
Abstract
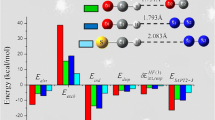
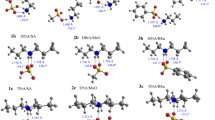
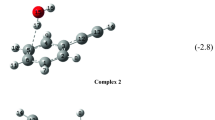
The structures, energies, and natural atomic charges of 2-dimethylaminophenol oxide, 2-Me2N-(O)C6H4OH, and 2-dimethylphosphinylphenol, 2-Me2P(O)C6H4OH, in three different conformations were computed at the ab initio MP2/6-31G* level. Computed natural charges indicate distributions of electron density in amine oxides and phosphine oxides that are quite different from what is normally assumed on the basis of the formal charges in the usual representations of these compounds. The charges on nitrogen and phosphorus in these compounds are typically computed to be approximately zero on nitrogen and +2 on phosphorus, and the oxygen is considerably more negative in the phosphine oxide than in the amino oxide. Electronegativity differences thus play a larger role and formal charges a smaller one in determining atomic charges in these compounds than is generally believed. Despite the more negative oxygen in phosphine oxides, amine oxides are computed to be considerably more basic when participating in hydrogen bonding. Calculations treating the computed natural charges on these six conformations as point charges for classical approximations of the coulombic energies support the idea that the quantum mechanically computed relative energies are largely determined by coulombic interactions.
Similar content being viewed by others
REFERENCES
Reed, A. E.; Schleyer, P. V. R. J. Am. Chem. Soc. 1990, 112, 1434.
Levy, J. B.; Hargittai, I., Molecular geometry: Bridging qualitative modeling and accurate computations. A comparative study of simple nitrogen and phosphorus derivatives. J. Mol. Struct. (Theochem), in press.
Borisenko, K. B.; Bock, C. W.; Hargittai, I. J. Phys. Chem. 1994, 98, 1442.
Levy, J. B.; Martin, N. H.; Hargittai, I.; Hargittai, M. J. Phys. Chem. A 1998, 102, 274.
Levy, J. B.; Hughes, S. V.; Esancy, M. K. Phosphorus, Sulfur, and Silicon 1993, 75, 75.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian 94, Revision E.1. Gaussian, Inc.: Pittsburgh PA, 1995.
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735.
Levy, J. B. Unpublished results.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Levy, J.B. Coulombic Basis for Relative Hydrogen-Bonding Basicities and Conformational Energies of Tertiary Amine Oxides and Phosphine Oxides. Structural Chemistry 9, 179–185 (1998). https://doi.org/10.1023/A:1022466913479
Issue Date:
DOI: https://doi.org/10.1023/A:1022466913479