
Abstract
Pharmacologically elevating brain endocannabinoids (eCBs) share anxiolytic and fear extinction-facilitating properties with classical therapeutics, including the selective serotonin reuptake inhibitor, fluoxetine. There are also known functional interactions between the eCB and serotonin systems and preliminary evidence that antidepressants cause alterations in brain eCBs. However, the potential role of eCBs in mediating the facilitatory effects of fluoxetine on fear extinction has not been established. Here, to test for a possible mechanistic contribution of eCBs to fluoxetine’s proextinction effects, we integrated biochemical, electrophysiological, pharmacological, and behavioral techniques, using the extinction-impaired 129S1/Sv1mJ mouse strain. Chronic fluoxetine treatment produced a significant and selective increase in levels of anandamide in the BLA, and an associated decrease in activity of the anandamide-catabolizing enzyme, fatty acid amide hydrolase. Slice electrophysiological recordings showed that fluoxetine-induced increases in anandamide were associated with the amplification of eCB-mediated tonic constraint of inhibitory, but not excitatory, transmission in the BLA. Behaviorally, chronic fluoxetine facilitated extinction retrieval in a manner that was prevented by systemic or BLA-specific blockade of CB1 receptors. In contrast to fluoxetine, citalopram treatment did not increase BLA eCBs or facilitate extinction. Taken together, these findings reveal a novel, obligatory role for amygdala eCBs in the proextinction effects of a major pharmacotherapy for trauma- and stressor-related disorders and anxiety disorders.
Similar content being viewed by others

INTRODUCTION
There is growing evidence that pharmacologically elevating brain endocannabinoids (eCBs) share anxiolytic and antidepressant properties with classical therapeutics, including the selective serotonin reuptake inhibitors (SSRIs) (Bortolato et al, 2007; Gunduz-Cinar et al, 2013a; Hill and McEwen, 2010; Micale et al, 2013; Neumeister et al, 2015; Papini et al, 2015). Additionally, there are known functional interactions between the eCB and serotonin systems and indications that antidepressants cause alterations in brain eCBs. The deletion of CB1 receptor (CB1R) located on serotonin neuron terminals (Haring et al, 2007) increases serotonin release (Aso et al, 2009) but has modest effects on behavioral responses to stress (Dubreucq et al, 2012). Prior studies have also found that chronic fluoxetine treatment increases CB1R binding, CB1R-mediated G-protein signaling, and cAMP pathway activity (PKA, pCREB) in the prefrontal cortex (PFC) (Mato et al, 2010; Zarate et al, 2008) (cf. Hill et al, 2008a). Furthermore, chronic fluoxetine increases hippocampal levels of calcineurin (Crozatier et al, 2007), a Ca(2+)-activated phosphatase that mediates eCB-dependent synaptic plasticity in limbic regions (Heifets et al, 2008). In turn, gene deletion of fatty acid amide hydrolase (FAAH), the principal enzyme responsible for the degradation of the eCB, anandamide (N-arachidonylethanolamine), excites serotonin neurons, increases extracellular serotonin, and desensitizes 5-HT2A and 5-HT2C receptors in the cortex (Bambico et al, 2010; Cassano et al, 2011; Gobbi et al, 2005). Behaviorally, blocking CB1 receptors (CB1Rs) prevents antidepressant- and anxiety-related effects of acutely administered fluoxetine (Umathe et al, 2011). This reciprocal relationship between eCBs and SSRIs suggests that there may be common neural mechanisms underlying their anxiolytic and antidepressant-related effects.
Fear extinction is a measure of learned inhibition of conditioned fear that has proven valuable as a translational assay for studying novel treatments for stress- and trauma-related disorders (Bukalo et al, 2014; Milad and Quirk, 2012; Singewald et al, 2015). Gene deletion or pharmacological antagonism of CB1R profoundly impairs extinction, whereas augmenting eCBs, or amygdala anandamide specifically, facilitates extinction (Bowers and Ressler, 2015; Gunduz-Cinar et al, 2013b; Micale et al, 2013). In parallel to this work on eCBs, a number of recent studies have demonstrated that chronic treatment with fluoxetine, but not certain other SSRIs, such as citalopram (Burghardt and Bauer, 2013), facilitates extinction (Camp et al, 2012; Deschaux et al, 2011, 2013; Hartley et al, 2012; Karpova et al, 2012; Norcross et al, 2008; Popova et al, 2014; Riddle et al, 2013; Spennato et al, 2008). Conversely, acute fluoxetine administration increases conditioned fear in rodents; reminiscent of the heighted anxiety experienced by some patients during the early stages of SSRI treatment (Burghardt et al, 2007).
The mechanisms underlying fluoxetine’s extinction-facilitating effects remain poorly understood. Recent work (Karpova et al, 2012; Popova et al, 2014) has shown that chronic fluoxetine increases long-term potentiation (LTP) and excitatory currents at lateral amygdala (LA) synapses, alters a range of amygdala plasticity-related proteins, including the extinction-consolidating brain-derived neurotrophic factor (BDNF) (Andero and Ressler, 2012; Peters et al, 2010), and decreases amygdala expression of fear memory-conserving perineuronal nets (PNNs) (Gogolla et al, 2009). However, despite the known interactions between the eCB and 5-HT systems and similarities in the effects of fluoxetine and eCB-augmenting drugs, the role of eCBs to fluoxetine’s effects on extinction and associated amygdala functions remains unexplored. Here, integrating biochemical, electrophysiological, and pharmacological approaches, we tested for a mechanistic contribution of eCBs to fluoxetine’s proextinction effects in a mouse strain (129S1/Sv1mJ) model of impaired extinction (Camp et al, 2012).
MATERIALS AND METHODS
Subjects
Subjects were male 8- to 12-week-old 129S1/Sv1mJ (S1) mice obtained from The Jackson Laboratory (Bar Harbor, ME). This mouse strain exhibits impaired fear extinction that is rescued by various pharmacological manipulations, including FAAH inhibitors (Holmes and Singewald, 2013). Mice were housed 2 per cage in a temperature- and humidity-controlled vivarium under a 12-h light–dark cycle (lights on 0600 hours). Experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the local NIAAA and Vanderbilt Animal Care and Use Committees. The number of mice used in each experiment is indicated in the figure legends.
Fluoxetine and Citalopram Treatment
Mice were provided with 120 mg/l fluoxetine hydrochloride (LKT Laboratories, St Paul, MN, USA) in (their only source of) drinking water, as described previously (Camp et al, 2012; Dulawa et al, 2004; Karpova et al, 2012; Jiao et al 2011). The dose and concentration was chosen based on previous data in mice (Holmes and Rodgers, 2003; Karlsson et al, 2008; Norcross et al, 2008) to achieve a dose of ~10 mg/kg. Non-treated controls received water alone. Solutions were refreshed weekly. Fluoxetine and water consumption was measured from bottle weights (corrected for evaporation and spillage) and converted to a daily dose expressed in mg/kg body weight. For chronic treatment, the drug was given for 21 days before killing/fear conditioning. The procedure was the same for subchronic fluoxetine treatment, with the exception that treatment was 7 days in duration. Chronic citalopram treatment was the same as for chronic fluoxetine treatment.
Brain and serum levels of fluoxetine and citalopram were determined in a cohort of mice by liquid chromatography/tandem mass spectrometry (LC-MS/MS) using chromatographic and mass spectrometer conditions based on previously described methods (Gunduz-Cinar et al, 2013b). The molecular ion and fragments were measured by multiple reactions monitoring as follows: m/z 310.1→148.2 and 310.1→91.1 for fluoxetine (CID-energy: 0 and 80 V, respectively) and m/z 325.2→109 and 325.2→262.1 for citalopram (CID-energy: 24 and 16 V, respectively). The amounts of fluoxetine and citalopram in the samples were then determined against standard curves, with values expressed as ng/g or ng/ml in wet brain tissue weight or serum volume and then converted into nM free concentration values (based on plasma binding for each drug; Aronoff et al, 1984; Milne and Goa, 1991). The calculated free concentration values were serum fluoxetine=20 nM, citalopram=307 nM, tissue fluoxetine=456 nM, and tissue citalopram=2136 nM.
Fluoxetine Effects on Amygdala eCBs
To examine the effects of fluoxetine on anandamide and 2-arachidonylglycerol (2-AG) levels, mice were treated with the drug for 21 days (as above) and killed the following day. Brains were removed and the BLA (see Figure 1a), ventromedial PFC, dorsal hippocampus (DH), and dorsolateral striatum were dissected on ice using 1- and 2-mm-diameter micropunches. Tissue was homogenized in 80–100 μl Tris (pH 8.0) buffer and protein concentrations determined using the Bradford assay with bovine serum albumin as a standard. Lipids were extracted and anandamide and 2-AG levels quantified by liquid chromatography/tandem mass spectrometry, using multiple reactions monitoring, as described previously (Gunduz-Cinar et al, 2013b). The mass spectrometer was set for electrospray ionization operated in positive ion mode. The molecular ion and fragments for each compound measured were as follows: m/z 352.3→66.1 and 352.3→91 for [2H4] anandamide (CID-energy: 12 and 56 V, respectively), m/z 348.3→62.1 and 348.3→91 for anandamide (CID-energy: 12 and 48 V, respectively), and m/z 379.3→91 and 379.3→67.1 for 2-AG (CID-energy: 64 and 56 V, respectively). Analytes were quantified using MassHunter Workstation LC/QQQ Acquisition and MassHunter Workstation Quantitative Analysis (Agilent Technologies). Levels of anandamide and 2-AG in each brain region were determined against standard curves and expressed as fmol/mg (anandamide) or pmol/mg (2-AG) of protein.

Fluoxetine augments amygdala anandamide levels and inhibits fatty acid amide hydrolase (FAAH) activity. Chronic fluoxetine treatment increased basolateral amygdala (BLA) levels of anandamide (a) and oleoylethanolamide (b). Anandamide levels were increased in the dorsal hippocampus (HC) and dorsal striatum, but not prefrontal cortex (PFC), after chronic fluoxetine (c). Chronic fluoxetine increased 2-arachidonylglycerol (2-AG) levels in the dorsal hippocampus but not in the other brain regions examined (d). BLA expression of a suite of genes involved in endocannabinoid (eCB) synthesis and degradation was normal after chronic fluoxetine (e). FAAH activity in the BLA was reduced after chronic fluoxetine (f). Data are means±SEM. *P<0.05. Flx, fluoxetine; Wtr, water.
To explore possible effects of fluoxetine on amygdala eCB production and degradation, mice were treated with the drug for 21 days and killed the following day to remove the BLA. BLA punches were stored in RNAlater and kept in −20 °C until RNA extraction. Total RNA was isolated with RNeasy Kit (Qiagen, Germantown, MD) followed by DNase I treatment (Invitrogen, Grand Island, NY, USA) to eliminate DNA, to purify RNA. Reverse transcription was performed with 0.1 μg of total RNA using the Iscript cDNA Synthesis Kit (Bio-Rad, Hercules, CA) and a C1000 Thermal Cycler (Bio-Rad). We measured gene expression of the eCB-synthesizing enzymes, diacylglycerol lipase alpha (Daglα) (Qiagen; cat. no.: QT00167706, lot. no.: 180289526), diacylglycerol lipase beta (Daglβ) (Qiagen; cat. no.: QT00173453, lot. no.: 180289525), phospholipase C beta 1 (Plcb1) (Qiagen; cat. no.: QT00173817, lot no.: 180289525), and N-acyl phosphatidylethanolamine phospholipase D (Napepld) (Qiagen; cat. no.: QT00165347, lot. no.: 180300989), and the anandamide-degrading enzymes, Faah (Qiagen; cat. no.: QT00149520, lot. no.: 180289529) and cyclooxygenase-2 (Cox2) (Qiagen; cat. no.: QT00165347, lot. no.: 180300989). Gene expression was quantified with QuantiTect Primer Assay and Power SYBR Green PCR master mix (Applied Biosystems, Grand Island, NY) using a StepOnePlus Real-Time PCR instrument (Applied Biosystems) and normalized to the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (Qiagen; cat. no.: QT01658692, lot. no.: 176907735) using the QuantiTect Primer Assay.
Fluoxetine Effects on Amygdala FAAH Activity
To examine the effects of fluoxetine on amygdala FAAH activity, mice were treated with the drug for 21 days and killed the following day. Brains were removed and the BLA dissected on ice using 1-mm-diameter micropunches. Tissue was homogenized in 10 mM (pH 7.6) Tris-HCl buffer containing 1 mM EDTA and centrifuged at 1000 g to remove cell debris. Tissue was assayed for FAAH activity via measurement of the release of [3H]ethanolamine from [3H]anandamide labeled on the ethanolamine moiety, as described previously (Gunduz-Cinar et al, 2013b). The tissue homogenate (30 μg) was mixed with radiolabeled [3H]anandamide (containing 10 mg/ml fatty acid-free bovine serum albumin) and unlabeled anandamide to produce samples of 200 μl total volume.
Labeled and unlabeled chemicals were obtained from American Radiolabeled Chemicals (St Louis, MO) and Cayman Chemical Company (Ann Arbor, MI), respectively. Samples were incubated at 37 °C with continuous shaking in a water bath. ‘Blank’ samples contained assay buffer, instead of the homogenate, were incubated in the same manner. Incubation was stopped after 15 min by placing the tubes on ice, and 400 μl of 1 : 1 chloroform/methanol was added to the samples. Samples were then vortexed 3 × followed by centrifugation to produce phase separation. Two hundred microliters of the upper aqueous phase was removed and analyzed for radioactivity by liquid scintillation counting. The blank sample values were subtracted from each count.
Fluoxetine Effects on In vitro FAAH Activity
To examine the direct effect of fluoxetine on FAAH activity in vitro, an FAAH inhibitor Screening Assay Kit (Cayman, Ann Arbor, MI) was used. This is a fluorescence-based method, where recombinant human FAAH enzyme hydrolyzes the fluorophore attached synthetic substrate, and the fluorescence release was analyzed by using an excitation wavelength of 340–360 nm and an emission wavelength of 450–465 nm as per the instructions given in the kit. The effect of fluoxetine (1 nM–10 μM) on FAAH inhibition was tested against human recombinant FAAH and compared with a reference FAAH inhibitor AM3506 (Godlewski et al, 2010).
Fluoxetine Effects on Amygdala Synaptic Transmission
To examine the effects of fluoxetine on inhibitory and excitatory (in separate cohorts of mice) transmission in the amygdala, mice were treated with the drug for 21 days and slice electrophysiological experiments were performed using previously described methods (Gunduz-Cinar et al, 2013b; Ramikie et al, 2014). To ensure reliable electrophysiological recordings, mice were drug-treated beginning at 5 weeks of age to perform recordings when they were still young (8-week-old) adults. Mice were deeply anesthetized with isoflurane, and then transcardially perfused with ice-cold high sucrose, low Na+-containing ACSF and killed by decapitation. The brain was quickly removed and a 3 mm coronal block containing the BLA was cut using an ice-chilled, coronal brain matrix (see Figure 2a). Hemisected coronal 250-μm-thick slices were cut using a Leica VT1000S vibratome (Leica Microsystems, Bannockburn, IL) in a 1–4 °C oxygenated (95% (v/v) O2, 5% (v/v) CO2) high sucrose, low Na+-containing ACSF comprised of (in mM): 208 sucrose, 2.5 KCl, 1.6 NaH2PO4, 1 CaCl2·2H2O, 4 MgCl2·6H2O, 4 MgSO4·7H2O, 26 NaHCO3, 1 ascorbate, 3 Na-pyruvate, and 20 glucose. Slices were transferred to a 32 °C oxygenated recovery buffer composed of (in mM): 100 sucrose, 60 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.1 CaCl2·2H2O, 3.2 MgCl2·6H2O, 2 MgSO4·7H2O, 22 NaHCO3, 1 ascorbate, 3 Na pyruvate, and 20 glucose for 20 min, followed by a minimum of 30 min in 24 °C, oxygenated ACSF (in mM): 113 NaCl, 2.5 KCl, 1.2 MgSO4·7H2O, 2.5 CaCl2·2H2O, 1 NaH2PO4, 26 NaHCO3, 1 ascorbate, and 3 Na pyruvate, and 20 glucose.

Fluoxetine amplifies endocannabinoid (eCB)-mediated suppression of inhibitory amygdala transmission. Cartoon depiction of recordings of inhibitory transmission in the basolateral amygdala (BLA), with example traces showing effects of the CB1 receptor (CB1R) antagonist, SR141716 (SR) (a). Application of SR141716 increased inhibitory postsynaptic potential (IPSP) amplitude after chronic fluoxetine, unmasking eCB-mediated suppression of inhibitory transmission (b, c). Depolarization-induced suppression of inhibition was unaltered after chronic fluoxetine treatment (scale bars: 100 pA, 10-ms, stimulation artifacts deleted from traces for clarity) (d, e). Data are means±SEM. *P<0.01. eIPSC, evoked inhibitory postsynaptic current; Flx, fluoxetine; GABA, γ-aminobutyric acid; Wtr, water.
Slices were then placed in a submerged recording chamber where they were continuously perfused with oxygenated ACSF (23–25 °C) at a flow rate of 2–3 ml/min. To pharmacologically isolate GABAergic transmission, the ACSF was supplemented with 50 μM of the NMDA receptor blocker, 2-amino-5-phosphonopentanoic acid sodium salt (AP-5), and 20 μM of the AMPA receptor blocker, 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX). To isolate glutamatergic transmission and to avoid population activity, the ACSF was supplemented with the GABAA receptor antagonist 50 μM picrotoxin (Abcam, Cambridge, MA) and 50 μM of AP5, respectively. For all experiments, 0.5 g/l of fatty acid-free bovine serum albumin (Sigma-Aldrich, St Louis, MO) was also added to the ACSF to increase solubility of the CB1R antagonist, SR141716, and minimize nonspecific binding of this lipophilic compound. AP-5, CNQX and SR141716 were generously provided by the NIH Drug Supply Program (Bethesda, MD).
Whole-cell recordings were obtained from BLA pyramidal cells visualized using a Nikon microscope equipped with differential interference contrast video microscopy. Recordings were performed using pipettes pulled from borosilicate glass (3–5 MΩ resistance). For IPSP recordings, pipettes were filled with K+-based high [Cl]i pipette solution containing (in mM): 70 K+ gluconate, 4 NaCl, 10 HEPES, 60 KCl, 4 Mg-ATP, 0.3 Na-GTP, 10 Na phosphocreatine, and 0.6 EGTA (285mOsm, adjusted to pH 7.30–7.35 with KOH). For EPSP recordings, pipette solution contained (in mM): 125 K+ gluconate, 4 NaCl, 10 HEPES, 20 KCl, 4 Mg-ATP, 0.3 Na-GTP, 10 Na phosphocreatine, and 10 QX-314 boromide to avoid action potentials. Whole-cell recordings were made using an Axopatch 700B amplifier (Molecular Devices, Sunnyvale, CA).
GABAergic and glutamatergic neurotransmission was elicited by current stimulation via an ACSF-filled glass electrode placed ~100 μm from the cell soma with stimulation intensities ranged from 10 to 40 μA. SR141716 (5 μM) application studies were performed under current-clamp conditions and the membrane potential was maintained near −70 mV by constant current injection. Input resistance was monitored throughout the experiment and the cell was rejected if the input resistance changed by > 20%. Current stimulation was used to evoke inhibitory or excitatory postsynaptic potentials (eIPSPs and eEPSPs, respectively) at a rate of 0.1 Hz. Evoked responses were elicited at 50–60% of maximal response amplitude and 6 consecutive responses were averaged to generate one data point per minute. Following a 6-min baseline, SR141716 was bath applied. The magnitude of the drug effect was calculated as a percentage of averaged responses relative to predrug baseline.
Depolarization-induced suppression of inhibition (DSI) was examined under voltage-clamp conditions where cells were recorded at a holding potential of −70 mV. Evoked inhibitory postsynaptic currents (eIPSCs) were elicited at a rate of 0.5 Hz. To induce DSI, a depolarizing pulse (−70 to 0 mV) was applied to the postsynaptic neuron for 10 s. Maximum DSI was classified as the first eIPSC following the depolarizing pulse. Within each DSI trial, eIPSC amplitudes were normalized to the averaged baseline response and data from 2 DSI trials, per cell, were averaged for analyses. Access resistance (Ra) was monitored online and cells that demonstrated a >20% change in Ra were excluded from analysis. Offline data analysis was performed using Clampfit 9 program (Molecular Devices, Sunnyvale, CA).
Fluoxetine Effects on Fear Extinction: eCB Dependence
To examine the effects of fluoxetine on fear extinction, mice were fear conditioned and then (beginning the next day) treated with the drug for 23 days (extinction training on day 22, retrieval on day 23), as described previously (Fitzgerald et al, 2014; Gunduz-Cinar et al, 2013b). Mice were conditioned in Context A (a 27 × 27 × 11 cm3 chamber with a metal-rod floor, cleaned with a 79.5% water/19.5% ethanol/1% vanilla extract solution). After a 180-s acclimation period, there were 3 × pairings (60–120 s interpairing interval) of the conditioned stimulus (CS; 30 s, 75 dB, 3 kHz tone) and the unconditioned stimulus (US; 2 s, 0.6 mA scrambled foot shock), in which the US was presented during the last 2 s of the CS. The session ended 120 s after the final CS–US pairing. Stimulus presentation was controlled by the Med Associates VideoFreeze system (Med Associates, Burlington, VT). Freezing (no visible movement except respiration) was scored every 5 s by an observer blind to condition/treatment and converted to a percentage ((freezing observations/total observations) × 100).
We next tested whether fluoxetine’s effects on extinction were eCB-dependent. Mice were fear conditioned and then chronically fluoxetine-treated, as above. Mice were then injected with the 1 mg/kg of the CB1R antagonist, SR14176 (provided by the NIH Drug Supply Program, Bethesda, MD), suspended in an 18 : 1 : 1 saline/Tween-80/DMSO solution and injected intraperitoneally in a volume of 10 ml/kg body weight), or vehicle, 50 min before extinction training. Extinction training was conducted in a novel context (context B cylinders with black/white-checkered walls and a solid Plexiglas opaque floor cleaned with a 1% acetic acid/99% water solution) housed in a different room from conditioning. After a 180-s acclimation period, the CS was presented 50 × (5-s inter-CS interval). Freezing during extinction was averaged to 5-trial blocks for analysis. Extinction retrieval was tested the following day in context B. After a 180-s acclimation period, there were 3 × CS presentations (5-s inter-CS interval). For schematic of the experimental design, see Figure 3a.

Fluoxetine (Flx) facilitates extinction through CB1 receptor (CB1R) signaling in the amygdala. Chronic fluoxetine treatment following conditioning and before extinction reduced freezing on extinction retrieval in a manner prevented by pre-extinction administration of the CB1R antagonist, SR141716 (SR) (a). Chronic fluoxetine after conditioning and before extinction reduced freezing on extinction retrieval in a manner prevented by pre-extinction bilateral microinfusion of the CB1R antagonist, SR141716, into the basolateral amygdala (BLA) (b). Pre-extinction administration of the fatty acid amide hydrolase (FAAH) inhibitor, AM3506, reduced freezing on extinction retrieval (c). Chronic fluoxetine treatment or administration of the FAAH inhibitor, AM3506, before extinction reduced freezing on extinction retrieval in a non-additive manner (d). Data are means±SEM. *P<0.05. AM, AM3506; Con, conditioning; Ext, extinction; Flx, fluoxetine; Ret, retrieval; Wtr, water.
Fluoxetine Effects on Amygdala-Mediated eCB-Dependent Fear Extinction
To test whether the effects of fluoxetine on extinction were dependent on eCBs, specifically within the BLA, mice were fear conditioned and then chronically fluoxetine-treated, as above. Two μg/μl SR14176, or vehicle, was bilaterally infused into the BLA 30 min before extinction training. For intra-BLA microinfusions, 26-gauge bilateral guide cannulae (Plastics One, Roanoke, VA) were stereotaxically targeted under isoflurane anesthesia to the BLA (−1.40 mm anterior–posterior, ±3.30 mm mediolateral, −3.90 mm ventral to Bregma) and held in place with dental cement. Mice were singly housed and given a 7-day postsurgery recovery period during which dummy cannulae were replaced daily to habituate them to handling of cannulae and prevent blocking. Drug was suspended as described above and infused via bilateral 33-gauge injectors (Plastics One, Wallingford, CT) projecting 1 mm past the guide cannula, in a volume of 0.5 μl per hemisphere over 2 min using a syringe pump (Harvard Apparatus PHD 22/2000; Harvard Apparatus Holliston, MA). Injectors were left in place for a further 3 min to allow drug diffusion into the tissue. For schematic of the experimental design, see Figure 3b.
To verify accurate BLA cannula placements at the completion of testing, mice were terminally overdosed with ketamine/xylazine and transcardially perfused with phosphate-buffered saline, and then 4% paraformaldehyde (PFA). After suspension in 4% PFA overnight and then 4 °C 0.1 M phosphate buffer for 1–2 days, 50 μm coronal sections were cut with a vibratome (Classic 1000 model; Vibratome, Bannockburn, IL). Cannula location was determined with the aid of an Olympus BX41 microscope (Olympus, Center Valley, PA) and mice without bilateral placements were removed from the analysis (for cannula placement estimates, see Figure 3b).
Fluoxetine and FAAH Inhibitor Effects on Fear Extinction
To test the effects of the FAAH inhibitor, AM3506, on fear extinction, mice were injected with 1 mg/kg AM3506 (synthesized at Northeastern University suspended in an 9 : 1 saline/DMSO solution and injected intraperitoneally in a volume of 10 ml/kg body weight), or vehicle, 1 h before extinction training. Retrieval was tested 10 days later (for schematic of the experimental design, see Figure 3c).
Next, to examine the effects of combined treatment with fluoxetine and AM3506, mice were fear conditioned and chronically fluoxetine-treated, as above: then injected with 1 mg/kg AM3506, or vehicle, 60 min before extinction training. Retrieval was tested, as above, 10 days later (for schematic of the experimental design, see Figure 3d). We repeated the same experimental design in a separate cohort of mice, but used subthreshold doses of fluoxetine (5 mg/kg) and AM3506 (0.1 mg/kg).
Citalopram Effects on Fear Extinction and Amygdala eCBs
To examine the effects of citalopram on fear extinction, mice were fear conditioned and then chronically citalopram-treated, using the same procedures described above for fluoxetine (for schematic of the experimental design, see Figure 4a). A separate cohort of mice was chronically treated with citalopram in the same manner and measured for brain eCB levels (for schematic of the experimental design, see Figure 4b).

Citalopram (Cit) does not facilitate fear extinction or elevate amygdala endocannabinoids (eCBs). Chronic citalopram treatment following conditioning and before extinction did not alter freezing on extinction retrieval (a). Chronic citalopram treatment did not alter basolateral amygdala (BLA) levels of anandamide (b) or 2-arachidonylglycerol (2-AG) (c). Data are means±SEM. Con, conditioning; Ext, extinction; Ret, retrieval; Wtr, water.
Statistical Analysis
Data were analyzed using t-tests or analysis of variance (ANOVA), followed by Newman–Keuls post hoc tests where appropriate. The threshold for statistical significance was set at P<0.05.
RESULTS
Fluoxetine Augments Amygdala Anandamide Levels and Inhibits FAAH Activity
We first assessed the potential effects of chronic fluoxetine treatment on brain eCBs. Using liquid chromatography/tandem mass spectrometry (LC/MS/MS), we found that levels of the eCB, anandamide, were significantly elevated in the BLA of fluoxetine-treated mice, as compared with water-drinking controls (t(29)=2.30, P<0.05, n=15–16) (Figure 1a). Levels of oleoylethanolamide, a lipid mediator and non-cannabinoid analogue of anandamide and substrate of FAAH, were also elevated in the BLA after fluoxetine treatment (t(29)=2.19, P<0.05, n=15–16) (Figure 1b). We also detected higher anandamide levels in the dorsal striatum (t(27)=2.25, P<0.05, n=12–17) and DH (t(26)=2.13, P<0.05, n=14) but not PFC (P>0.05), in the fluoxetine-treated mice (Figure 1c). By contrast, with the exception of a modest increase in the DH (t(26)=2.10, P<0.05, n=14), fluoxetine treatment did not affect levels of the other major eCB, 2-AG, in any of the brain regions examined (Figure 1d). Moreover, subchronic (7-day) fluoxetine treatment failed to alter eCB levels in any brain region examined, including the BLA (Supplementary Figure S1).
Given the prominent role of FAAH In the regulation of brain anandamide levels, we determined the effects of fluoxetine treatment on FAAH gene expression and FAAH activity. No changes were seen in Faah gene expression (t-tests: P>0.05; Figure 1e), or other eCB-synthesizing enzymes, Daglα, Daglβ, Plcb1, or Napepld, and anandamide-degrading enzymes, Cox2, (all t-tests: P>0.05; Figure 1e). To rule out the possibility that fluoxetine increases AEA levels via direct inhibition of FAAH enzymatic activity, we showed that fluoxetine did not inhibit hFAAH activity in vitro under conditions in which the FAAH inhibitor AM3506 markedly inhibited hFAAH activity in a concentration-dependent manner (EC50 value=35 nM) (Supplementary Figure S2). However, the catalytic activity of FAAH was significantly reduced in the BLA of the fluoxetine-treated mice (69.5±2.8 fmol/mg/min), relative to water controls (77.5±2.3 fmol/mg/min) (t(13)=2.18, P<0.05, n=6–9) (Figure 1f). These data suggest that chronic fluoxetine treatment could indirectly augment BLA anandamide levels, possibly via post-translational inhibition of FAAH activity.
Fluoxetine Amplifies eCB-Mediated Suppression of Inhibitory Amygdala Transmission
Prior work has shown that anandamide regulates certain forms of synaptic transmission in the BLA and causes long-term depression of inhibitory transmission (Gunduz-Cinar et al, 2013b) via CB1Rs located on GABAergic terminals (Kano et al, 2009). We therefore next assessed whether fluoxetine affected eCB-mediated inhibitory transmission in the BLA, by recording GABAergic synaptic potentials in brain slices of fluoxetine-treated mice.
Recordings showed that, application of the CB1R antagonist, SR141716, caused a significant increase in IPSP amplitude in fluoxetine-treated mice, but not water controls (treatment × time interaction: F1,35=5.04, P<0.01, n=4–6 cells from n=4–6 mice) (Figure 2b). The maximum IPSP amplitude in the presence of SR141716 was ~50% higher in fluoxetine-treated mice (t(9)=3.67, P<0.01) (Figure 2c). Conversely, when we measured excitatory transmission, SR141716 had no effect of EPSP amplitude in either water or fluoxetine-treated mice (n=9–12 cells from n=3–4 mice) (Supplementary Figure S3). Taken together, these data reveal an amplification of CB1R-mediated tonic suppression of inhibitory, but not excitatory, transmission after fluoxetine treatment that is unmasked by CB1R blockade. As our LC/MS/MS data indicated that fluoxetine increased levels of anandamide and not 2-AG, we reasoned that there would be relatively limited alterations in BLA synaptic functions after fluoxetine treatment and, specifically, that 2-AG-mediated functions would not be affected. To test this proposition, we measured DSI: a 2-AG, but not anandamide (Hashimotodani et al, 2007), dependent form of short-term plasticity previously observed in the BLA (Patel et al, 2009; Zhu and Lovinger, 2005; Shonesy et al, 2014). We showed that a depolarizing pulse from −70 to 0 mV significantly reduced eIPSC amplitude for ~20 s, indicating DSI. However, and consistent with our prediction, fluoxetine treatment affected neither the duration (t-test: P>0.05, n=6 cells from 3 mice) (Figure 2d) nor maximum extent (t-test: P>0.05, n=6 cells from 3 mice) (Figure 2e) of DSI.
The results of these electrophysiological experiments show that fluoxetine-induced increases in anandamide are associated with the amplification of eCB-mediated tonic constraint of inhibitory transmission in the BLA.
Fluoxetine Facilitates Extinction Through CB1R Signaling
Prior work has shown that the depression of inhibitory synaptic transmission in the BLA, which is produced by increasing anandamide levels by administration of an FAAH inhibitor, is associated with the facilitation of fear extinction (Gunduz-Cinar et al, 2013b). On this basis, our next step, therefore, was to ask whether fluoxetine produced an extinction-facilitating effect via eCB signaling in the BLA.
We first examined whether systemic blockade of CB1Rs prevented the extinction-facilitating effects of chronic fluoxetine treatment, by injecting fluoxetine-treated mice with SR141716 before extinction training. Results indicated a significant increase in freezing across conditioning trials (F1,43=11.14, P<0.01, n=9–15). Drug treatment did not affect freezing during the initial trial block of extinction training (ANOVA effect for treatment effect: P>0.05)—a measure of fear expression. There was a small but significant decrease in freezing across extinction training (F1,43=452.72, P<0.01) that is typical of the impaired extinction shown by the S1 mouse strain (Gunduz-Cinar et al, 2013b). In line with previous findings, neither fluoxetine (Fitzgerald et al, 2014) nor SR141716 (Gunduz-Cinar et al, 2013b) affected freezing on extinction training in this strain (P>0.05) (Supplementary Figure S4a). During extinction retrieval, however, fluoxetine-treated mice froze significantly less than water controls, and this effect was absent in mice that had been injected with SR141716 (ANOVA effect for treatment effect: F3,43=2.82, P<0.05; post hoc comparisons: water vs fluoxetine P<0.05, fluoxetine vs fluoxetine+SR141716 P>0.05, fluoxetine vs SR141716 P>0.05) (Figure 3a).
Our systemic pharmacological experiment showed that fluoxetine’s pro-extinction efficacy is dependent on CB1Rs, but did not localize this effect to the brain region (BLA) where we found that fluoxetine exerted its effects on FAAH activity and synaptic functions. We therefore next blocked CB1R specifically within the BLA before extinction training via microinfusions of SR141716. Irrespective of treatment group, there was a significant increase in freezing across conditioning trials (ANOVA effect of trial: F1,42=295.33, P<0.01, n=9–14). Drug treatment did not affect freezing during the initial trial block of extinction training (ANOVA effect for treatment effect: P>0.05). There was significant increase in freezing across extinction trial blocks (ANOVA effect of trial block: F1,42=75.06, P<0.01), but no effect of treatment (P>0.05) (Supplementary Figure S4b). On extinction retrieval, fluoxetine-treated mice showed the expected reduction in freezing, relative to water controls, but this facilitatory effect was absent in mice that had received SR141716 infusions (ANOVA effect for treatment F3,42=3.33, P<0.05; post hoc comparisons: water vs fluoxetine P<0.05, water vs fluoxetine P<0.05, fluoxetine vs fluoxetine+SR141716 P>0.05, fluoxetine vs SR141716 P>0.05) (Figure 3b), confirming that CB1R signaling in the BLA is necessary for fluoxetine’s pro-extinction effects.
Fluoxetine and FAAH Inhibition have Comparable, Non-Additive Effects on Extinction
The results of our experiments up to this point suggest a scheme by which fluoxetine inhibits FAAH to elevate BLA anandamide, activates CB1R, and thereby promotes extinction. If fluoxetine and FAAH inhibition exert their pro-extinction effects by recruiting similar downstream mechanisms (e.g., activate CB1R), then combined treatment with the two drugs might not be able to exert additive effects on extinction.
To test for this, we first sought to replicate the earlier finding that pre-extinction administration of the selective FAAH inhibitor, AM3506 (Gunduz-Cinar et al, 2013b), facilitated extinction retrieval. There was a significant increase in freezing across conditioning trials (ANOVA effect of trial: F1,16=67.85, P<0.01, n=6–12). Drug treatment did not affect freezing during the initial trial block of extinction training (ANOVA effect for treatment effect: P>0.05). There was no change in freezing across extinction trial blocks (ANOVA effect of trial block: P>0.05) and no effect of drug treatment (P>0.05) (Supplementary Figure S4c). On extinction retrieval, mice injected with AM3056 froze significantly less than vehicle-alone controls (t-test treatment effect: t(16)=4.50, P<0.05) (Figure 3c), confirming a pro-extinction effect of the FAAH inhibitor.
We next examined the effects of cotreating mice with fluoxetine and AM3506. There was a significant increase in freezing across conditioning trials (ANOVA effect of trial: F1,63=718.31, P<0.01, n=15–18). Drug treatment did not affect freezing during the initial trial block of extinction training (ANOVA effect for treatment effect: P>0.05). There was a significant decrease in freezing across extinction trial blocks (ANOVA effect of trial block: F1,58=19.73, P<0.01), regardless of treatment group (P>0.05) (Supplementary Figure S4d). On extinction retrieval, there was significantly less freezing in mice that had been treated with fluoxetine or AM3506, or the combination of the two, as compared with water, but the reduction in freezing was no greater in the combined-treatment group (ANOVA effect for treatment F3,63=2.85, P<0.05, followed by post hoc comparisons) (Figure 3d), demonstrating the absence of an additive drug effect.
We also tested whether combined treatment with subthreshold doses of fluoxetine and AM3506 would produce demonstrable behavioral effects, but found that none of the single or combined treatments differed from untreated controls on conditioning, extinction, or extinction retrieval (all statistical tests: P>0.05; Supplementary Figure S5).
Citalopram does not Facilitate Fear Extinction or Elevate Amygdala eCBs
The finding that fluoxetine facilitates extinction by augmenting eCB activity in the BLA suggests a novel mechanism by which antidepressants produce therapeutic efficacy in disorders, such as trauma- and stressor-related disorders (formerly classified as post-traumatic stress disorder). However, in contrast to fluoxetine, previous studies have found that chronic administration of other SSRIs, such as citalopram, does not exert extinction facilitating activity in rats (Burghardt and Bauer, 2013). This led us to ask whether citalopram may differ from fluoxetine in not altering eCBs.
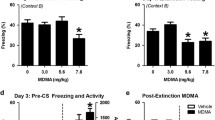
To test for this, we first sought to confirm and extend the published data in rats by showing that citalopram fails to facilitate extinction in the S1 mouse strain. Regardless of treatment, we found a significant increase in freezing across conditioning trials (ANOVA effect of trial: F1,34=414.82, P<0.01, n=18). Drug treatment did not affect freezing during the initial trial block of extinction training (ANOVA effect for treatment effect: P>0.05). There was a decrease in freezing across extinction trial blocks (ANOVA effect of trial block: F1,34=7.60, P<0.01) (Supplementary Figure S4e). In contrast to the profile seen with fluoxetine treatment, citalopram-treated mice froze at similar levels as water controls during extinction retrieval (t-test effect of treatment: P>0.05) (Figure 4a).
We then performed a parallel neurochemical experiment, to test whether the negative behavioral effects of citalopram treatment were associated with the absence of changes in BLA eCBs. We found that citalopram-treated mice indeed showed levels of both anandamide and 2-AG comparable to levels in water controls in the BLA (Figures 4b and c), as well as DH, dorsal striatum, and PFC (all t-test effects: P>0.05, n=18) (Supplementary Figure S6).
DISCUSSION
The results of the current study uncover a previously unappreciated contribution of eCBs to the fear extinction promoting actions of fluoxetine, and identify the BLA as a critical locus underlying these effects.
An enhancement in extinction retrieval following chronic treatment with fluoxetine has been reported in numerous studies in rats and various mouse strains (Camp et al, 2012; Deschaux et al, 2011, 2013; Hartley et al, 2012; Karpova et al, 2012; Norcross et al, 2008; Popova et al, 2014; Riddle et al, 2013; Spennato et al, 2008). In prior work, this behavioral effect has been linked to alterations in the BLA expression of PNNs and plasticity-regulating proteins, such as BDNF, and the enhancement of excitatory synaptic currents and LTP (Andero and Ressler, 2012; Gogolla et al, 2009; Karpova et al, 2012; Peters et al, 2010; Popova et al, 2014). Our current findings demonstrate that chronic fluoxetine’s pro-extinction effects are also associated with marked changes in BLA eCBs and eCB-mediated synaptic plasticity. More specifically, our data suggest a model in which fluoxetine elevates BLA anandamide levels, through the inhibition of FAAH activity, to suppress inhibitory transmission via tonic CB1 receptor activation. We postulate that the consequence of this decrease in inhibitory transmission is to relieve a functional constraint on extinction-generating plasticity at excitatory BLA pyramidal neurons (Bissiere et al, 2003; Herry et al, 2008).
The eCB-related effects on extinction we report here may occur in parallel to the other reported plasticity-related changes in the BLA produced by fluoxetine, or may be functionally interlinked with them. For example, BDNF is known to stimulate eCBs and promote eCB-dependent plasticity in the cortex and midbrain (Lemtiri-Chlieh and Levine, 2010; Zhong et al, 2015), suggesting the two systems could interact to mediate fluoxetine’s effects on extinction. Whether eCBs could also affect extinction-constraining interneuronal PNNs in the BLA (Gogolla et al, 2009; Karpova et al, 2012) is currently less clear. While prior work shows that fluoxetine reduces the number of PNNs on parvalbumin-positive (but not calbindin or calretinin) BLA interneurons (Karpova et al, 2012), the drug’s effects on the cholecystokinin (CCK)-positive interneuronal subtype on which CB1R is almost exclusively expressed (Katona et al, 1999; Yoshida et al, 2011) remain unknown. The potential effects of fluoxetine on CCK-PNNs is particularly intriguing in light of recent evidence that extinction training increases both CB1R expression and the number of perisomatic synapses at CCK-positive interneurons located proximal to BLA pyramidal cells active during training (Trouche et al, 2013) and, more generally, known interactions between CCK and eCB signaling in the context of extinction (Bowers and Ressler, 2015). However, it still remains unclear whether FAAH is indeed expressed on extinction-mediating BLA principal neurons receiving synaptic contacts from CCK/CB1R-positive interneurons.
Other aspects of fluoxetine’s effects on BLA eCBs also require clarification. The current results do not establish the precise mechanism by which fluoxetine elevates anandamide levels in the BLA. A number of plausible candidates exist, including N-arachidonoyl-serotonin (AA-5-HT). AA-5HT has been shown to inhibit FAAH (and the TRPV1 receptor) (Bisogno et al, 1998; Rose et al, 2014) and exert anxiolytic-like and antistress effects following systemic or direct intra-BLA administration (John and Currie, 2012; Micale et al, 2009; Navarria et al, 2014). However, given the technical difficulty of accurately measuring AA-5-HT in neural tissue, it is unclear whether there are functionally relevant increases after fluoxetine treatment. It should also be noted that the fluoxetine-induced decrease in FAAH activity, whether occurring via AA-5HT or other molecules, was modest by comparison with FAAH inhibitors, such as AM3506 and URB597 (Godlewski et al, 2010; Gunduz-Cinar et al, 2013b; Piomelli et al, 2006). Moreover, despite a modest effect of chronic fluoxetine on the activity of FAAH in the BLA, we did not detect fluoxetine-induced changes in hFAAH enzymatic activity (in vitro), the mRNA expression (ex vivo) of Faah, or a suite of other enzymes involved in anandamide synthesis and catabolism. This raises the possibility that, in addition to the inhibitory effects on FAAH catalytic activity we observed here, fluoxetine exerts actions on the production and degradation of anandamide that are yet to be identified. Another consideration is that the eCB-dependent effects of fluoxetine on extinction are not necessarily mediated solely via anandamide, but could involve alternate mechanisms that result in the increased CB1R sensitivity that is seen in other brain regions following chronic fluoxetine treatment (Mato et al, 2010).
Whatever the precise neural basis of fluoxetine’s effects on extinction and eCBs, they do not appear to be a characteristic that is broadly shared by SSRIs. In line with prior work showing that chronic citalopram impairs extinction in rats (Burghardt and Bauer, 2013), we found that chronic treatment with citalopram in extinction-deficient S1 mice failed to facilitate extinction and did not increase levels of anandamide in the BLA. These differences are likely attributable to distinct pharmacological profiles of the two drugs (Sanchez and Hyttel, 1999). Although both drugs fall into SSRI class, citalopram is highly selective for the 5-HT transporter, whereas fluoxetine has activity at the norepinephrine transporter and various receptors, including the 5-HT2A and 5-HT2C subtypes (Sanchez and Hyttel, 1999). Thus, one or more of these off-target actions could potentially contribute to fluoxetine’s eCB-mediated extinction-promoting effects, and their absence with citalopram. In this context, trace fear extinction in mice is facilitated or disrupted by systemic agonism or antagonism, respectively, of 5-HT2A receptors (Zhang et al, 2013). The norepinephrine system is also implicated in fear and extinction (Bukalo et al, 2014; Holmes and Quirk, 2010) and eCB effects on fear memory have been shown to require β-adrenoceptors in the BLA (Atsak et al, 2015; Roozendaal et al, 2009). A previous study found that the antidepressant-related effects of fluoxetine, but not citalopram, were absent in (dopamine β-hydroxylase knockout) mice lacking endogenous norepinephrine mutant (Cryan et al, 2004). These findings suggest the interesting possibility of norepinephrine recruitment in the eCB-mediated effects of fluoxetine on extinction.
SSRIs including fluoxetine (Prozac) remain a front-line treatment for anxiety disorders, including trauma- and stressor-related disorders, but there has been surprisingly little investigation of their effects on fear extinction and its clinical analogue, exposure therapy (Singewald et al, 2015), although one recent study did report that chronic escitalopram treatment facilitated fear extinction in healthy human subjects (Bui et al, 2013). The current findings add to growing preclinical evidence that fluoxetine may be an effective adjunct to exposure therapy for anxiety disorders. They also predict that, given their eCB mode of action, these effects could be therapeutically mimicked by directly targeting anandamide (e.g., with FAAH inhibitors (Gunduz-Cinar et al, 2013a; Neumeister et al, 2015). Lastly, they suggest that individual differences in eCB levels (Dlugos et al, 2012; Hill et al, 2008b, 2013) or variation in eCB amygdala function because of gene variation in FAAH or CB1R (Dincheva et al, 2015; Hariri et al, 2009; Neumeister et al, 2013) could significantly modify the efficacy of fluoxetine as an adjunct to exposure therapy. In this regard, the current study used a single mouse model of impaired extinction and although the pro-extinction effects of chronic fluoxetine are seen in rats and other genetic mouse strains, it would prove valuable to generalize the role of eCBs in the drug’s effects to other rodent models.
In conclusion, the current study reveals a novel contribution of eCBs in the extinction-facilitating effects of fluoxetine. We demonstrate that chronic treatment with fluoxetine modestly inhibits the activity of FAAH, the major catabolic enzyme for the eCB anandamide, and elevates anandamide levels in the BLA, a key neural locus for fear extinction (Bukalo et al, 2015; Tovote et al, 2015). Moreover, we found these neurochemical changes produce a tonic suppression of inhibitory transmission in the BLA to positively gate the activity of extinction-generating pyramidal neurons in this brain region. Demonstrating the functional necessity of BLA eCB signaling for fluoxetine’s extinction-promoting actions, we were able to prevent these effects via either systemic or BLA-specific blockade of CB1R. We also found that administration of an FAAH inhibitor mimicked the extinction-facilitating effects of fluoxetine, but combined treatment with both drugs did not produce a greater effect than either alone. Finally, we showed that the highly selective SSRI, citalopram, failed to affect extinction or brain eCBs. Collectively, these findings identify a critical neural mechanism mediating fluoxetine’s effects, with implications for the selection and future development of compounds for the treatment of anxiety disorders.
Funding and Disclosure
This research was supported by the NIAAA Intramural Research Program and NIH Grants MH100096 and MH09012 (to SP). SP receives research contract support from Lundbeck Pharmaceuticals; however, the current work was supported entirely by the NIH and Vanderbilt University. The authors declare no conflict of interest.
References
Andero R, Ressler KJ (2012). Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav 11: 503–512.
Aronoff GR, Bergstrom RF, Pottratz ST, Sloan RS, Wolen RL, Lemberger L (1984). Fluoxetine kinetics and protein binding in normal and impaired renal function. Clin Pharmacol Ther 36: 138–144.
Aso E, Renoir T, Mengod G, Ledent C, Hamon M, Maldonado R et al (2009). Lack of CB1 receptor activity impairs serotonergic negative feedback. J Neurochem 109: 935–944.
Atsak P, Hauer D, Campolongo P, Schelling G, Fornari RV, Roozendaal B (2015). Endocannabinoid signaling within the basolateral amygdala integrates multiple stress hormone effects on memory consolidation. Neuropsychopharmacology 40: 1485–1494.
Bambico FR, Cassano T, Dominguez-Lopez S, Katz N, Walker CD, Piomelli D et al (2010). Genetic deletion of fatty acid amide hydrolase alters emotional behavior and serotonergic transmission in the dorsal raphe, prefrontal cortex, and hippocampus. Neuropsychopharmacology 35: 2083–2100.
Bisogno T, Melck D, De Petrocellis L, Bobrov M, Gretskaya NM, Bezuglov VV et al (1998). Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun 248: 515–522.
Bissiere S, Humeau Y, Luthi A (2003). Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci 6: 587–592.
Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A et al (2007). Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry 62: 1103–1110.
Bowers ME, Ressler KJ (2015). Interaction between the cholecystokinin and endogenous cannabinoid systems in cued fear expression and extinction retention. Neuropsychopharmacology 40: 688–700.
Bui E, Orr SP, Jacoby RJ, Keshaviah A, LeBlanc NJ, Milad MR et al (2013). Two weeks of pretreatment with escitalopram facilitates extinction learning in healthy individuals. Hum Psychopharmacol 28: 447–456.
Bukalo O, Pinard CR, Holmes A (2014). Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol 171: 4690–4718.
Bukalo O, Pinard C, Silverstein S, Brehm C, Hartley N, Whittle N et al (2015). Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv 1: e1500251.
Burghardt NS, Bush DE, McEwen BS, LeDoux JE (2007). Acute selective serotonin reuptake inhibitors increase conditioned fear expression: blockade with a 5-HT(2C) receptor antagonist. Biol Psychiatry 62: 1111–1118.
Burghardt NS, Bauer EP (2013). Acute and chronic effects of selective serotonin reuptake inhibitor treatment on fear conditioning: implications for underlying fear circuits. Neuroscience 247: 253–272.
Camp MC, Macpherson KP, Lederle L, Graybeal C, Gaburro S, Debrouse LM et al (2012). Genetic strain differences in learned fear inhibition associated with variation in neuroendocrine, autonomic, and amygdala dendritic phenotypes. Neuropsychopharmacology 37: 1534–1547.
Cassano T, Gaetani S, Macheda T, Laconca L, Romano A, Morgese MG et al (2011). Evaluation of the emotional phenotype and serotonergic neurotransmission of fatty acid amide hydrolase-deficient mice. Psychopharmacology (Berl) 214: 465–476.
Crozatier C, Farley S, Mansuy IM, Dumas S, Giros B, Tzavara ET (2007). Calcineurin (protein phosphatase 2B) is involved in the mechanisms of action of antidepressants. Neuroscience 144: 1470–1476.
Cryan JF, O’Leary OF, Jin SH, Friedland JC, Ouyang M, Hirsch BR et al (2004). Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc Natl Acad Sci USA 101: 8186–8191.
Deschaux O, Spennato G, Moreau JL, Garcia R (2011). Chronic treatment with fluoxetine prevents the return of extinguished auditory-cued conditioned fear. Psychopharmacology (Berl) 215: 231–237.
Deschaux O, Zheng X, Lavigne J, Nachon O, Cleren C, Moreau JL et al (2013). Post-extinction fluoxetine treatment prevents stress-induced reemergence of extinguished fear. Psychopharmacology (Berl) 225: 209–216.
Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC et al (2015). FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nat Commun 6: 6395.
Dlugos A, Childs E, Stuhr KL, Hillard CJ, de Wit H (2012). Acute stress increases circulating anandamide and other N-acylethanolamines in healthy humans. Neuropsychopharmacology 37: 2416–2427.
Dubreucq S, Matias I, Cardinal P, Haring M, Lutz B, Marsicano G et al (2012). Genetic dissection of the role of cannabinoid type-1 receptors in the emotional consequences of repeated social stress in mice. Neuropsychopharmacology 37: 1885–1900.
Dulawa SC, Holick KA, Gundersen B, Hen R (2004). Effects of chronic fluoxetine in animal models of anxiety and depression. Neuropsychopharmacology 29: 1321–1330.
Fitzgerald PJ, Whittle N, Flynn SM, Graybeal C, Pinard CR, Gunduz-Cinar O et al (2014). Prefrontal single-unit firing associated with deficient extinction in mice. Neurobiol Learn Mem 113: 69–81.
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M et al (2005). Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102: 18620–18625.
Godlewski G, Alapafuja SO, Batkai S, Nikas SP, Cinar R, Offertaler L et al (2010). Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol 17: 1256–1266.
Gogolla N, Caroni P, Luthi A, Herry C (2009). Perineuronal nets protect fear memories from erasure. Science 325: 1258–1261.
Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A (2013a). Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci 34: 637–644.
Gunduz-Cinar O, Macpherson KP, Cinar R, Gamble-George J, Sugden K, Williams B et al (2013b). Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Mol Psychiatry 18: 813–823.
Haring M, Marsicano G, Lutz B, Monory K (2007). Identification of the cannabinoid receptor type 1 in serotonergic cells of raphe nuclei in mice. Neuroscience 146: 1212–1219.
Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F et al (2009). Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry 66: 9–16.
Hartley CA, McKenna MC, Salman R, Holmes A, Casey BJ, Phelps EA et al (2012). Serotonin transporter polyadenylation polymorphism modulates the retention of fear extinction memory. Proc Natl Acad Sci USA 109: 5493–5498.
Hashimotodani Y, Ohno-Shosaku T, Kano M (2007). Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J Neurosci 27: 1211–1219.
Heifets BD, Chevaleyre V, Castillo PE (2008). Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci USA 105: 10250–10255.
Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A (2008). Switching on and off fear by distinct neuronal circuits. Nature 454: 600–606.
Hill MN, Bierer LM, Makotkine I, Golier JA, Galea S, McEwen BS et al (2013). Reductions in circulating endocannabinoid levels in individuals with post-traumatic stress disorder following exposure to the World Trade Center attacks. Psychoneuroendocrinology 38: 2952–2961.
Hill MN, Ho WS, Hillard CJ, Gorzalka BB (2008a). Differential effects of the antidepressants tranylcypromine and fluoxetine on limbic cannabinoid receptor binding and endocannabinoid contents. J Neural Transm 115: 1673–1679.
Hill MN, McEwen BS (2010). Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry 34: 791–797.
Hill MN, Miller GE, Ho WS, Gorzalka BB, Hillard CJ (2008b). Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry 41: 48–53.
Holmes A, Rodgers RJ (2003). Prior exposure to the elevated plus-maze sensitizes mice to the acute behavioral effects of fluoxetine and phenelzine. Eur J Pharmacol 459: 221–230.
Holmes A, Quirk GJ (2010). Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders—the case of yohimbine. Trends Pharmacol Sci 31: 2–7.
Holmes A, Singewald N (2013). Individual differences in recovery from traumatic fear. Trends Neurosci 36: 23–31.
Jiao J, Nitzke AM, Doukas DG, Seiglie MP, Dulawa SC (2011). Antidepressant response to chronic citalopram treatment in eight inbred mouse strains. Psychopharmacology (Berl) 213: 509–520.
John CS, Currie PJ (2012). N-arachidonoyl-serotonin in the basolateral amygdala increases anxiolytic behavior in the elevated plus maze. Behav Brain Res 233: 382–388.
Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89: 309–380.
Karlsson RM, Choe JS, Cameron HA, Thorsell A, Crawley JN, Holmes A et al (2008). The neuropeptide Y Y1 receptor subtype is necessary for the anxiolytic-like effects of neuropeptide Y, but not the antidepressant-like effects of fluoxetine, in mice. Psychopharmacology (Berl) 195: 547–557.
Karpova NN, Pickenhagen A, Lindholm J, Tiraboschi E, Kulesskaya N, Agustsdottir A et al (2012). Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334: 1731–1734.
Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K et al (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19: 4544–4558.
Lemtiri-Chlieh F, Levine ES (2010). BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. J Neurophysiol 104: 1923–1932.
Mato S, Vidal R, Castro E, Diaz A, Pazos A, Valdizan EM (2010). Long-term fluoxetine treatment modulates cannabinoid type 1 receptor-mediated inhibition of adenylyl cyclase in the rat prefrontal cortex through 5-hydroxytryptamine 1A receptor-dependent mechanisms. Mol Pharmacol 77: 424–434.
Micale V, Cristino L, Tamburella A, Petrosino S, Leggio GM, Drago F et al (2009). Anxiolytic effects in mice of a dual blocker of fatty acid amide hydrolase and transient receptor potential vanilloid type-1 channels. Neuropsychopharmacology 34: 593–606.
Micale V, Di Marzo V, Sulcova A, Wotjak CT, Drago F (2013). Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol Ther 138: 18–37.
Milad MR, Quirk GJ (2012). Fear extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol 63: 129–151.
Milne RJ, Goa KL (1991). Citalopram. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs 41: 450–477.
Navarria A, Tamburella A, Iannotti FA, Micale V, Camillieri G, Gozzo L et al (2014). The dual blocker of FAAH/TRPV1 N-arachidonoylserotonin reverses the behavioral despair induced by stress in rats and modulates the HPA-axis. Pharmacol Res 87: 151–159.
Neumeister A, Normandin MD, Pietrzak RH, Piomelli D, Zheng MQ, Gujarro-Anton A et al (2013). Elevated brain cannabinoid CB receptor availability in post-traumatic stress disorder: a positron emission tomography study. Mol Psychiatry 18: 1034–1040.
Neumeister A, Seidel J, Ragen BJ, Pietrzak RH (2015). Translational evidence for a role of endocannabinoids in the etiology and treatment of posttraumatic stress disorder. Psychoneuroendocrinology 51: 577–584.
Norcross M, Mathur P, Enoch AJ, Karlsson RM, Brigman JL, Cameron HA et al (2008). Effects of adolescent fluoxetine treatment on fear-, anxiety- or stress-related behaviors in C57BL/6J or BALB/cJ mice. Psychopharmacology (Berl) 200: 413–424.
Papini S, Sullivan GM, Hien DA, Shvil E, Neria Y (2015). Toward a translational approach to targeting the endocannabinoid system in posttraumatic stress disorder: a critical review of preclinical research. Biol Psychol 104C: 8–18.
Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG (2009). Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology 34: 2699–2709.
Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ (2010). Induction of fear extinction with hippocampal-infralimbic BDNF. Science 328: 1288–1290.
Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR et al (2006). Pharmacological profile of the selective FAAH inhibitor KDS-4103 (URB597). CNS Drug Rev 12: 21–38.
Popova D, Agustsdottir A, Lindholm J, Mazulis U, Akamine Y, Castren E et al (2014). Combination of fluoxetine and extinction treatments forms a unique synaptic protein profile that correlates with long-term fear reduction in adult mice. Eur Neuropsychopharmacol 24: 1162–1174.
Ramikie TS, Nyilas R, Bluett RJ, Gamble-George JC, Hartley ND, Mackie K et al (2014). Multiple mechanistically distinct modes of endocannabinoid mobilization at central amygdala glutamatergic synapses. Neuron 81: 1111–1125.
Riddle MC, McKenna MC, Yoon YJ, Pattwell SS, Santos PM, Casey BJ et al (2013). Caloric restriction enhances fear extinction learning in mice. Neuropsychopharmacology 38: 930–937.
Roozendaal B, McEwen BS, Chattarji S (2009). Stress, memory and the amygdala. Nat Rev Neurosci 10: 423–433.
Rose TM, Reilly CA, Deering-Rice CE, Brewster C (2014). Inhibition of FAAH, TRPV1, and COX2 by NSAID–serotonin conjugates. Bioorg Med Chem Lett 24: 5695–5698.
Sanchez C, Hyttel J (1999). Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cell Mol Neurobiol 19: 467–489.
Shonesy BC, Bluett RJ, Ramikie TS, Báldi R, Hermanson DJ, Kingsley PJ et al (2014). Genetic disruption of 2-arachidonoylglycerol synthesis reveals a key role for endocannabinoid signaling in anxiety modulation. Cell Rep 9: 1644–1653.
Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ (2015). Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149: 150–190.
Spennato G, Zerbib C, Mondadori C, Garcia R (2008). Fluoxetine protects hippocampal plasticity during conditioned fear stress and prevents fear learning potentiation. Psychopharmacology (Berl) 196: 583–589.
Tovote P, Fadok JP, Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16: 317–331.
Trouche S, Sasaki JM, Tu T, Reijmers LG (2013). Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses. Neuron 80: 1054–1065.
Umathe SN, Manna SS, Jain NS (2011). Involvement of endocannabinoids in antidepressant and anti-compulsive effect of fluoxetine in mice. Behav Brain Res 223: 125–134.
Yoshida T, Uchigashima M, Yamasaki M, Katona I, Yamazaki M, Sakimura K et al (2011). Unique inhibitory synapse with particularly rich endocannabinoid signaling machinery on pyramidal neurons in basal amygdaloid nucleus. Proc Natl Acad Sci USA 108: 3059–3064.
Zarate J, Churruca I, Echevarria E, Casis L, Lopez de Jesus M, Saenz del Burgo L et al (2008). Immunohistochemical localization of CB1 cannabinoid receptors in frontal cortex and related limbic areas in obese Zucker rats: effects of chronic fluoxetine treatment. Brain Res 1236: 57–72.
Zhang G, Asgeirsdottir HN, Cohen SJ, Munchow AH, Barrera MP, Stackman RW Jr (2013). Stimulation of serotonin 2A receptors facilitates consolidation and extinction of fear memory in C57BL/6J mice. Neuropharmacology 64: 403–413.
Zhong P, Liu Y, Hu Y, Wang T, Zhao YP, Liu QS (2015). BDNF interacts with endocannabinoids to regulate cocaine-induced synaptic plasticity in mouse midbrain dopamine neurons. J Neurosci 35: 4469–4481.
Zhu PJ, Lovinger DM (2005). Retrograde endocannabinoid signaling in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurosci 25: 6199–6207.
Acknowledgements
We thank Ms Judy Harvey-White for her excellent assistance during the lipid extractions for LC-MS/MS measurements.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Gunduz-Cinar, O., Flynn, S., Brockway, E. et al. Fluoxetine Facilitates Fear Extinction Through Amygdala Endocannabinoids. Neuropsychopharmacol 41, 1598–1609 (2016). https://doi.org/10.1038/npp.2015.318
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2015.318
- Springer Nature Switzerland AG
This article is cited by
-
Effects of cannabidiol on fear conditioning in anxiety disorders: decreased threat expectation during retention, but no enhanced fear re-extinction
Psychopharmacology (2024)
-
The effect of SSRIs on fear learning: a systematic review and meta-analysis
Psychopharmacology (2023)
-
Reproductive experience alters the effects of diazepam and fluoxetine on anxiety-like behaviour, fear extinction, and corticosterone levels in female rats
Psychopharmacology (2023)
-
Local modulation by presynaptic receptors controls neuronal communication and behaviour
Nature Reviews Neuroscience (2022)
-
Additive effect of 5-HT2C and CB1 receptor blockade on the regulation of sleep–wake cycle
BMC Neuroscience (2019)