
Abstract
The polycystic kidney and hepatic disease 1 (PKHD1) gene located on chromosome 6p12 encodes for a large transmembrane protein called fibrocystin. Biallelic pathogenic variants in this gene cause autosomal recessive polycystic kidney disease (ARPKD). ARPKD often leads to both early-onset polycystic kidney disease as well as congenital hepatic fibrosis. In addition to the early onset phenotypes, some patients present much later with adult-onset liver involvement which is often labeled as Caroli’s syndrome. The kidney phenotype can resemble medullary sponge kidney disease with nephrolithiasis as well as atypical cystic kidney disease. Here, we present two families, each with 2 affected siblings, where the presenting liver and kidney features were variable among the siblings, with presentations including late-onset liver phenotypes, kidney features which had been labeled as medullary sponge kidney, and cystic kidney disease. Molecular genetic investigations identified biallelic pathogenic variants in PKHD1 in the affected siblings, including a novel nonsense allele. These cases emphasize the adult-onset and variable and sometimes discordant phenotypes that may be observed with PKHD1 biallelic pathogenic variants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The PKHD1 gene is located on chromosome 6p21.1-p12 and encodes for a large transmembrane protein called fibrocystin (also referred to as polyductin) consisting of 4074 amino acids and presenting in multiple isoforms. It is believed to be one of the largest genes in the human body spanning almost 470 kb of genomic DNA [1]. The exact function of fibrocystin remains unclear, but it is known to localize to the primary cilia in the cortical and medullary collecting ducts and the thick ascending limb of the kidneys along with the epithelial cells of the hepatic bile ducts [2, 3]. This suggests that fibrocystin might play a central role in the development and maintenance of the normal tubular architecture of epithelial cells present in the renal tubules and intrahepatic ducts. Biallelic variants in PKHD1 lead to abnormal proliferation in both the renal and biliary epithelial cells that manifest as ARPKD, which is typically an early-onset polycystic kidney disease phenotype with congenital hepatic fibrosis (CHF) [4, 5] with an estimated incidence of 1 in 20,000 live births [6]. Indeed, patients who present at a young age tend to have severe kidney disease and low survival rates. Such severe cases are generally identified on prenatal ultrasound scanning at 18–20 weeks gestation but can be confirmed much later in the pregnancy since nephrogenesis is only completed at around 34 weeks. The affected fetuses display a “Potter” phenotype with characteristic facial appearances; contracted limbs with club feet and bilaterally, symmetrically enlarged and hyper-echogenic kidneys with multiple small cysts; and loss of corticomedullary differentiation. This loss of the normal architecture of the kidneys causes a decrease in fetal urinary output leading to oligohydramnios that decrease the expansion of the lungs in utero. Also, the enlargement of the kidneys pushes the diaphragm upwards that decreases the volume of the thoracic cavity contributing to pulmonary hypoplasia. Studies report that approximately 40% of neonatal patients have to be supported by mechanical ventilation, and in spite of that, 30% die due to pulmonary insufficiency. Patients who survive beyond the perinatal period suffer from associated comorbidities such as systemic hypertension, repeated episodes of urinary tract infection (UTI), and chronic kidney disease (CKD). Over time, one-half of the patients progress to kidney failure (KF), usually within the first decade of their life, and require dialysis or a renal transplant [7, 8].
ARPKD may also present later in life, either in adolescence or adulthood, and these patients usually have a milder disease course, with symptoms mostly related to CHF and its complications, including portal hypertension (PT) [9]. Although histological evidence of hepatic fibrosis can be present at birth, it may not manifest clinically in newborns. Some patients present with non-obstructive dilatation of intrahepatic bile ducts called Caroli’s disease (CD) [10] along with CHF. This is collectively known as Caroli’s syndrome (CS). Patients presenting with CS have a 7–10% risk of developing dysplasia or cholangiocarcinoma [11]. Patients with CS have been observed to have fusiform or cystic dilatation of the renal tubules particularly the collecting ducts that closely resemble medullary sponge kidney (MSK) [12]. MSK is a renal malformation with cystic anomalies of the precalyceal ducts which is associated with nephrocalcinosis and nephrolithiasis as well as urinary tract infections. It is diagnosed radiologically with urography images showing the ectatic papillary ducts, giving a blush appearance. In addition to these morphological changes in the kidney, MSK is associated with distal renal tubular acidosis, hypocitraturia, and urinary concentration defects. However, MSK is generally considered a sporadic disorder with only a small percentage having an autosomal dominant pattern of familial inheritance [13]. The cystic kidney lesions associated with ARPKD typically lead to an increase in medullary echogenicity due to collecting duct dilatation, and renal imaging may not easily distinguish the two conditions. It therefore remains important to look for additional phenotypic features, including liver pathology and features of portal hypertension that may point to a diagnosis of ARPKD.
Case presentation
Family 1
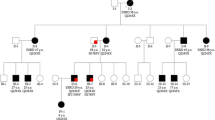
A 47-year-old female patient (family 1, II:2) (Fig. 1A) was admitted to the emergency department following an episode of melena. She had a previous history of hematemesis, a previous diagnostic label of MSK that had presented as recurrent urinary tract infections. There was evidence of hepatomegaly and splenomegaly on physical examination, but the kidneys were not enlarged. Blood results showed only a mild rise in bilirubin and a normal platelet count. Upper gastrointestinal endoscopy revealed erosive gastritis and grade II varices. Abdominal ultrasound scans showed a slightly coarse texture of the liver, and the portal vein was patent with normal flow. An abdominal CT scan showed hepatomegaly and splenomegaly as well as bilateral non-obstructing renal calculi and simple renal cysts (Fig. 1B). There was evidence of CKD stage G2A1 (eGFR 82 mL/min/1.73 m2) (Fig. 1C). A liver biopsy was performed which showed features of CHF and cholestasis. Magnetic resonance cholangiopancreatography (MRCP) showed numerous tiny saccular dilatations of the biliary tree consistent with CS.

Clinical, imaging, and molecular genetic findings in 2 families with ARPKD. Family 1 (A–D). A Pedigree diagram. Circles represent females, and squares represent males; red-filled symbols denote the presence of a kidney phenotype, and blue-filled symbols denote the presence of a liver phenotype. B CT abdomen of II:2 showing hepatomegaly and splenomegaly (top) and bilateral non-obstructing renal calculi (blue arrow) and simple renal cysts and biliary stent in situ (red arrow) (below). MRCP of II:3 revealed intrahepatic small cysts, multiple varices in the upper abdomen, splenomegaly, and multiple renal cysts. C Plot of eGFR (mL/min/1.73 m2) vs. age in siblings II:2 and II:3. D The two heterozygous PKHD1 pathogenic variants detected. Sequence traces (nucleotides and respective codons) are shown for affected individuals. Family 2 (E–H). E Pedigree diagram. Circles represent females, and squares represent males; red-filled symbols denote the presence of a kidney phenotype, and blue-filled symbols denote the presence of a liver phenotype. F CT abdomen of II:1 reveals pneumobilia and multiple fluid- and gas-filled cavities in the liver. The kidneys showed normal appearances aside from a left-sided renal cyst and non-obstructing right renal calculi (blue arrow). CT abdomen of II:2 revealed normal appearances of the liver, normal-sized spleen, and multiple renal calyceal calculi (blue arrows). G Plot of eGFR (mL/min/1.73 m2) vs. age in siblings II:1 and II:2. H The two heterozygous PKHD1 pathogenic variants detected. Sequence traces (nucleotides and respective codons) are shown for the affected individuals
Family history revealed that her younger sister (family 1, II:3) (Fig. 1A) had also been diagnosed with features of MSK at the age of 25 years. She had also a history of recurrent cholangitis. MRCP examination in this patient revealed numerous intrahepatic small cysts, multiple varices in the upper abdomen, splenomegaly, and multiple renal cysts (Fig. 1B). She had CKD stage G3bA1 (eGFR 36 mL/min/1.73 m2) (Fig. 1C).
In this family, there was also an older brother, who had died at the age of 42 years following a cerebral hemorrhage, but further details were not available. Both parents were clinically unaffected. Molecular genetic studies (a next-generation sequencing panel for cystic ciliopathy genes (Illumina TruSight One) which included 102 kidney disease genes (Table S1) in both the sisters (family 1, II2 and II:3)) identified biallelic pathogenic variants in PKHD1 (NM_138694 c.6046A>T p.S2016C and c.10637delT; p.V3546Afs*22) (Fig. 1D, Table 1) suggesting a unifying diagnosis of ARPKD leading to congenital hepatic fibrosis and polycystic kidney disease in both siblings.
Family 2
A 40-year-old lady (family 2, II:2) (Fig. 1E) was referred for the investigation of multiple episodes of loin pain. She had a previous history of presumed MSK which was diagnosed at the age of 29 years and since then had recurrent pain in both the flanks with fever. An abdominal CT scan revealed normal appearances of the liver, normal-sized spleen, and multiple renal calyceal calculi (Fig. 1F). Kidney function was preserved, CKD G1A1 (Fig. 1G).
Family history revealed that her older sister (family 2: II:1) (Fig. 1E) had been diagnosed with CS at the age of 35 years following multiple admissions to the hospital due to recurrent episodes of cholangitis and bile duct stones. MRCP of the liver showed multifocal cystic dilatation of the intrahepatic ducts and evidence of cholangitis. CT abdomen showed pneumobilia and multiple fluid- and gas-filled cavities in the liver (Fig. 1F). Following a progressive decline in liver function, she received an orthototic liver transplant at the age of 49 years.
The kidneys showed normal appearances aside from a left-sided renal cyst and non-obstructing right renal calculi (Fig. 1F), and kidney function was preserved, CKD G1A1 (Fig. 1G). There was no other family history of kidney or liver disease. Molecular genetic testing was performed in both siblings (NGS SureSelect Polycystic Disease Panel containing 17 genes) and identified biallelic variants in PKHD1 (NM_138694 c.10658T>C; p.I3553T and c.11026G>T; p.G3676*) (Fig. 1H, Table 1) allowing a unifying diagnosis of ARPKD to be reached.
Discussion
The wide spectrum of clinical presentations due to biallelic pathogenic variants in PKHD1 (Table 2) can lead to underrecognition and delayed diagnosis. Modern NGS approaches however allow the PKHD1 gene to be analyzed and provide a more precise diagnosis for patients presenting with diverse kidney and liver phenotypes. The prompt recognition of ARPKD and its various presentations has important implications for patient management including the prevention of the complications of CHF and progressive CKD.
Here, we report two unrelated families where the pairs of siblings were found to have biallelic pathogenic variants in PKHD1. In both the pairs of siblings, one of the sisters was diagnosed with adult-onset CS whereas the other had more prominent kidney features including nephrocalcinosis and nephrolithiasis resembling MSK.
CS is a congenital disorder characterized by segmental dilatation of the intrahepatic ducts and hepatic fibrosis. The incidence of CS is estimated to be 1 per million of the population [13]. CS presents with a combination of features of both CD and CHF mainly in the form of cholangitis, hepatomegaly, choledocholithiasis, PT, and the development of esophageal varices leading to hematemesis. Clinical progression and presentation of CS are highly variable, and symptoms may appear early or late during life. CS is a descriptive liver phenotype and has been associated with various kidney pathologies including MSK, ARPKD, and rarely autosomal dominant polycystic kidney disease (ADPKD) [19,20,21].
MSK was thought to be a sporadic condition and is a renal malformation associated with medullary nephrocalcinosis and renal stones. The only convincing genetic link is secondary to heterozygous pathogenic variants in GDNF, encoding the glial cell-derived neurotrophic factor [22] and its receptor rearranged during transfection (RET) that was recognized to play a crucial role in MSK in roughly 12% of the cases [13]. GDNF produced by the metanephric blastema and RET induces ureteric bud outgrowth and branching from the Wolffian duct during nephrogenesis. Failure of ureteric bud growth due to a pathogenic variant in GDNF may result in abnormalities leading to branching morphogenesis defects leading to renal hypoplasia or dysplasia. The precise mechanism resulting in the distinct tubular defects seen in MSK such as distal renal tubular acidosis, proximal tubular dysfunction, hypercalciuria, and hypocitraturia remains unclear. A possible association between PKHD1 and MSK has been previously postulated, but some caution is needed as radiological nephrocalcinosis may be secondary to both histopathological nephrocalcinosis and MSK [23]. It is possible that PKHD1 variants (both biallelic and in some cases monoallelic) lead to a spectrum of kidney phenotypes that may resemble MSK as well as cause cystic kidney disease [24]. The initial stages of ARPKD might therefore be pathologically misdiagnosed as MSK. Supporting this hypothesis is the fact that both are characterized by cystic dilatation of the collecting ducts, although the changes involved in MSK are confined to the renal medullary and intrapapillary collecting ducts. Shan et al. also reported that heterozygous Pkhd1 mutant mice develop cystic liver disease and tubule ectasia mimicking MSK [19]. Nephrolithiasis is a well-recognized complication of ADPKD [25], and risk factors include both distorted anatomy and metabolic factors. Calcification may also occur in the cyst wall and renal parenchyma [26]. ARPKD is also a recognized, but perhaps less often reported cause of nephrolithiasis and may occur in all age ranges of patients [27].
According to the Department of Human Genetics, RWTH Aachen University, Germany (www.humgen.rwth-aachen.de), 748 PKHD1 variants have been mentioned in their genetic database out of which approximately 60% are truncating and 40% are missense alleles. The presence of two truncating PKHD1 pathogenic variants is most often lethal, causing neonatal demise. Hence, survival beyond the neonatal period requires the presence of at least one missense variant in PKHD1 [15]. The most commonly observed pathogenic missense variant in the PKHD1 gene is c.107C>T (p.Thr36Met) which is estimated to constitute 20% of all PKHD1 pathogenic variants [6, 28]. Although many genetic variants of PKHD1 have been described, there is no conclusive evidence that determines the genetic contribution of missense alterations to the patient phenotypes observed clinically. Like our cases, interfamilial phenotypic variability has also been reported in patients with biallelic PKHD1 pathogenic variants [29].
The two families reported here had a combination of a loss of function allele and a missense allele. In family 1, the variants in PKHD1 where c.10637delT caused a frameshift result in a predicted truncated protein together with a missense change, p.S2016C. In family 2, the PKHD1 variants were a missense change (p.Ile3553Thr) and a nonsense pathogenic variant. The nonsense allele (c.11026G>T; p.Gly3676*) is novel but is highly likely to be pathogenic and clinically disease-causing.
Despite the identification of many families with ARPKD, a complete understanding of the genotype-phenotype relationship is lacking. In particular, the age of onset of disease and the renal versus kidney presentations remain unexplained. Additional investigations into other factors such as modifier genes, epigenetic factors, and environmental influences are required to explain the diverse clinical spectrum of disease and allow further understanding of this genetic disorder. We conclude that a recognition of the different ages that ARPKD may present is useful and the adult manifestations, including nephrolithiasis and nephrocalcinosis, and its potential to mimic or phenocopy MSK deserved renewed attention, especially as the molecular genetic diagnosis is now affordable, reliable, and will lead to a greater understanding of the ARPKD disease spectrum.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
Onuchic LF, Furu L, Nagasawa Y, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–17.
Wang S, Zhang J, Nauli SM, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007;27:3241–52.
Lea WA, Ward CJ. A new epitope-tagged Pkhd1 allele sheds light on fibrocystin signaling. Kidney Int. 2017;92:1041–3.
Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–69.
Masyuk TV, Huang BQ, Ward CJ, et al. Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat. Gastroenterology. 2003;125:1303–10.
Bergmann C, Küpper F, Dornia C, Schneider F, Senderek J, Zerres K. Algorithm for efficient PKHD1 mutation screening in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2005;25:225–31.
Guay-Woodford LM, Desmond RA. Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics. 2003;111:1072–80.
Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11:302–6.
Rossetti S, Harris PC. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J Am Soc Nephrol. 2007;18:1374–80.
Wang ZX, Li YG, Wang RL, et al. Clinical classification of Caroli’s disease: an analysis of 30 patients. HPB (Oxford). 2015;17:278–83.
Dayton MT, Longmire WP Jr, Tompkins RK. Caroli’s disease: a premalignant condition? Am J Surg. 1983;145:41–8.
Gambaro G, Feltrin GP, Lupo A, Bonfante L, D’Angelo A, Antonello A. Medullary sponge kidney (Lenarduzzi-Cacchi-Ricci disease): a Padua Medical School discovery in the 1930s. Kidney Int. 2006;69:663–70.
Fabris A, Anglani F, Lupo A, Gambaro G. Medullary sponge kidney: state of the art. Nephrol Dial Transplant. 2013;28:1111–9.
Bergmann C, Senderek J, Windelen E, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005;67:829–48.
Denamur E, Delezoide AL, Alberti C, et al. Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 2010;77:350–8.
Tong YQ, Liu B, Fu CH, et al. Genetic analysis of the PKHD1 gene with long-rang PCR sequencing. J Huazhong Univ Sci Technolog Med Sci. 2016;36:758–66.
Szabó T, Orosz P, Balogh E, et al. Comprehensive genetic testing in children with a clinical diagnosis of ARPKD identifies phenocopies. Pediatr Nephrol. 2018;33:1713–21.
Obeidova L, Seeman T, Elisakova V, Reiterova J, Puchmajerova A, Stekrova J. Molecular genetic analysis of PKHD1 by next-generation sequencing in Czech families with autosomal recessive polycystic kidney disease. BMC Med Genet. 2015;16:116.
Shan D, Rezonzew G, Mullen S, et al. Heterozygous Pkhd1(C642*) mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am J Physiol Renal Physiol. 2019;316:F463–f472.
Sinha RJ, Sharma A, Singh V, Pandey S. Medullary sponge kidney and Caroli’s disease in a patient with stricture urethra: look for the hidden in presence of the apparent. BMJ Case Rep. 2018;11:bcr-2018.
Kumar A, Akselrod D, Prikis M. Caroli disease revisited: a case of a kidney transplant patient with autosomal polycystic kidney disease and recurrent episodes of cholangitis. Transplant Proc. 2019;51:541–4.
Torregrossa R, Anglani F, Fabris A, et al. Identification of GDNF gene sequence variations in patients with medullary sponge kidney disease. Clin J Am Soc Nephrol. 2010;5:1205–10.
Gunay-Aygun M, Turkbey BI, Bryant J, et al. Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol Genet Metab. 2011;104:677–81.
Letavernier E, Schwoehrer M, Livrozet M, et al. Atypical clinical presentation of autosomal recessive polycystic kidney mimicking medullary sponge kidney disease. Kidney Int Rep. 2022;7:916–9.
Nishiura JL, Neves RF, Eloi SR, Cintra SM, Ajzen SA, Heilberg IP. Evaluation of nephrolithiasis in autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol. 2009;4:838–44.
Koorevaar IW, Gansevoort RT, Leliveld AM, Casteleijn NF. Renal stones in patients with autosomal dominant polycystic kidney disease, a treatment challenge? Clin Exp Nephrol. 2020;24:1088–9.
Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine. 2006;85:1–21.
Al Alawi I, Molinari E, Al Salmi I, Al Rahbi F, Al Mawali A, Sayer JA. Clinical and genetic characteristics of autosomal recessive polycystic kidney disease in Oman. BMC Nephrol. 2020;21:347.
Deget F, Rudnik-Schöneborn S, Zerres K. Course of autosomal recessive polycystic kidney disease (ARPKD) in siblings: a clinical comparison of 20 sibships. Clin Genet. 1995;47:248–53.
Acknowledgements
Not applicable
Funding
J.A.S. is supported by Kidney Research UK (Paed_RP_20180925) and the Northern Counties Kidney Research Fund.
Author information
Authors and Affiliations
Contributions
JAS conceived the manuscript. AD and PM were major contributors in writing the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the North East - Newcastle & North Tyneside 1 Research Ethics Committee (18/NE/350). All included individuals provided informed and written consent.
Consent for publication
Patients have given written consent for publication.
Competing interests
Professor John Sayer is a co-author of this study and an editorial board member of the journal. He was not involved in handling this manuscript during the review process. The rest of the authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
NGS Cystic Ciliopathy Gene Panel. Table S2. SureSelect Polycystic Kidney and Liver Disease full panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Das, A., Mead, P. & Sayer, J.A. Adult presentations of variable kidney and liver phenotypes secondary to biallelic PKHD1 pathogenic variants. J Rare Dis 2, 1 (2023). https://doi.org/10.1007/s44162-022-00002-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-022-00002-7