion and metastatic progression in cells and animal models of breast cancer64.
Another environmental factor, such as stress, activates G protein-coupled receptors (GPCRs), such as β2-adrenergic receptor (β2-AR) and δ-opioid receptor205. β2-AR agonists stimulate Aβ production via (1) the association with PS1, (2) the endocytosis of the receptor, and (3) the trafficking of γ-secretase to late endosomes and lysosomes205. High-throughput functional genomics screening identified another GPCR, orphan GPR3, that modulates Aβ production92. Overexpression of GPR3 increased (1) the expression of mature γ-secretase complexes at 440 kDa, (2) the localization of γ-secretase complexes to lipid rafts, and (3) Aβ and AICD production, but (4) did not change Notch cleavage92. Crossing APP/PS1 mice with GPR3 KO mice also decreased Aβ production, and GPR3 was expressed in the brains of SAD patients92.
GPCRs require adaptor proteins such as arrestins to prevent further G protein-mediated signaling206. β-Arrestin1 is highly expressed in the brain206, and β-arrestin1 KO mice exhibit reduced Aβ production and spared Notch cleavage207. β-arrestin1 interacts only with Aph-1 in γ-secretase, and overexpression of β-arrestin1 enhanced mature γ-secretase complex formation at 440 kDa207. KO of β-arrestin1 in APP/PS1 mice decreased Aβ production and improved memory deficits207. Stress-associated endoplasmic reticulum protein 1 (SERP1) was also reported to regulate the assembly of γ-secretase complexes and contribute to Aβ pathogenesis208. SERP1 interacts with the Aph-1a/Nct subcomplex of γ-secretase and increases γ-secretase activity for Aβ generation but reduces Notch processing208.
GSMP in neuroinflammation
Recently, IFITM3 (interferon-induced transmembrane protein 3, also known as fragilis) was identified as an imidazole GSM, E2012, binding protein158. Photolabeling with E2012-BPyne (an E2012-based photoaffinity probe) followed by LC-MS/MS identified IFITM3 at 15 kDa as a GSMP158. IFITM3 plays a role in innate immunity as an antiviral protein that restricts viral protein entry into host cell membranes by inhibiting membrane fusion209. IFITM3 KO mice are susceptible to viral infections210. Previously, microarray analysis and RT-PCR showed 19.9- and 3.4-fold increases in IFITM3 in SAD brains211. Hur et al.158 showed that IFITM3 binds to PS1-NTF in active γ-secretase complexes and regulates γ-secretase activity for Aβ production (Aβ40 and Aβ42) (Fig. 6). KD or KO of IFITM3 decreased Aβ production, and overexpression of IFITM3 increased Aβ levels in IFITM3 KO cells158. Moreover, crossing IFITM3 KO mice with 5XFAD Tg mice decreased Aβ production and amyloid plaque formation in the cortex and hippocampus158. Aging mouse models also showed increased IFITM3 levels, γ-secretase activity, and active IFITM3-γ-secretase complex formation levels by aging158. A positive correlation between the amount of active IFITM3-γ-secretase complexes and the high γ-secretase activity resulting in high Aβ production was shown in the subsets of SAD patient brains expressing high IFITM3 protein levels158. Proinflammatory cytokines such as Type I IFN or Type II IFN can induce IFITM3 protein expression, increase the engagement of IFITM3 in active γ-secretase complexes, and increase Aβ production in mouse primary cortical neurons158. This result shows the direct link between inflammation and Aβ production via IFITM3-γ-secretase in neurons158,212,213. IFITM3 modulates γ-secretase under inflammation in neurons and astrocytes and may contribute to aging and the pathogenesis of AD158. The “antimicrobial protection hypothesis of AD” proposes that Aβ is beneficial as an antimicrobial peptide and that Aβ fibrilization entraps bacteria and viruses as an innate immune response to pathogens214. The involvement of IFITM3 in Aβ production might suggest the role of the “neuronal innate immune response” against pathogens, and Aβ has resulted as a protective pathway against infection212. At the same time, the accumulation of Aβ poses a risk of developing AD158. Further studies on regulating other γ-secretase substrates by IFITM3-γ-secretase complexes are needed to understand possible adverse effects when targeting IFITM3 in AD.

Normally, active γ-secretase cleaves its substrate to release Aβ. Under inflammatory conditions such as aging and infection, proinflammatory cytokines are induced by microglia and astrocytes. These cytokines upregulate IFITM3 protein expression in astrocytes and neurons, which in turn increases the processing of APP-CTF (C99) by active IFITM3-γ-secretase complexes to produce Aβ40 and Aβ42. The accumulation of amyloid leads to amyloid build-ups in the brain. Note that less than 14% of γ-secretase complexes are enzymatically active, while the rest are inactive.
Conclusion
Aβ is the key driver in AD according to the amyloid cascade hypothesis. Since γ-secretase cleaves its immediate substrate APP-CTF to release Aβ, which causes AD, and its unique biology as a transmembrane protein complex enzyme is still much to be learned, γ-secretase is still interesting to study. How γ-secretase cleaves over 100 substrates and how those signaling cascades could result in different physiological functions remain to be determined in the future. To validate γ-secretase as an Aβ modifying drug, further studies on the regulation/modulation of γ-secretase by GSMs and transiently binding GSMPs are needed. In addition, the effects of GSMs and GSMPs on different substrate processing need to be elucidated. This knowledge could advance the development of AD-modifying drugs by selectively inhibiting APP processing by γ-secretase.
References
Terry, R. D. & Davies, P. Dementia of the Alzheimer type. Annu. Rev. Neurosci. 3, 77–95 (1980).
St George-Hyslop, P. H. Piecing together Alzheimer’s. Sci. Am. 283, 76–83 (2000).
Perl, D. P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 77, 32–42 (2010).
Bateman, R. J. et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804 (2012).
De Strooper, B., Vassar, R. & Golde, T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 6, 99–107 (2010).
Sevigny, J. et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56 (2016).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Bettens, K., Sleegers, K. & Van Broeckhoven, C. Current status on Alzheimer disease molecular genetics: from past, to present, to future. Hum. Mol. Genet. 19, R4–R11 (2010).
Selkoe, D. J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 81, 741–766 (2001).
Xu, X. Gamma-secretase catalyzes sequential cleavages of the AbetaPP transmembrane domain. J. Alzheimers Dis. 16, 211–224 (2009).
Gu, Y. et al. Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J. Biol. Chem. 276, 35235–35238 (2001).
Sastre, M. et al. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2, 835–841 (2001).
Weidemann, A. et al. A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41, 2825–2835 (2002).
Yu, C. et al. Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in gamma -secretase processing of the APP and Notch1 transmembrane domains. J. Biol. Chem. 276, 43756–43760 (2001).
Qi-Takahara, Y. et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J. Neurosci. 25, 436–445 (2005).
Zhao, G. et al. Identification of a new presenilin-dependent zeta-cleavage site within the transmembrane domain of amyloid precursor protein. J. Biol. Chem. 279, 50647–50650 (2004).
Takami, M. et al. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 (2009).
Matsumura, N. et al. gamma-Secretase associated with lipid rafts: multiple interactive pathways in the stepwise processing of beta-carboxyl-terminal fragment. J. Biol. Chem. 289, 5109–5121 (2014).
Olsson, F. et al. Characterization of intermediate steps in amyloid beta (Abeta) production under near-native conditions. J. Biol. Chem. 289, 1540–1550 (2014).
Welander, H. et al. Abeta43 is more frequent than Abeta40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 110, 697–706 (2009).
Portelius, E. et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Mol. Neurodegener. 5, 2 (2010).
Portelius, E. et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 120, 185–193 (2010).
Iwatsubo, T. et al. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13, 45–53 (1994).
Roher, A. E. et al. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 10836–10840 (1993).
Roher, A. E., Palmer, K. C., Yurewicz, E. C., Ball, M. J. & Greenberg, B. D. Morphological and biochemical analyses of amyloid plaque core proteins purified from Alzheimer disease brain tissue. J. Neurochem. 61, 1916–1926 (1993).
Wolfe, M. S. & Kopan, R. Intramembrane proteolysis: theme and variations. Science 305, 1119–1123 (2004).
Laudon, H. et al. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 280, 35352–35360 (2005).
Levy-Lahad, E. et al. A familial Alzheimer’s disease locus on chromosome 1. Science 269, 970–973 (1995).
Kimberly, W. T. & Wolfe, M. S. Identity and function of gamma-secretase. J. Neurosci. Res. 74, 353–360 (2003).
Thinakaran, G. et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190 (1996).
De Strooper, B. et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 (1998).
Herreman, A. et al. Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2, 461–462 (2000).
Zhang, Z. et al. Presenilins are required for gamma-secretase cleavage of beta-APP and transmembrane cleavage of Notch-1. Nat. Cell Biol. 2, 463–465 (2000).
Esler, W. P. et al. Transition-state analogue inhibitors of gamma-secretase bind directly to presenilin-1. Nat. Cell Biol. 2, 428–434 (2000).
Li, Y. M. et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694 (2000).
Yu, G. et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54 (2000).
Francis, R. et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev. Cell 3, 85–97 (2002).
Goutte, C., Tsunozaki, M., Hale, V. A. & Priess, J. R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl Acad. Sci. USA 99, 775–779 (2002).
Leem, J. Y. et al. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J. Biol. Chem. 277, 19236–19240 (2002).
Arawaka, S. et al. The levels of mature glycosylated nicastrin are regulated and correlate with gamma-secretase processing of amyloid beta-precursor protein. J. Neurochem. 83, 1065–1071 (2002).
Edbauer, D., Winkler, E., Haass, C. & Steiner, H. Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc. Natl Acad. Sci. USA 99, 8666–8671 (2002).
Kimberly, W. T. et al. Complex N-linked glycosylated nicastrin associates with active gamma-secretase and undergoes tight cellular regulation. J. Biol. Chem. 277, 35113–35117 (2002).
Steiner, H. et al. PEN-2 is an integral component of the gamma-secretase complex required for coordinated expression of presenilin and nicastrin. J. Biol. Chem. 277, 39062–39065 (2002).
Hebert, S. S. et al. Coordinated and widespread expression of gamma-secretase in vivo: evidence for size and molecular heterogeneity. Neurobiol. Dis. 17, 260–272 (2004).
Donoviel, D. B. et al. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 13, 2801–2810 (1999).
Herreman, A. et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl Acad. Sci. USA 96, 11872–11877 (1999).
Nguyen, V. et al. Loss of nicastrin elicits an apoptotic phenotype in mouse embryos. Brain Res. 1086, 76–84 (2006).
Serneels, L. et al. Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proc. Natl Acad. Sci. USA 102, 1719–1724 (2005).
Campbell, W. A. et al. Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen-2) demonstrate excessive p53-dependent apoptosis and neuronal loss. J. Neurochem. 96, 1423–1440 (2006).
Edbauer, D. et al. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 5, 486–488 (2003).
Hayashi, I. et al. Selective reconstitution and recovery of functional gamma-secretase complex on budded baculovirus particles. J. Biol. Chem. 279, 38040–38046 (2004).
Kimberly, W. T. et al. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl Acad. Sci. USA 100, 6382–6387 (2003).
Takasugi, N. et al. The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441 (2003).
Zhang, L. et al. Characterization of the reconstituted gamma-secretase complex from Sf9 cells co-expressing presenilin 1, nicastrin [correction of nacastrin], aph-1a, and pen-2. Biochemistry 44, 4450–4457 (2005).
Farmery, M. R. et al. Partial purification and characterization of gamma-secretase from post-mortem human brain. J. Biol. Chem. 278, 24277–24284 (2003).
Capell, A. et al. Gamma-secretase complex assembly within the early secretory pathway. J. Biol. Chem. 280, 6471–6478 (2005).
Beher, D. et al. In vitro characterization of the presenilin-dependent gamma-secretase complex using a novel affinity ligand. Biochemistry 42, 8133–8142 (2003).
Ahn, K. et al. Activation and intrinsic gamma-secretase activity of presenilin 1. Proc. Natl Acad. Sci. USA 107, 21435–21440 (2010).
Fraering, P. C. et al. Purification and characterization of the human gamma-secretase complex. Biochemistry 43, 9774–9789 (2004).
Escamilla-Ayala, A. A. et al. Super-resolution microscopy reveals majorly mono- and dimeric presenilin1/gamma-secretase at the cell surface. Elife 9, e56679 (2020).
Sato, T. et al. Active gamma-secretase complexes contain only one of each component. J. Biol. Chem. 282, 33985–33993 (2007).
Chen, F. et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature 440, 1208–1212 (2006).
He, G. et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98 (2010).
Villa, J. C. et al. Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell Rep. 8, 1077–1092 (2014).
Greenfield, J. P. et al. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl Acad. Sci. USA 96, 742–747 (1999).
Small, S. A. & Gandy, S. Sorting through the cell biology of Alzheimer’s disease: intracellular pathways to pathogenesis. Neuron 52, 15–31 (2006).
Cataldo, A. M. et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 25, 1263–1272 (2004).
Takahashi, R. H. et al. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 161, 1869–1879 (2002).
Sisodia, S. S. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl Acad. Sci. USA 89, 6075–6079 (1992).
Koo, E. H. & Squazzo, S. L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 269, 17386–17389 (1994).
Zhang, J. et al. Subcellular distribution and turnover of presenilins in transfected cells. J. Biol. Chem. 273, 12436–12442 (1998).
Lah, J. J. & Levey, A. I. Endogenous presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol. Cell. Neurosci. 16, 111–126 (2000).
Chyung, J. H., Raper, D. M. & Selkoe, D. J. Gamma-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J. Biol. Chem. 280, 4383–4392 (2005).
Frykman, S. et al. Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS One 5, e8948 (2010).
Beher, D. et al. Proteolytic fragments of Alzheimer’s disease-associated presenilin 1 are present in synaptic organelles and growth cone membranes of rat brain. J. Neurochem. 72, 1564–1573 (1999).
Efthimiopoulos, S. et al. Enrichment of presenilin 1 peptides in neuronal large dense-core and somatodendritic clathrin-coated vesicles. J. Neurochem. 71, 2365–2372 (1998).
Lah, J. J. et al. Light and electron microscopic localization of presenilin-1 in primate brain. J. Neurosci. 17, 1971–1980 (1997).
Ribaut-Barassin, C. et al. Alzheimer’s disease proteins in cerebellar and hippocampal synapses during postnatal development and aging of the rat. Neuroscience 120, 405–423 (2003).
Jutras, I. et al. Gamma-secretase is a functional component of phagosomes. J. Biol. Chem. 280, 36310–36317 (2005).
Pasternak, S. H. et al. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 278, 26687–26694 (2003).
Chun, J., Yin, Y. I., Yang, G., Tarassishin, L. & Li, Y. M. Stereoselective synthesis of photoreactive peptidomimetic gamma-secretase inhibitors. J. Org. Chem. 69, 7344–7347 (2004).
Hansson, C. A. et al. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J. Biol. Chem. 279, 51654–51660 (2004).
Crump, C. J. et al. Development of sulfonamide photoaffinity inhibitors for probing cellular gamma-secretase. ACS Chem. Neurosci. 7, 1166–1173 (2016).
Simons, K. & Ikonen, E. Functional rafts in cell membranes. Nature 387, 569–572 (1997).
Ehehalt, R., Keller, P., Haass, C., Thiele, C. & Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123 (2003).
Urano, Y. et al. Association of active gamma-secretase complex with lipid rafts. J. Lipid Res. 46, 904–912 (2005).
Vetrivel, K. S. et al. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 280, 25892–25900 (2005).
Hattori, C. et al. BACE1 interacts with lipid raft proteins. J. Neurosci. Res. 84, 912–917 (2006).
Wahrle, S. et al. Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol. Dis. 9, 11–23 (2002).
Hur, J. Y. et al. Active gamma-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 275, 1174–1187 (2008).
Osenkowski, P., Ye, W., Wang, R., Wolfe, M. S. & Selkoe, D. J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 283, 22529–22540 (2008).
Thathiah, A. et al. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science 323, 946–951 (2009).
Cheng, H. et al. Mechanisms of disease: new therapeutic strategies for Alzheimer’s disease-targeting APP processing in lipid rafts. Nat. Clin. Pract. Neurol. 3, 374–382 (2007).
Rajendran, L., Knolker, H. J. & Simons, K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 9, 29–42 (2010).
Rajendran, L. et al. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 320, 520–523 (2008).
Fraering, P. C. Structural and functional determinants of gamma-secretase, an intramembrane protease implicated in Alzheimer’s disease. Curr. Genomics 8, 531–549 (2007).
Kopan, R. & Ilagan, M. X. Gamma-secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504 (2004).
Lazarov, V. K. et al. Electron microscopic structure of purified, active gamma-secretase reveals an aqueous intramembrane chamber and two pores. Proc. Natl Acad. Sci. USA 103, 6889–6894 (2006).
Bai, X. C. et al. An atomic structure of human gamma-secretase. Nature 525, 212–217 (2015).
Yang, G. et al. Structural basis of Notch recognition by human gamma-secretase. Nature 565, 192–197 (2019).
Zhou, R. et al. Recognition of the amyloid precursor protein by human gamma-secretase. Science 363, eaaw0930 (2019).
Zhou, R., Yang, G. & Shi, Y. Macromolecular complex in recognition and proteolysis of amyloid precursor protein in Alzheimer’s disease. Curr. Opin. Struct. Biol. 61, 1–8 (2020).
Bolduc, D. M., Montagna, D. R., Gu, Y., Selkoe, D. J. & Wolfe, M. S. Nicastrin functions to sterically hinder gamma-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl Acad. Sci. USA 113, E509–E518 (2016).
Wolfe, M. S. gamma-Secretase in biology and medicine. Semin. Cell Dev. Biol. 20, 219–224 (2009).
Wolfe, M. S. Substrate recognition and processing by gamma-secretase. Biochim. Biophys. Acta Biomembr. 1862, 183016 (2020).
Guner, G. & Lichtenthaler, S. F. The substrate repertoire of gamma-secretase/presenilin. Semin. Cell Dev. Biol. 105, 27–42 (2020).
Lee, S. H. et al. APP family regulates neuronal excitability and synaptic plasticity but not neuronal survival. Neuron 108, 676–690 (2020).
Struhl, G. & Adachi, A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 6, 625–636 (2000).
Griciuc, A. & Tanzi, R. E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 34, 228–236 (2021).
Wunderlich, P. et al. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036 (2013).
Bentahir, M. et al. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem. 96, 732–742 (2006).
Yang, G. et al. Structural basis of gamma-secretase inhibition and modulation by small molecule drugs. Cell 184, 521–533 (2021).
De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146 (2007).
Sun, L., Zhou, R., Yang, G. & Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl Acad. Sci. USA 114, E476–E485 (2017).
Shearman, M. S. et al. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid beta-protein precursor gamma-secretase activity. Biochemistry 39, 8698–8704 (2000).
Esler, W. P. et al. Activity-dependent isolation of the presenilin- gamma -secretase complex reveals nicastrin and a gamma substrate. Proc. Natl Acad. Sci. USA 99, 2720–2725 (2002).
Dovey, H. F. et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 (2001).
Lai, M. T. et al. Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J. Biol. Chem. 278, 22475–22481 (2003).
Nie, P., Vartak, A. & Li, Y. M. gamma-Secretase inhibitors and modulators: mechanistic insights into the function and regulation of gamma-secretase. Semin. Cell Dev. Biol. 105, 43–53 (2020).
Lanz, T. A. et al. The gamma-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester reduces A beta levels in vivo in plasma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice. J. Pharmacol. Exp. Ther. 305, 864–871 (2003).
Wong, G. T. et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 279, 12876–12882 (2004).
Mitani, Y. et al. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 32, 2037–2050 (2012).
Martone, R. L. et al. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 331, 598–608 (2009).
Doody, R. S. et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350 (2013).
Coric, V. et al. Safety and tolerability of the gamma-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 69, 1430–1440 (2012).
Siemers, E. R. et al. Effects of a gamma-secretase inhibitor in a randomized study of patients with Alzheimer disease. Neurology 66, 602–604 (2006).
Gillman, K. W. et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable gamma-secretase inhibitor. ACS Med. Chem. Lett. 1, 120–124 (2010).
Crump, C. J. et al. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 51, 7209–7211 (2012).
Zhao, J., Liu, X., Xia, W., Zhang, Y. & Wang, C. Targeting amyloidogenic processing of APP in Alzheimer’s disease. Front. Mol. Neurosci. 13, 137 (2020).
Lanz, T. A., Hosley, J. D., Adams, W. J. & Merchant, K. M. Studies of Abeta pharmacodynamics in the brain, cerebrospinal fluid, and plasma in young (plaque-free) Tg2576 mice using the gamma secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-di hydro-5H-dibenzo[b,d]azepin-7-yl]-L-alaninamide (LY-411575). J. Pharmacol. Exp. Ther. 309, 49–55 (2004).
Lanz, T. A. et al. Concentration-dependent modulation of amyloid-beta in vivo and in vitro using the gamma-secretase inhibitor, LY-450139. J. Pharmacol. Exp. Ther. 319, 924–933 (2006).
Crump, C. J., Johnson, D. S. & Li, Y. M. Development and mechanism of gamma-secretase modulators for Alzheimer’s disease. Biochemistry 52, 3197–3216 (2013).
Weggen, S. et al. A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216 (2001).
Page, R. M. et al. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J. Biol. Chem. 283, 677–683 (2008).
Kretner, B. et al. Attenuated Abeta42 responses to low potency gamma-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J. Biol. Chem. 286, 15240–15251 (2011).
Li, T. et al. Gamma-secretase modulators do not induce Abeta-rebound and accumulation of beta-C-terminal fragment. J. Neurochem. 121, 277–286 (2012).
Hawkins, J. et al. Dynamics of Abeta42 reduction in plasma, CSF and brain of rats treated with the gamma-secretase modulator, GSM-10h. Neurodegener. Dis. 8, 455–464 (2011).
Borgegard, T. et al. First and second generation gamma-secretase modulators (GSMs) modulate amyloid-beta (Abeta) peptide production through different mechanisms. J. Biol. Chem. 287, 11810–11819 (2012).
Green, R. C. et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564 (2009).
Yu, Y. et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel gamma-secretase modulator, E2212, in healthy human subjects. J. Clin. Pharmacol. 54, 528–536 (2014).
Nakano-Ito, K. et al. E2012-induced cataract and its predictive biomarkers. Toxicol. Sci. 137, 249–258 (2014).
Ahn, J. E. et al. Pharmacokinetic and pharmacodynamic effects of a gamma-secretase modulator, PF-06648671, on CSF amyloid-beta peptides in randomized phase I studies. Clin. Pharmacol. Ther. 107, 211–220 (2020).
Sivilia, S. et al. Multi-target action of the novel anti-Alzheimer compound CHF5074: in vivo study of long term treatment in Tg2576 mice. BMC Neurosci. 14, 44 (2013).
Imbimbo, B. P. et al. Pharmacokinetics and pharmacodynamics of CHF5074 after short-term administration in healthy subjects. Alzheimer Dis. Assoc. Disord. 27, 278–286 (2013).
Mekala, S., Nelson, G. & Li, Y. M. Recent developments of small molecule gamma-secretase modulators for Alzheimer’s disease. RSC Med. Chem. 11, 1003–1022 (2020).
Brendel, M. et al. Amyloid-PET predicts inhibition of de novo plaque formation upon chronic gamma-secretase modulator treatment. Mol. Psychiatry 20, 1179–1187 (2015).
Xu, Y. et al. Molecular imaging of Alzheimer’s disease-related gamma-secretase in mice and nonhuman primates. J. Exp. Med. 217, e20182266 (2020).
Rynearson, K. D. et al. Preclinical validation of a potent gamma-secretase modulator for Alzheimer’s disease prevention. J. Exp. Med. 218, e20202560 (2021).
Crump, C. J. et al. Piperidine acetic acid based gamma-secretase modulators directly bind to Presenilin-1. ACS Chem. Neurosci. 2, 705–710 (2011).
Pozdnyakov, N. et al. gamma-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. J. Biol. Chem. 288, 9710–9720 (2013).
Ohki, Y. et al. Phenylpiperidine-type gamma-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824 (2011).
Chau, D. M., Crump, C. J., Villa, J. C., Scheinberg, D. A. & Li, Y. M. Familial Alzheimer disease presenilin-1 mutations alter the active site conformation of gamma-secretase. J. Biol. Chem. 287, 17288–17296 (2012).
Bateman, R. J. et al. Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 12, 856–861 (2006).
Roberts, B. R. et al. Biochemically-defined pools of amyloid-beta in sporadic Alzheimer’s disease: correlation with amyloid PET. Brain 140, 1486–1498 (2017).
Holtzman, D. M., Morris, J. C. & Goate, A. M. Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr1 (2011).
Zhou, S., Zhou, H., Walian, P. J. & Jap, B. K. CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer’s disease amyloid beta-peptide production. Proc. Natl Acad. Sci. USA 102, 7499–7504 (2005).
Wakabayashi, T. et al. Analysis of the gamma-secretase interactome and validation of its association with tetraspanin-enriched microdomains. Nat. Cell Biol. 11, 1340–1346 (2009).
Hur, J. Y. et al. The innate immunity protein IFITM3 modulates gamma-secretase in Alzheimer’s disease. Nature 586, 735–740 (2020).
Teranishi, Y. et al. Affinity pulldown of gamma-secretase and associated proteins from human and rat brain. J. Cell. Mol. Med. 14, 2675–2686 (2010).
Muramatsu, T. & Miyauchi, T. Basigin (CD147): a multifunctional transmembrane protein involved in reproduction, neural function, inflammation and tumor invasion. Histol. Histopathol. 18, 981–987 (2003).
Naruhashi, K. et al. Abnormalities of sensory and memory functions in mice lacking Bsg gene. Biochem. Biophys. Res. Commun. 236, 733–737 (1997).
Winkler, E. et al. Purification, pharmacological modulation, and biochemical characterization of interactors of endogenous human gamma-secretase. Biochemistry 48, 1183–1197 (2009).
Vetrivel, K. S. et al. Evidence that CD147 modulation of beta-amyloid (Abeta) levels is mediated by extracellular degradation of secreted Abeta. J. Biol. Chem. 283, 19489–19498 (2008).
Blum, R. et al. Tmp21 and p24A, two type I proteins enriched in pancreatic microsomal membranes, are members of a protein family involved in vesicular trafficking. J. Biol. Chem. 271, 17183–17189 (1996).
Jenne, N., Frey, K., Brugger, B. & Wieland, F. T. Oligomeric state and stoichiometry of p24 proteins in the early secretory pathway. J. Biol. Chem. 277, 46504–46511 (2002).
Dolcini, V. et al. TMP21 regulates Abeta production but does not affect caspase-3, p53, and neprilysin. Biochem. Biophys. Res. Commun. 371, 69–74 (2008).
Pardossi-Piquard, R. et al. TMP21 transmembrane domain regulates gamma-secretase cleavage. J. Biol. Chem. 284, 28634–28641 (2009).
Vetrivel, K. S. et al. Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol. Neurodegener. 2, 4 (2007).
Hur, J. Y. et al. Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. J. Biol. Chem. 287, 11991–12005 (2012).
Vetrivel, K. S. et al. Localization and regional distribution of p23/TMP21 in the brain. Neurobiol. Dis. 32, 37–49 (2008).
Zhang, X. et al. A novel alzheimer-associated SNP in Tmp21 increases amyloidogenesis. Mol. Neurobiol. 55, 1862–1870 (2018).
Vetrivel, K. S. et al. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279, 44945–44954 (2004).
Levy, S. & Shoham, T. Protein-protein interactions in the tetraspanin web. Physiol. (Bethesda) 20, 218–224 (2005).
Arduise, C. et al. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J. Immunol. 181, 7002–7013 (2008).
Seipold, L. & Saftig, P. The emerging role of tetraspanins in the proteolytic processing of the amyloid precursor protein. Front. Mol. Neurosci. 9, 149 (2016).
Chen, A. C. et al. Physical and functional interaction between the alpha- and gamma-secretases: a new model of regulated intramembrane proteolysis. J. Cell Biol. 211, 1157–1176 (2015).
Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 41, 425–431 (2009).
Menard, G., Evrard, B., Bureau, M. & Trottier, S. Cerebral distribution of the B-36 VDAC protein in rat, cow and man brain: immunocytochemical study. Cell. Mol. Biol. 40, 295–300 (1994).
Ramirez, C. M. et al. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol. Cell. Neurosci. 42, 172–183 (2009).
Sultana, R. et al. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 22, 76–87 (2006).
Rios, J. C. et al. Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J. Neurosci. 20, 8354–8364 (2000).
Bai, Y. et al. The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteom. 7, 15–34 (2008).
Bamford, R. A. et al. The interaction between contactin and amyloid precursor protein and its role in Alzheimer’s disease. Neuroscience 424, 184–202 (2020).
Fan, L. F. et al. Caspr interaction with Amyloid Precursor Protein reduces amyloid-beta generation in vitro. Neurosci. Lett. 548, 255–260 (2013).
Teranishi, Y. et al. Erlin-2 is associated with active gamma-secretase in brain and affects amyloid beta-peptide production. Biochem. Biophys. Res. Commun. 424, 476–481 (2012).
Inoue, M. et al. Human brain proteins showing neuron-specific interactions with gamma-secretase. FEBS J. 282, 2587–2599 (2015).
Smith, S. K. et al. Identification of syntaxin 1A as a novel binding protein for presenilin-1. Brain Res. Mol. Brain Res. 78, 100–107 (2000).
Teranishi, Y. et al. Proton myo-inositol cotransporter is a novel gamma-secretase associated protein that regulates Abeta production without affecting Notch cleavage. FEBS J. 282, 3438–3451 (2015).
Frykman, S. et al. Identification of two novel synaptic gamma-secretase associated proteins that affect amyloid beta-peptide levels without altering Notch processing. Neurochem. Int. 61, 108–118 (2012).
Adolfsson, R., Gottfries, C. G., Oreland, L., Wiberg, A. & Winblad, B. Increased activity of brain and platelet monoamine oxidase in dementia of Alzheimer type. Life Sci. 27, 1029–1034 (1980).
Schedin-Weiss, S. et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with gamma-secretase and regulates neuronal amyloid beta-peptide levels. Alzheimers Res. Ther. 9, 57 (2017).
Netzer, W. J. et al. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc. Natl Acad. Sci. USA 100, 12444–12449 (2003).
Wong, E. et al. GSAP modulates gamma-secretase specificity by inducing conformational change in PS1. Proc. Natl Acad. Sci. USA 116, 6385–6390 (2019).
Chu, J., Lauretti, E., Craige, C. P. & Pratico, D. Pharmacological modulation of GSAP reduces amyloid-beta levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. J. Alzheimers Dis. 41, 729–737 (2014).
Hussain, I. et al. The role of gamma-secretase activating protein (GSAP) and imatinib in the regulation of gamma-secretase activity and amyloid-beta generation. J. Biol. Chem. 288, 2521–2531 (2013).
Olsson, B. et al. Imatinib treatment and Abeta42 in humans. Alzheimers Dement. 10, S374–S380 (2014).
Satoh, J., Tabunoki, H., Ishida, T., Saito, Y. & Arima, K. Immunohistochemical characterization of gamma-secretase activating protein expression in Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 38, 132–141 (2012).
Zhu, M. et al. Common GSAP promoter variant contributes to Alzheimer’s disease liability. Neurobiol. Aging 35, 2656.e1–2656.e7 (2014).
Lu, T. et al. Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891 (2004).
Pendlebury, S. T. & Rothwell, P. M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018 (2009).
Jendroska, K. et al. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathol. 90, 461–466 (1995).
Sun, X., Bromley-Brits, K. & Song, W. Regulation of beta-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 120, 62–70 (2012).
Sharp, F. R. & Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 5, 437–448 (2004).
Gustafsson, M. V. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 9, 617–628 (2005).
Ni, Y. et al. Activation of beta2-adrenergic receptor stimulates gamma-secretase activity and accelerates amyloid plaque formation. Nat. Med. 12, 1390–1396 (2006).
Latapy, C. & Beaulieu, J. M. beta-Arrestins in the central nervous system. Prog. Mol. Biol. Transl. Sci. 118, 267–295 (2013).
Liu, X. et al. beta-arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res. 23, 351–365 (2013).
Jung, S. et al. SERP1 is an assembly regulator of gamma-secretase in metabolic stress conditions. Sci. Signal. 13, eaax8949 (2020).
Zani, A. & Yount, J. S. Antiviral protection by IFITM3. Vivo. Curr. Clin. Microbiol. Rep. 5, 229–237 (2018).
Everitt, A. R. et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523 (2012).
Ricciarelli, R. et al. Microarray analysis in Alzheimer’s disease and normal aging. IUBMB Life 56, 349–354 (2004).
Wang, X. A bridge between the innate immunity system and amyloid-beta production in Alzheimer’s disease. Neurosci. Bull. 37, 898–901 (2021).
Yao, A. Y. & Yan, R. Activity of Alzheimer’s gamma-secretase is linked to changes of interferon-induced transmembrane proteins (IFITM) in innate immunity. Mol. Neurodegener. 15, 69 (2020).
Kumar, D. K., Eimer, W. A., Tanzi, R. E. & Moir, R. D. Alzheimer’s disease: the potential therapeutic role of the natural antibiotic amyloid-beta peptide. Neurodegener. Dis. Manag. 6, 345–348 (2016).
Acknowledgements
The author thanks Dr. Yue-Ming Li and Dr. Eitan Wong for valuable comments on the manuscript and Dr. Pengju Nie for the critical reading of the manuscript. Some parts of the figures were made with Motifolio.com and BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hur, JY. γ-Secretase in Alzheimer’s disease. Exp Mol Med 54, 433–446 (2022). https://doi.org/10.1038/s12276-022-00754-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00754-8
- Springer Nature Limited