
Abstract
KRAS is the most frequently mutated oncogene in human malignancies, observed in approximately two in five colorectal cancers (CRC). KRAS mutations were historically considered “undruggable” ten years ago and associated with resistance to EGFR targeted therapy. The success of finding allele-specific covalent KRASG12C inhibitors recently has made markedly breakthrough in KRAS targeted therapy, and has accelerated the discovery of agents targeting other KRAS mutants, such as G12D and G12V. Evidence in preclinical and clinical settings has proved excellent efficacy of several inhibitors in KRAS mutant CRC. Sotorasib and Adagrasib are currently changing the treatment paradigm for patients with metastatic CRC harboring KRASG12C mutation. The phenomenon that KRASG12C inhibition shows inferior efficacy in patients with CRC compared with non-small cell lung cancer has been observed in clinic due to drug resistance, and combination strategies to overcome the resistance are now being investigated in clinical trials. Here, we review the development of KRAS targeted treatment in CRC, mechanisms of resistance and potential combination strategies to improve efficacy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Colorectal cancer (CRC) is the third most common malignancy and ranks as the second in mortality worldwide [1]. According to the statistics of the National Cancer Center, the incidence and mortality of CRC have been on the rise in recent years [2]. Nearly 20% of patients have advanced disease when diagnosed with CRC (https://seer.cancer.gov/statfacts/html/ colorect.html), and about 30–50% patients who present with early-stage or localized disease will later develop metastases even after systemic treatment [3, 4]. Compared with early and localized CRC, the prognosis of advanced CRC is poor, with a 5-year survival rate of 15% (https://seer.cancer.gov/statfacts/html/ colorect.html). Therefore, it’s urgent to develop new treatment strategies to prolong survival period of CRC patients. With the rapid development of molecular biology, the understanding of tumorigenesis is further deepened. At present, it is widely believed that the occurrence and development of CRC involve complex pathways and gene changes, and targeted therapy brings new hope for patients with advanced CRC.
As the most important member of RAS family, KRAS is one of the most commonly mutated oncogenes in CRC, with an incidence of 37.8% in China [5]. Majority of KRAS mutations are single-base missense mutations and more than 95% occurred in three major hotspots including residues G12, G13 and Q61 [6]. The G12D missense mutation results in the replacement of glycine with aspartic acid, accounting for about 37% of KRAS mutations, while G12C (~ 5%) mutation represents a nucleotide substitution at c.34G > T, coding for the amino acid cysteine instead of glycine [5, 7].
RAS, a kind of guanylate binding protein with GTP hydrolase (GTPase) activity located on the cell membrane, is in inactive state when binding to GDP. Once activated by upstream signaling, RAS oncoproteins cycle to active GTP-bound state, followed by activation of downstream signaling. At the same time, GTP hydrolysis prevents the continuous activation of the RAS pathway. However, oncogenic KRAS mutations diminish GTPase activity and prevent KRAS deactivation, thus leading to persistent downstream signaling, along with abnormal cell proliferation and oncogenesis [8]. In CRC patients, KRAS mutations are found that tend to occurred in the right colon and have a negative impact on the prognosis [9]. And a worse outcome is observed to be associated with KRASG12C mutation [10]. Furthermore, the abnormal activation of KRAS disturbs the upstream regulatory signals to transmit, leading to resistance to EGFR targeted therapies [11]. However, the development of drugs targeting KRAS is confronted with many obstacles, and it was considered as the “undruggable gene” mainly for the following reasons [12, 13]: (1) the differences between KRAS mutant and the wild type are too small to selectivity target; (2) the picomolar affinity of KRAS protein for GTP and high intracellular concentrations of GTP make it difficult for antagonists to compete. (3) mutant KRAS protein lacks a pharmacologically actionable small-molecule-binding pocket. Fortunately, the recent successful identification of compounds that covalently target KRASG12C oncoprotein makes it possible to directly inhibit KRAS mutation, providing new viable therapeutic strategies for advanced CRC, and also bringing insight toward the rational development of agents targeting other mutations of KRAS.
In this article, we review the advances in targeted therapy for CRC with KRASG12C mutation. Furthermore, we try to explore the potential mechanisms of resistance to KRASG12C inhibition and propose combination strategies to improve the efficacy of KRASG12C inhibitors. Finally, we report the development of other agents targeting non-KRASG12C.
2 Advances in targeting KRASG12C with small molecule inhibitors
2.1 ARS-853
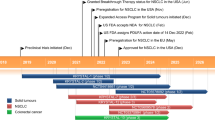
Although Ostrem et al. identified a new allosteric pocket, namely switch II pocket (S-IIP) in the G12C mutant of KRAS oncoprotein in 2013, making it possible to develop irreversible and mutant-specific inhibitors of KRAS, it’s about 5 years later when the first covalent compound with high potency and selectivity for KRASG12C emerged and showed promising therapeutic potential both in vitro and vivo [14, 15]. Mechanically, the small compound irreversibly binds to KRASG12C relying on the mutant cysteine 12 sits which close to both the pocket and effector binding switch region according to crystallographic studies, and then impairs RAS function through two distinct mechanisms: (1) shift the relative nucleotide affinity of RAS to favor GDP over GTP, leading to accumulation of inactive RAS-GDP; (2) occupy the S-IIP region to disrupt interactions with downstream effectors [14].
Focused on the novel S-IIP, Patricelli and colleagues subsequently discovered a covalent compound, ARS-853, with robust cellular activity against KRASG12C in a low micromolar range. After treated with ARS-853, H358 cells (KRASG12C cell lines) performed significant loss of KRAS-CRAF interactions, and consistent inhibition of downstream both MAPK and PI3K signaling pathways. Increased apoptosis and growth inhibition were also observed in H358 cells, and all of the aforementioned inhibitory effects of ARS-853 were not observed in other KRAS mutant oncoprotein (like G12S). Interestingly, the researchers further investigated KRASG12C mutant function in vitro by using ARS-853 as a pharmacologic tool and demonstrated that KRASG12C is not constitutively active but rather cycles rapidly between inactive GDP-bound and active GTP-bound state, and the KRASG12C GTP fraction is modulated by upstream signaling factors. The combination of EGFR inhibitor increases compound accessible GDP-bound KRASG12C engagement, consistent with complete inhibition of MAPK/PI3K signaling, leading to high levels of apoptosis in H358 cells [16]. Consistent with these results of this study, Lito et al. further confirmed that KRASG12C mutant oncoprotein retains GTPase activity and drug-bound KRASG12C-GDP exits the cycle due to insensitivity to nucleotide exchange, thus decreasing the level of KRASG12C-GTP [17]. These findings provide insight for exploring synergistic combinations and mechanisms of resistance to KRAS mutation inhibitors. Unfortunately, the short plasma metabolic stability and poor oral bioavailability in mice of the ARS-853 series made them unsuitable for translation into in vivo exploration [15].
2.2 ARS-1620
Following optimizing covalent binding activity and pharmacologic properties of previously reported S-IIP KRASG12C inhibitors through designing a structurally distinct quinazoline scaffold, researchers reported ARS-1620 as the first small molecule inhibitor covalently targeting KRASG12C mutant in vitro and in vivo. In p.G12C mutant cell lines, ARS-1620 exhibited rapid target occupancy of G12C in a dose- and time-dependent manner. Transcriptome profiling and clustering analysis revealed a significant downregulation of KRAS regulated network enriched of genes, such as E2F transcription factors, MYC regulatory network and ERK activation following ARS-1620 treatment. In addition, ARS-1620 exhibited high oral bioavailability and blood stability with capacity to elicit more than 70% KRASG12C target occupancy for an extended period of time at a single dose in vivo, along with marked tumor regression in subcutaneous xenograft models bearing KRAS p.G12C at a dose of 200 mg/kg once a day. What’s more, ARS-1620 was well tolerated and exhibited excellent antitumor effects in KRASG12C mutated patient-derived tumor xenograft model with NSCLCs and pancreatic adenocarcinoma. And no clinical signs or toxicity were observed in CD-1 mice over a 3-week period of treatment [15]. However, the small volume of the pocket occupied by ARS-1620, which was confirmed by the X-ray co-crystal structure of KRASG12C bound to ARS-1620, results in its suboptimal potency and failed to extent ARS-1620 to clinical testing [18].
2.3 AMG-510/sotorasib
Canon et al. discovered a surface His95 groove of KRASG12C which could enhance interactions with inhibitors. AMG 510 was identified as the preferred candidate of His95 groove-binding molecules, and performed an approximately 10-fold increase in potency with GDP-bound KRASG12C compared to ARS-1620. A rapid and sustained inhibiting effect of AMG 510 treatment selectively on KRASG12C signaling was demonstrated by measuring the level of phosphorylation of ERK. And it significantly impaired the growth of KRASG12C cell lines, as well as KRASG12C-mutant colorectal-cancer patient-derived xenografts. Further studies suggested an enhanced antitumor effect in the combination with MEK inhibitors. Intriguingly, AMG 510 markedly increased immune cell infiltration including proliferating CD3 + T cells, CD8 + T cells and innate immune cells (like macrophages and dendritic cells). Analysis of immune-associated genes from CT-26 KRASG12C tumors after 2 days of AMG 510 treatment showed that AMG 510 enhanced the gene expression of interferon signaling, chemokines including CXCL10 and CXCL11, antigen processing, cytotoxic and natural killer cell, thereby promoting the formation of a pro-inflammatory microenvironment. In contrast to MEK inhibition, AMG 510 promoted T cell priming, and antigen recognition of tumor cell. Furthermore, combination with anti-PD-1 therapy promoted the establishment of long-term anti-tumor T cell response in mice with CT-26 KRASG12C tumors [18]. All these preclinical findings strongly suggest that AMG 510 might be an effective anti-tumor agent targeting KRASG12C tumors as monotherapy or combined with other treatments.
AMG510, also named Sotorasib is the first covalent inhibitor specifically targeting KRAS to enter clinical trials. In a phase 1 trial of Sotorasib for patients with KRASG12C mutant advanced solid tumors (CodeBreaK100, NCT03600883), 42 CRC were enrolled in a dose escalation and expansion cohort with a median follow-up time of 12.8 months. No dose-limiting toxic effects or treatment-related deaths occurred. Although the efficacy of Sotorasib was more prominent in NSCLC subgroup, most patients (73.8%) in the CRC cohort had disease control, with a duration of stable disease of 5.4 months (range, 2.5 + to 11.1+) and median progression-free survival (PFS) of 4.0 months (range, 0.0 + to 11.1+) [19]. In the single-arm, phase 2 CodeBreaK100 clinical trial, Sotorasib showed modest anti-tumor activity and manageable safety in 62 patients with previously treated and refractory KRASG12C mutant CRC [20]. Only six (9.7%) patients received objective response (all with partial response). 51 patients (82.3%) achieved disease control (complete response, partial response, or stable disease), with a median percentage of best tumor shrinkage of 46.3% in 41 (66%) responders. The median PFS was 4.0 months and median overall survival (OS) was 10.6 months. No fatal events were observed. A total of 7 (12%) patients had grade 3/4 treatment-related adverse events (TRAEs), and two (3%) patients suffered serious treatment-related adverse events (back pian and acute kidney injury).
2.4 MRTX849/adagrasib
By optimizing the structure of candidate compounds that inhibit KRASG12C, investigators identified MRTX849 as another potent, selective, orally bioavailable and irreversible covalent inhibitor binding to the inactive GDP-bound form of KRASG12C [21]. In H358 xenograft-bearing mice, MRTX849 treatment demonstrated dose-dependent modification of KRAS, inhibition of KRAS pathway, and antitumor activity. Furthermore, in 26 human KRASG12C -mutant cell line-derived xenograft or patient-derived xenograft models including lung, colon, pancreas, cervix and esophagus cancer, MRTX849 elicited more than 30% volume shrinkage in 65% models after 3 weeks of treatment at a dose projected to achieve maximal target-inhibition in most models, suggesting that MRTX849 has a broad antitumor efficacy through a KRASG12C-dependent mechanism. RNA sequencing analysis showed that not individual genetic alteration, such as KRAS-mutant allele frequency, TP53, STK11, or CDKN 2 A, but baseline expression of RTKs and of regulators of early cell-cycle transition might predict the antitumor response of MRTX849 [22].
Consistent with the preclinical results, adagrasib (MRTX849) has demonstrated well toleration and excellent antitumor activity for advanced solid tumors harboring the KRASG12C mutation in a multicohort phase I/IB study (KRYSTAL-1). In the dose escalation cohort, 4 CRC were enrolled. Among them, 2 patients received 600-mg twice-a-day dose, one of which obtained confirmed partial response with a duration of response (DOR) of 4.2 months. PFS durations of the 2 patients were 3.3 and 5.5 months, and OS durations are 9.5 and 10.5 months, respectively [23]. Based on these encouraging results, investigators are developing further clinical trials. Preliminary results have reported promising clinical activity in heavily pretreated patients with KRASG12C-mutant CRC. As of Jun 16 2022, 44 patients had received adagrasib monotherapy and 32 patients were treated with adagrasib + cetuximab. In the monotherapy cohort with a median follow-up of 20.1 months, 43 patients were evaluable for clinical activity. The objective response rate (ORR) was 19%, and disease control rate (DCR) was 86%. Median DOR was 4.3 months and median PFS was 5.6 months. In the combined treatment group, ORR was 46% (13/28) and DCR was 100% (28/28). Median DOR was 7.6 months and median PFS was 6.9 months [24]. Another multicenter, open-label, randomized phase 3 study evaluating the efficacy of adagrasib + cetuximab vs. chemotherapy in patients with previously treated advanced CRC with KRASG12C-mutation is ongoing now [25].
2.5 Other KRASG12C inhibitors
Several other KRASG12C inhibitors which work through a similar mechanism have moved forward into clinical trials recently. GDC-6036, a highly potent and selective KRASG12C inhibitor and also known as RG6330, is currently in clinical development as monotherapy and in combination with anti-PD-1 antibody, anti-EGFR inhibitor, SHP2 inhibitor or PI3Kα inhibitor in solid tumors (NCT04449874). JAB-21,822 is also being investigated alone (NCT05009329) and in combination with anti-EGFR inhibitor (NCT05002270, NCT05194995) as well as SHP2 inhibitor (NCT05288205) in phase I/II trials for patients with solid tumor harboring KRASG12C. Other compounds targeting KRASG12C which are currently undergoing clinical investigation include YL-15,293, HS-10,370, HBI-2438, GFH925, D3S-001, JNJ-74,699,157, RMC-6291 and RMC 6336.
3 Mechanisms of resistance to KRASG12C inhibitors
With the breakthrough of KRASG12C mutation from undruggable to druggable, patients with KRASG12C mutant solid tumor, which was more common in NSCLC and CRC, had benefited from KRASG12C targeted therapy a lot. However, the efficacy of KRASG12C inhibitor monotherapy such as adagrasib and sotorasib was limited, especially for CRC patients [19, 20, 23, 26]. Finding out the exact mechanisms of resistance met urgent need in clinical.
As we all know, KRASG12C inhibitors were designed to combine an allosteric regulatory site of switch II binding pocket covalently and therefore impaired the function of RAS. Destruction of the covalent binding might confer resistance [27]. Awad et al. collected blood and tissue samples before therapy and after disease progression from 38 KRASG12C mutant patients (containing 10 CRC patients) who had received adagrasib and performed genomic and histologic analyses to explore resistance mechanism. Putative gene mutations that might cause resistance to adagrasib were found in 17 patients, and seven patients were found to have incorporate multiple resistance mechanisms. It was worth noting that novel H95Q and H95R mutations within the adagrasib-binding pocket were found in two CRC patients, which might disrupt the covalent binding of adagrasib. Moreover, acquired activating mutations in KRAS, such as G12D/R/V, G13D, Q61H, and Y96C, and high levels of KRASG12C allele amplification were found in CRC, which might also confer resistance [28]. Interestingly, mutations at different allele sites of KRAS seemed to have different effects on adagrasib and sotorasib, indicating the existence of drug-specific resistance mutations [29].
Actually, RAS is at the center of the complex RTK-RAS-MAPK signaling cascade, therefore acquired mutations or reactivation of bypass or upstream pathways might also impact the efficacy of KRASG12C inhibitors [22, 30]. Acquired bypass activating mutations of the RTK-RAS-MAPK pathway such as NRAS, BRAF, and MAP2K1 that conferred resistance to adagrasib or sotorasib in CRC were detected in prior studies [28, 31]. It was worth to note that KRASG12C mutant CRC was found to have a high level of basal RTK activation and maintain sensitivity to upstream RTK signaling, particularly EGFR. Stimulation of EGF resulted in activation of KRAS downstream effectors in KRASG12C mutant CRC cell lines, whereas similar phenomenon was not observed in NSCLC models, which might partly explain the gap in efficacy to KRASG12C inhibitors in NSCLC and CRC. Concomitant EGFR and KRASG12C inhibition showed synergistic antitumor activity compared to monotherapy both in cell lines and in patient-derived models [32]. Moreover, EGFR was found to promote the transition of KRAS from inactivate (KRAS-GDP) to activate (KRAS-GTP) conformation [33], which further supported that EGFR signaling might be the dominant mechanism of resistance to KRASG12C inhibitors in CRC.
Other potential resistance mechanisms published in CRC included rearrangement or fusion of oncogene (CCDC6-RET, EML4-ALK, FGFR3-TACC3, AKAP9-BRAF) [28], inhibition of cell cycle genes [22], and so on.
4 Combination strategies to improve the efficacy of KRASG12C inhibitors
4.1 Combination with EGFR inhibition
Preclinical study had identified EGFR signaling as the dominant mechanism of resistance to KRASG12C inhibitor in CRC, and combinatorial targeting of EGFR and KRASG12C showed promising efficacy in CRC models in vivo and vitro [32]. Several clinical trials of combination of KRASG12C and EGFR inhibitor are ongoing (Table 1). In a cohort of phase Ib study CodeBreak 101 with KRASG12C mutation CRC patients (n = 40), combination of sotorasib and panitumumab showed an ORR of 30% and DCR of 90%. Althouth TRAEs are observed in 92.5% patients, no TRAE was Grade > 3 or led to the discontinuation of treatment [34]. Similarly, in KRSTAL-1 study, CRC patients harboring KRASG12C mutation (n = 32) were treated with adagrasib and cetuximab. The ORR was 46% among 28 evaluable patients and DCR was 100%, and median PFS was 6.9 month (median prior lines of systemic therapy was 3) [24]. And a phase III study comparing the efficacy of adagrasib in combination with cetuximab versus FOLFOX or FOLFIRI in second-line treatment setting in patients with KRASG12C CRC is ongoing (NCT04793958).
4.2 Combination with SHP2 inhibition
SHP2, a non-receptor protein tyrosine phosphatase, is required for full activation of RAS. Preclinical study has shown promising anticancer activity from KRASG12C inhibitor combination with SHP2 inhibitor, which could increase GDP-bound KRASG12C [35, 36]. However, the report of this combination in patients was limited. In a phase Ib study of sotorasib in combination with RMC-4630 (a SHP2 inhibitor) in KRASG12C mutated NSCLC, preliminary results showed that ORR in all enrolled patients was 27% (n = 11) and in KRASG12C inhibitor-naive patients (n = 6) was 50%. More evidence from clinical trials is eagerly awaited as more data are reported and more patients are enrolled (Table 1).
4.3 Combination with immunotherapy
Although KRAS mutation is not associated with efficacy of immunotherapy, KRAS mutation CRC patients do have increased Tregs, while decreased macrophage M1 and activated CD4 memory T cell compared with KRAS wild patients [37]. The immunosuppressive effect has also been seen in KRAS mutation NSCLC and pancreatic cancer [38, 39]. However, KRASG12C inhibitor have showed the ability to recovery the tumor immune microenvironment [18, 40]. In preclinical trial, the combination of KRASG12C inhibitor with anti-PD-1 therapy increased CD8 + T cell infiltration and long-term tumor-specific immune responses [18]. And the response in patients with this combination regimen is currently being evaluated in clinical trials (Table 1). In the CodeBreaK 100/101 Ib study, prelimary results about the safety and tolerability of sotorasib in combination with pembrolizumab or atezoliumab have been reported in KRASG12C mutation NSCLC (n = 58). the ORR was 29%, DCR was 83%, and median OS was 15.7 month. However, grade 3 or higher TRAE with this combination was reported in 65.8% of patients, mainly hepatotoxicity [41]. More data on safety, tolerability and efficacy of combination therapy in patients with CRC are eagerly awaited.
4.4 Other combination strategies
Combined inhibition of KRASG12C and other components of the RAS pathway has been investigated to enhance the effect of KRASG12C inhibition. The protein Son of Sevenless (SOS) 1 is a key guanine nucleotide exchange factor that can interact with RAS and stimulate it to cycle from the inactive GDP-bound state to the active GTP-bound state. Inhibition of SOS1 has been proved to show synergistic antiproliferative activity with KRASG12C inhibitor in KRASG12C tumor cell lines [42]. The safety, tolerability, molecular effects and clinical activity of BI 1,701,963, a SOS1 inhibitor, in combination with KRASG12C inhibitor are being investigated in clinical trials (NCT04973163, NCT04975256). Combinations of inhibitors targeting two main downstream signaling pathways, RAF-MEK-ERK and PI3K-AKT-mTOR pathway, are currently undergoing clinical investigation. Cell-cycle inhibition offers another potential combination strategy for synergy with KRASG12C inhibitors (Table 1). Future results of these clinical trials are promising to provide alternative targeted therapy strategies for patients with KRAS-mutated CRC.
5 Advances in inhibitors beyond KRASG12C
The success in targeting KRASG12C inspires the development of efficient therapeutic agents for other KRAS mutations. However, targeting KRAS non-G12C mutation is far more challenging as other mutants lack an allosteric switch II binding pocket that can be covalently bound by small-molecular drugs and have a lower GTP hydrolysis rate compared with G12C mutant. Mutant-selective and pan-KRAS inhibitors are the two major directions of KRAS-targeting therapy. Of the selectively targeting agents, KRASG12D inhibitors are in the leading position. MRTX1133 is a highly selective noncovalent inhibitor of KRASG12D mutation which binds to the switch-II pocket of both inactive and active states of KRASG12D protein with a > 500-fold selectivity compared with wild-type KRAS [43]. MRTX1133 has demonstrated cytoreductive antitumor efficacy across a panel of cell- and patient-derived xenograft tumor models including pancreatic cancer, colorectal cancer, and gastric cancer [44]. It will be exciting to see MRTX1133 enter clinical trials shortly. RMC-9805(RM-036) is a novel agent using a tri-complex platform that enables selective covalent targeting of KRASG12D “on” status. RMC-9805 showed a remarkable MAPK pathway suppression and regression of KRASG12D mutant cancer in vivo [45]. A similar strategy is also used in the development of a KRASG13C inhibitor, RMC-8839 which is currently in the IND-enabling process [46]. The tri-complex platform that exploits the surface of a chaperone protein and a target protein to form a new ligand-binding pocket for the small molecule inhibitor is supposed to unlock the opportunity for selective inhibitors across all KRAS mutants [7].
Unselective pan-KRAS inhibition by directly binding RAS protein has also been intensively investigated. RMC-6236 is a first-in-class potent tri-complex KRASG12X (ON) inhibitor that was demonstrated to induce dose-dependent and durable suppression of the MAPK pathway in preclinical models [47]. Significant tumor regression was observed in KRASG12D, KRASG12V and KRASG12R mutant patient-derived xenografts, and the phase I clinical trial of RMC-6236 is ongoing (NCT5379985) [47]. And BI-pan-KRAS3, a pan-KRAS probe compound also showed promising clinical activity in KRASG12D and KRASG13D mutant colorectal cancer [48]. Furthermore, small-molecule drugs bounding to Switch I/II pocket of all RAS isoforms, exemplified by BI-2852 and JAM20, were also demonstrated to reduce oncogenic RAS-mediates tumorigenesis in vivo [49, 50].
Inhibition of key downstream effectors of KRAS, such as MRK and ERK which are members of the RAF-MEK-ERK, has shown limited clinical activity due to the activation of feedback networks, among which SOS1 is an important regulator of the negative feedback network [51]. In preclinical studies, combination of SOS1 (BI 17-1963) and MEK (trametinib) inhibition resulted robust antitumor efficacy and attenuated adaptive resistance to MEK inhibitor in a broad spectrum of KRAS-mutated CRC PDX models [52]. A phase I clinical trial currently undergoing in patients with advanced KRAS-mutated cancers to evaluate safety, tolerability and preliminary efficacy of BI 1,701,963 in combination with trametinib (NCT04111458). Table 2 has shown clinical trials of other non-KRAS-targeted therapies in patients with KRAS mutant solid tumor including CRC. In near the future, therapeutically active KRAS and non-KRAS inhibitors are expected to address extensive unmet clinical demands for patients with KRAS mutant solid tumors.
Proteolysis-targeted chimeras (PROTACS) are emerged as new class of agents for KRAS inhbitation. These bifunctional molecules simultaneously bind to a protein of interest and an E3 ligase to triger proteasomal degradation of the interested protein by ubiquitin-proteasome systerm [53, 54]. Bond MJ et al. designed the first PROTAC, LC-2, that successfully degraded endogenous KRASG12C. LC-2 colvalently binds KRASG12C and recruits VHL by conjugation of the covalent KRASG12C inhibitor MRTX849 to the E3 ligase VHL, and then the mutant KRAS is broken up rapidly along with a sustaind suppression of MAPK signaling in KRASG12C cell lines [55]. Compound 17f is another PROTAC that indirectly inhibits KRAS by efficiently inducing the degradation of PDEδ, a key shuttling factor which promotes KRAS diffusion, proper localization and signal transduction. Significant tumor growth inhibition of 17f has been observed in a KRAS mutated colorectal cancer xenograft model [56]. Compared to classical small-molecule inhibitors, PROTACs may only need to bind the targets with low or moderate-affinity small-molecule ligands to induce degratation, which provides opportunities to degrade “undruggable” proteins [57].
The KRAS mutant proteins are highly immunogenic and could be used to develop precision imunoreagents to promote anti-tumor inmmue response. mRNA-5671/V941 is a lipid nanoparticle-formulated mRNA-based cancer vaccine that targets four of the most commonly occurring KRAS mutations (G12C, G12D, G12V and G13D). After administration, the vaccine V941 is taken up and translated by antigen presenting cells (APCs), and then the epitopes are presented on the surface of the APCs via major histocompatibility complex (MHC) molecules, stimulating both the cytotoxic T-lymphocyte (CTL) and memory T-cell-dependent immune responses against tumor cells harboring these specific KRAS mutations [57]. A phase 1 clinical trial (NCT03948763) is ongoing to evaluate the safety and tolerability of V941 as a monotherapy and in combination with pembrolizumab among patients with KRAS mutant advanced or metastatic solid tumors including CRC. ELI-002 2P is another tumor vaccine comprised of a lymph-node targeted amphiphile (AMP)-modified G12D and G12R mutant KRAS petides together with an AMP-modified CpG oligonucleotide adjuvant. Preliminary antitumor acitivity in KRAS mutated pancreatic ductal adenocarcinoma, CRC and other solid tumors will be described in a phase 1 dose escalation study (AMPLIFY-201, NCT04853017) [58]. KRAS mutants are also promising to be targeted specific antigens for the adoptive cell therapy (ACT). ACT is a form of immunotherapy in which a patient’s own immune cells reactive to neoantigens are expanded in vitro and then re-introduced into the patient, ultimately eliciting an immune response against any cancer cells expressing the targeted antigens [59]. Tran E et al. had successfully identified a polyclonal CD8 + T-cell response that specifically against mutant KRAS G12D in tumor-infiltrating lymphocytes from a paitent with metastatic CRC. Objective regression of all seven lung metastases was observed after infusiong approximately 1.11 × 1011 CD8 + T cells which specifically targeted KRAS 12D and HLA-C*08:02 [60]. Althouth the use of vaccines and ACT has not achieved the expected results, they have emerged as another promising strategies to target KRAS directly.
6 Conclusion
Based on intense efforts and rapid progress in targeting KRAS, many clinical trials related to KRAS inhibitors are currently undergoing in solid tumors. Although the direct-targeted KRASG12C inhibitors, adagrasib and sotorasib have shown preliminary clinical efficacy in colorectal cancer, acquired resistance should be considered and addressed to increase the efficacy of KRASG12C inhibition. Improved understanding of KRAS signaling and complexed resistance mechanisms to G12C inhibition has provided potential combination therapy strategies. Combination of EGFR and KRASG12C inhibitors has shown a synergistic response in CRC. What’s more, exciting results of combined treatment of G12C inhibitors with immunotherapy or other inhibitors targeting the key effectors of KRAS signaling (like SHP2, SOS1, CD4/6) are well worth to wait. Besides, significant breakthrough in targeting KRAS non-G12C has been noted in preclinical studies. PROTACS, vaccines, adoptive cell therapy are emerging strategies to expand the scope of targeted therapy for KRAS mutant CRC. In conclusion, the development of KRAS inhibitors is providing alternative therapeutic strategies and changing the treatment paradigm for advanced CRC with KRAS mutation.
Availability of data and materials
Not applicable.
Abbreviations
- CRC:
-
Colorectal cancer
- KRAS:
-
Kirsten rats arcomaviral oncogene homolog
- RAS:
-
Rat sarcoma
- GTP:
-
Guanosine triphosphate
- GDP:
-
Guanosine diphosphate
- EGFR:
-
Epidermal growth factor receptor
- S-IIP:
-
Switch II pocket
- MAPK:
-
Mitogen-activated protein kinase
- PI3K:
-
Phosphoinositide 3-kinase
- ERK:
-
Extracellular-signal-regulated kinase
- NSCLC:
-
Non-small cell lung carcinoma
- MEK:
-
Mitogen-activated extracellular signal-regulated kinase
- CXCL:
-
Chemokine (C-X-C motif) ligand protein
- PD-1:
-
Programmed death 1
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- TRAEs:
-
Treatment-related adverse events
- RTKs:
-
Receptor tyrosine kinase
- DOR:
-
Duration of response
- ORR:
-
Objective response rate
- DCR:
-
Disease control rate
- SOS1:
-
Son of Sevenless SOS 1
- mTOR:
-
Mammalian target of rapamycin
- PROTACs:
-
Proteolysis-targeted chimeras
- APCs:
-
Antigen presenting cells
- MHC:
-
Major histocompatibility complex
- CTL:
-
Cytotoxic T-lymphocyte
- ACT:
-
Adoptive cell therapy
- mCRC:
-
Metastatic colorectal cancer
- VEGF:
-
Vascular endothelial growth factor receptor
- CDK:
-
Cyclin dependent kinase
- NTRK:
-
Neurotrophic receptor tyrosine kinase
- PLK1:
-
Polo like kinase 1
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Zheng R, Zhang S, Zeng H, et al. Cancer incidence and mortality in China, 2016. J Natl Cancer Cent. 2022;2:1–9.
Scheer A, Auer RAC. Surveillance after curative resection of colorectal cancer. Clin Colon Rectal Surg. 2009;22:242–50.
Guraya SY. Pattern, stage, and time of recurrent colorectal cancer after curative surgery. Clin Colorectal Cancer. 2019;18:e223-8.
Li Y, Gao J, Ji C, et al. Infrequent gene mutations of KRAS, NRAS and BRAF in colorectal cancer and their clinical significance: a report of 1 513 cases. Chin J Dig Surg. 2020;19:315–23.
Dienstmann R, Connor K, Byrne AT. Precision therapy in RAS mutant colorectal cancer. Gastroenterology. 2020;158:806–11.
Parikh K, Banna G, Liu SV, et al. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol. 2022;15:152.
Punekar SR, Velcheti V, Neel BG, Wong K-K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. 2022;19:637–55.
Haigis KM. KRAS alleles: the devil is in the detail. Trends Cancer. 2017;3:686–97.
Modest DP, Ricard I, Heinemann V, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol. 2016;27:1746–53.
Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65.
Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–51.
Ledford H, Cancer. The ras renaissance. Nature. 2015;520:278–80.
Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–51.
Janes MR, Zhang J, Li L-S, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell. 2018;172:578-89.
Patricelli MP, Janes MR, Li L-S, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–29.
Lito P, Solomon M, Li L-S, et al. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–8.
Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Hong DS, Fakih MG, Strickler JH, et al. KRAS inhibition with Sotorasib in advanced solid tumors. N Engl J Med. 2020;383:1207–17.
Fakih MG, Kopetz S, Kuboki Y, et al. Sotorasib for previously treated colorectal cancers with KRAS mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022;23:115–24.
Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate, a covalent KRAS inhibitor for the treatment of cancer. J Med Chem. 2020;63:6679–93.
Hallin J, Engstrom LD, Hargis L, et al. The KRAS inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71.
Ou S-HI, Jänne PA, Leal TA, et al. First-in-human phase I/IB dose-finding study of Adagrasib (MRTX849) in patients with advanced solid tumors (KRYSTAL-1). J Clin Oncol. 2022;40:2530–8.
Klempner SJ, Weiss J, Pelster M, et al. LBA24 KRYSTAL-1: updated efficacy and safety of adagrasib (MRTX849) with or without cetuximab in patients with advanced colorectal cancer (CRC) harboring a KRASG12C mutation. Ann Oncol. 2022;33:1391.
Tabernero J, Bendell J, Corcoran R, et al. P-71 KRYSTAL-10: a randomized phase 3 study of adagrasib (MRTX849) in combination with cetuximab vs chemotherapy in patients with previously treated advanced colorectal cancer with KRASG12C mutation. Ann Oncol. 2021;32:121.
Skoulidis F, Li BT, Dy GK, et al. Sotorasib for Lung cancers with p.G12C mutation. N Engl J Med. 2021;384:2371–81.
Tanaka N, Lin JJ, Li C, et al. Clinical Acquired Resistance to KRAS Inhibition through a Novel KRAS Switch-II Pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021;11:1913–22.
Awad MM, Liu S, Rybkin II, et al. Acquired Resistance to KRAS Inhibition in Cancer. N Engl J Med. 2021;384:2382–93.
Koga T, Suda K, Fujino T, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, Sotorasib and Adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. 2021;16:1321–32.
Tsai YS, Woodcock MG, Azam SH, et al. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J Clin Invest. 2022;132:e155523.
Li BT, Velcheti V, Price TJ, et al. Largest evaluation of acquired resistance to sotorasib in KRAS p.G12C-mutated non–small cell lung cancer (NSCLC) and colorectal cancer (CRC): plasma biomarker analysis of CodeBreaK100. J Clin Oncol. 2022;40:102–2.
Amodio V, Yaeger R, Arcella P, et al. EGFR Blockade reverts resistance to KRAS inhibition in colorectal cancer. Cancer Discov. 2020;10:1129–39.
Xue JY, Zhao Y, Aronowitz J, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577:421–5.
Kuboki Y, Yaeger R, Fakih M, et al. 45MO Sotorasib in combination with panitumumab in refractory KRAS G12C-mutated colorectal cancer: safety and efficacy for phase ib full expansion cohort. Ann Oncol. 2022;33:1445–S1446.
Liu C, Lu H, Wang H, et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res. 2021;27:342–54.
Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021;218:e20201414.
Liu J, Huang X, Liu H, et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J Transl Med. 2021;19:27.
Ricciuti B, Alessi Jv, Elkrief A, et al. Dissecting the clinicopathologic, genomic, and immunophenotypic correlates of KRASG12D mutated non-small cell lung cancer. Ann Oncol. 2022;33:1029-40.
Gu M, Gao Y, Chang P. KRAS Mutation Dictates the Cancer Immune Environment in Pancreatic Ductal Adenocarcinoma and Other Adenocarcinomas. Cancers. 2021;13:2429.
Briere DM, Li S, Calinisan A, et al. The KRAS inhibitor MRTX849 reconditions the Tumor Immune Microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther. 2021;20:975–85.
Li BT, Falchook GS, Durm GA, et al. OA03.06 CodeBreaK 100/101: first report of safety/efficacy of Sotorasib in combination with Pembrolizumab or Atezolizumab in advanced KRAS p.G12C NSCLC. J Thorac Oncol. 2022;17:10-S11.
Hillig RC, Sautier B, Schroeder J, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A. 2019;116:2551–60.
Zheng Q, Peacock DM, Shokat KM. Drugging the Next Undruggable KRAS Allele-Gly12Asp. J Med Chem. 2022;65:3119–22.
Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS inhibitor. J Med Chem. 2022;65:3123–33.
Knox JE, Jiang J, Burnett GL, et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, tri-complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Res. 2022;82:3596–6.
Schulze CJ, Cregg J, Seamon KJ, et al. Abstract 3598: a first-in-class tri-complex KRASG13C(ON) inhibitor validates therapeutic targeting of KRASG13Cand drives tumor regressions in preclinical models. Cancer Res. 2022;82:3598–8.
Koltun ES, Rice MA, Gustafson WC, et al. Abstract 3597: direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. 2022;82:3597–7.
Hofmann MH, Gerlach D, Misale S, et al. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov. 2022;12:924–37.
Wallon L, Khan I, Teng KW, et al. Inhibition of RAS-driven signaling and tumorigenesis with a pan-RAS monobody targeting the switch I/II pocket. Proc Natl Acad Sci U S A. 2022;119:e2204481119.
Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 2019;116:15823–9.
Lake D, Corrêa SAL, Müller J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci. 2016;73:4397–413.
Hofmann MH, Gmachl M, Ramharter J, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK Inhibition. Cancer Discov. 2021;11:142–57.
Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13:50.
Li X, Pu W, Zheng Q, et al. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol Cancer. 2022;21:99.
Bond MJ, Chu L, Nalawansha DA, et al. Targeted degradation of oncogenic KRASG12C by VHL-Recruiting PROTACs. ACS Cent Sci. 2020;6:1367–75.
Cheng J, Li Y, Wang X, et al. Discovery of novel PDEδ Degraders for the treatment of KRAS mutant colorectal cancer. J Med Chem. 2020;63:7892–905.
Nagasaka M, Potugari B, Nguyen A, et al. KRAS inhibitors- yes but what next? Direct targeting of KRAS- vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev. 2021;101:102309.
Pant S, Furqan M, Abdul-Karim RM, et al. First-in-human phase 1 trial of ELI-002 immunotherapy as treatment for subjects with Kirsten rat sarcoma (KRAS)-mutated pancreatic ductal adenocarcinoma and other solid tumors. 2022;40:TPS2701.
Yu I, Dakwar A, Takabe K. Immunotherapy: Recent Advances and Its Future as a Neoadjuvant, Adjuvant, and Primary Treatment in Colorectal Cancer. Cells. 2023;12:258.
Tran E, Robbins PF, Lu Y-C, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375:2255–62.
Acknowledgements
Thank the anonymous reviewers for their suggestions on this article.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Idea for the article: Lin Shen, Jian Li; Researching literature: Chunhua Wu, Wenfei Li, Mifen Chen, Qi Zhang, Ting Xu, Yao Ma, Wanyi Liu, Zhenghang Wang; Drafting the work: Chunhua Wu, Wenfei Li, Mifen Chen, Qi Zhang, Ting Xu; Revising the article: Zhenghang Wang, Xicheng Wang, Jian Li, Tanios Bekaii-Saab, Lin Shen. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, C., Li, W., Chen, M. et al. Advances in KRAS mutation inhibition in metastatic colorectal cancer. Holist Integ Oncol 2, 14 (2023). https://doi.org/10.1007/s44178-023-00032-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-023-00032-1