
Abstract
Background
There is a high prevalence of rare genetic disorders in the Middle East, and their study provides unique clinical and genetic insights. Autosomal recessive polycystic kidney disease (ARPKD) is one of the leading causes of kidney and liver-associated morbidity and mortality in Oman. We describe the clinical and genetic profile of cohort of ARPKD patients.
Methods
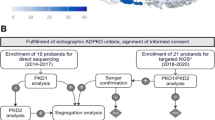
We studied patients with a clinical diagnosis of ARPKD (n = 40) and their relatives (parents (n = 24) and unaffected siblings (n = 10)) from 32 apparently unrelated families, who were referred to the National Genetic Centre in Oman between January 2015 and December 2018. Genetic analysis of PKHD1 if not previously known was performed using targeted exon PCR of known disease alleles and Sanger sequencing.
Results
A clinical diagnosis of ARPKD was made prenatally in 8 patients, 21 were diagnosed during infancy (0–1 year), 9 during early childhood (2–8 years) and 2 at later ages (9–13 years). Clinical phenotypes included polycystic kidneys, hypertension, hepatic fibrosis and splenomegaly. Twenty-four patients had documented chronic kidney disease (median age 3 years). Twenty-four out of the 32 families had a family history suggesting an autosomal recessive pattern of inherited kidney disease, and there was known consanguinity in 21 families (66%). A molecular genetic diagnosis with biallelic PKHD1 mutations was known in 18 patients and newly identified in 20 other patients, totalling 38 patients from 30 different families. Two unrelated patients remained genetically unsolved. The different PKHD1 missense pathogenic variants were: c.107C > T, p.(Thr36Met); c.406A > G, p.(Thr136Ala); c.4870C > T, p.(Arg1624Trp) and c.9370C > T, p.(His3124Tyr) located in exons 3, 6, 32 and 58, respectively. The c.406A > G, p.(Thr136Ala) missense mutation was detected homozygously in one family and heterozygously with a c.107C > T, p.(Thr36Met) allele in 5 other families. Overall, the most commonly detected pathogenic allele was c.107C > T; (Thr36Met), which was seen in 24 families.
Conclusions
Molecular genetic screening of PKHD1 in clinically suspected ARPKD cases produced a high diagnostic rate. The limited number of PKHD1 missense variants identified in ARPKD cases suggests these may be common founder alleles in the Omani population. Cost effective targeted PCR analysis of these specific alleles can be a useful diagnostic tool for future cases of suspected ARPKD in Oman.
Similar content being viewed by others


Background
Autosomal recessive polycystic kidney disease (ARPKD) is one of the most frequent cystic kidney diseases in infants and children, causing kidney and liver associated mortality and morbidity. The estimated incidence of ARPKD is 1:6000 to 1:55,000 births [1]. Classically, ARPKD manifests prenatally as enlarged echogenic kidneys with Potter’s syndrome or postnatally during childhood or adolescence with polycystic kidney disease and congenital hepatic fibrosis. It is characterized by bilateral echogenic cystic kidneys caused by dilatation of the renal tubular collecting ducts and congenital hepatic fibrosis secondary to malformation of the liver biliary ducts. ARPKD is also associated with systemic and portal hypertension. Current therapies focus on treating ARPKD symptoms [2].
ARPKD is caused by mutations in the polycystic kidney and hepatic disease 1 (PKHD1) (OMIM 606702), which is located on chromosome 6 (p12.3-p12.2). Consistent with the disease phenotype, high expression of PKHD1 is found in fetal and adult kidney, with low levels detected in the liver, pancreas and arterial walls [3]. The longest open reading frame transcript (NM_138694.4) contains 67 exons and encodes a 4074 amino acid protein called fibrocystin / polyductin. Fibrocystin is an integral membrane protein that is localized to the basal bodies of primary cilia in epithelial cells and hypothesized to control kidney tubular formation by modifying polycystin-2 expression [4]. It was shown that the severity of ARPKD is determined by the type of mutations rather than the location on the PKHD1 gene [5]. There are over 700 different mutations associated with an ARPKD phenotype [6].
The importance of early diagnosis and management of ARPKD through genetic testing is widely recognized [7]. Precise molecular genetic diagnosis can improve the clinical management of patients, avoiding the exposure to unnecessary and invasive investigations and enhance early detection of kidney and extrarenal complications. The size of PKHD1 and its heterogeneous mutational spectrum has previously been an obstacle to molecular genetic diagnosis. However, recent advances in massive parallel sequencing / next generation sequencing (NGS) have facilitated routine large scale screening for pathogenic mutations in PKHD1.
In addition to mutations in PKHD1, mutations in DZIP1L (OMIM 671570) may underlie ARPKD. Pathogenic variants have been identified in multiple unrelated consanguineous Turkish, Palestinian and Egyptian patients with ARPKD and a moderate renal phenotype [8]. DZIP1L encodes for DAZ-interacting zinc finger protein 1-like protein (DZIP1L) that is located in the centrioles and the distal ends of basal bodies [8].
Inherited kidney disease is a leading cause of end stage kidney disease (ESKD) in children in Oman [7]. In one Omani hospital-based study, the observed birth incidence of ARPKD was evaluated to be much higher than typical, estimated to be 1 in 12,000 births [9]. However, this number was from a selected hospital population and there are no population-based studies evaluating the incidence of ARPKD in the Omani population as a whole. A recent study from Oman showed that hereditary kidney disease accounts for 32% of etiologies causing chronic kidney disease (CKD) in children and ARPKD is the leading cause, accounting for 12% of CKD [10]. The objective of this study was to investigate the clinical features and the molecular genetic diagnosis in patients with suspected ARPKD in an Omani population.
Methods
Patients sampling and ethical approval
A group of 40 affected patients, 10 unaffected siblings and 24 parents, from 32 unrelated families were referred to the National Genetic Centre in Oman between January 2015 and December 2018 for the genetic study of ARPKD from the Royal Hospital (n = 31), Sohar Hospital (n = 8), Rustaq Hospital (n = 1) and Al Buraimi Hospital (n = 1). Clinical information, including age at clinical diagnosis, use of neonatal ventilation, presence of cystic kidney disease, stage of CKD, splenomegaly, hypertension, urinary concentration defect, urinary tract infection (UTI), pulmonary hypoplasia, oesophageal varices and renal replacement modalities were extracted from the patient’s medical files at the time of genetic testing and were analyzed. Congenital hepatic fibrosis was diagnosed based on clinical parameters, abdominal ultrasonography and where available, pathological results from liver biopsy specimens. The system used for CKD staging is according to that published by the National Kidney Foundation’s Kidney Disease Outcome Quality Initiative KDOQI in 2003 [11].
Additional demographic information including familial history of ARPKD, parental consanguinity and geographical distribution were also obtained. Family pedigrees described in this study were drawn using invitae online tool (https://familyhistory.invitae.com) (Supplementary Figure S1). We have previously reported family structures (but not clinical details) for 5 of these families with genetically proven ARPKD [12].
This study was ethically approved by the Research and Ethical Review and Approval Committee, Ministry of Health in Oman and by the Ethics Committee, College of Medicine and Health Sciences, Sultan Qaboos University in Oman (MREC#1096). All study participants provided written informed consent.
Mutational analysis
DNA was extracted manually from the peripheral blood lymphocytes of patients and family members using QIAamp DNA mini kit (QIAGEN). For 18 unrelated patients, molecular study had been previously performed through NGS using targeted gene panel that includes cystic kidney disease genes (including PKD1, PKD2, PKHD1, HNF1B) (Table S1) [12]. These 18 patients all had biallelic variants in PKHD1 but mutations were limited to 4 alleles [12]. Based on these previous NGS results, Sanger sequencing screening of the PKHD1 exons 3, 6, 32 and 58 was performed for another 22 patients from 14 different families with clinically suspected ARPKD. For the confirmation of segregation of variants within families, the available samples of parents, siblings and relatives were also screened using Sanger sequencing (Table 2, Supplementary Figure S1).
For Sanger sequencing, target regions were amplified using AmpliTaq Gold 360 Master Mix kit (Applied Biosystems) and oligonucleotide primers, which were designed using the Primer3 program (http://primer3.ut.ee/). Oligonucleotide primers were designed to cover the whole exon sequence at which variant is located. For large exons, a set of overlapping fragments were amplified using multiple oligonucleotide primer pairs (Supplementary Table S2).
PCR was carried out using the following conditions: denaturation at 95 °C for 15 min; followed by 13 cycles of 94 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s; followed by 8 cycles of 94 °C for 30 s, 46.5 °C for 30 s and 72 °C for 30 s; followed by 16 cycles of 94 °C for 30 s, 54.5 °C for 30 s and 72 °C for 30 s, amplification for a total of 37 cycles, and then elongation at 72 °C for 5 min. PCR products were purified using BigDye Terminator Cycle Sequencing kit (PE Applied Biosystems, Massachusetts, USA) and sequenced (automated ABI 3130).
Sequencing results were aligned and analyzed by comparison with the human reference sequence of the PKHD1 gene (NM_138694.3) using the SequencePilot 4.2.2 software (JSI Medical Systems GmbH). For genetic analysis all PKHD1 variants were classified with regard to pathogenicity according to the revised criteria of the American College of Medical Genetics [13].
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 20, with the results expressed as frequencies and percentage for categorical variables and as mean and median for continuous variables, as appropriate.
Results
Clinical characteristics
In total, 32 unrelated families were included in this study (Table 1). The cohort includes 40 clinically suspected ARPKD patients (n = 19 males and n = 21 females), 24 parents and 10 unaffected siblings.
Molecular genetic diagnosis of biallelic PKHD1 mutations was previously known in 18 patients described here and newly identified in a further 20 patients from a total of 30 different families, giving an overall mutation detection rate of 94% in our cohort of clinically suspected ARPKD (Table 2). Twenty-four families (75%) reported a known family history of kidney disease consistent with an autosomal recessive inheritance pattern. Two families remained genetically unsolved, both had no family history of kidney or liver disease. Parental consanguinity was known in 21 families (66%) in keeping with high rates of autosomal recessive disease in Oman.
The initial clinical diagnosis was made prenatally in eight patients (20%), during infancy (0–1 year) in 21 patients (52.5%), during early childhood (1–8 years) in 9 patients (22.5%), and in later years (9–13 years) in 2 patients (5%) (Table 1). At the time of molecular diagnosis, clinical features were documented, including systemic hypertension (72.5%), suspected congenital hepatic fibrosis (CHF) (77.5%), CKD (60%) with a median age of onset 3 years, splenomegaly (47.5%), pulmonary hypoplasia (17.5%) and perinatal / post-neonatal death due to respiratory failure (15%) (Table 1). Twelve of patients (27.5%) developed ESKD at median age of 13 years.
Genetics of Omani ARPKD patients
Our previous NGS study of inherited cystic kidney diseases had identified 4 PKHD1 missense pathogenic mutations [12] in 18 patients with suspected ARPKD. These included c.107C > T, p.(Thr36Met); c.406A > G, p.(Thr136Ala); c.4870C > T, p.(Arg1624Trp) and c.9370C > T, p.(His3124Tyr) located in exons 3, 6, 32 and 58, respectively. These alleles were found either homozygously or heterozygously in 20 other patients with clinically suspected ARPKD (Table 2). In total, twenty-one families (66%) carried homozygous alleles, while 9 (28%) carried compound heterozygous alleles. The most commonly detected mutation in PKHD1 was p.(Thr36Met), where 16 families carried it in homozygous state, while 8 families were compound heterozygous for this allele combined with the p.(Thr136Ala) allele (5 families) or the p.(Arg1624Trp) allele (3 families). Four families carried the homozygous mutation p.(Arg1624Trp), whilst one family carried the homozygous mutation p.(Thr136Ala). One family had compound heterozygous mutations p.(Arg1624Trp) and p.(His3124Tyr). The clinical presentations and genotypes are summarized in Table 2. Sanger sequencing chromatograms of the four different mutations in the PKHD1 gene are shown in Supplementary Figure S2A. The p. (Thr136Ala) has not been reported outside Oman and is absent from reference databases (Table 3).
Geographic distribution of PKHD1 mutations in Oman
The p.(Thr36Met) allele was detected in patients from all governorates of the country except, Musandam, Al Wusta and Dhofar (Fig. 1). The missense p.(Thr136Ala) allele was detected in its homozygous state in one family from Al Batinah South and in heterozygous state in families from Ad Dakhiliyah (n = 1), Al Dhahirah (n = 2) and Al Batinah south (n = 2) (Fig. 1). We postulate that this mutation may be a founder allele in this population.

Geographical distribution of the 4 missense PKHD1 mutations in ARPKD patients from Oman. Each family is presented by plot. Each different mutation is indicated by a different colour. Map sourced through www.freeusandworldmaps.com and modified using www.vectorstock.com software.
Discussion
Inherited kidney diseases, including ARPKD are leading causes of CKD and ESKD in children in Oman, leading to significant morbidity and mortality. Previous studies from Oman have provided ARPKD-associated morbidity data but lacked molecular genetic data [10, 23]. In this study we have provided a clinical and molecular genetic analysis of PKHD1 in a cohort of 40 patients, obtaining a molecular genetic diagnosis in 38 patients with clinically suspected ARPKD.
Most study patients had early onset ARPKD disease reflected by age at initial diagnosis. Five were diagnosed prenatally, 24 before their first year of life and 11 during childhood. These early-onset phenotypes are in agreement with that reported from other studies [22]. Clinical analysis of our ARPKD patients showed that the frequently associated morbidities were also common in our patients including systemic hypertension, congenital hepatic fibrosis, splenomegaly, pulmonary hypoplasia and CKD. 15% of studied patients died during the perinatal or neonatal period due to respiratory deficiency, which is similar to the death rate during the first year of life reported by Bergmann [22]. It is estimated that 30–50% of ARPKD patients die shortly after birth due to respiratory failure, whereas kidney failure is a rare cause of neonatal death [24]. With the advancement in renal replacement therapy modalities, the survival rate of neonates and children with ARPKD is improved. In our patients, 12 (30%) developed ESKD by median age of 13 years and hence required either renal replacement therapy (n = 8) or kidney transplantation (n = 4) (Table 2).
As the PKHD1 is a large gene and in order to identify the causative mutations in our ARPKD patients, we had previously applied targeted NGS gene panel for 18 unrelated patients (Table 2) [12]. Four missense mutations in PKHD1 were identified as genetic causes of ARPKD in this cohort, with the mutations identified within exons 3, 6, 32 and 58. Therefore, we proceeded with a targeted exon PCR diagnostics approach with Sanger sequencing of these exons alone for the molecular diagnosis of other ARPKD patients (n = 20) from 14 different families. In total, 30 out of 32 suspected ARPKD families were solved with biallelic changes in PKHD1, achieving a diagnostic rate of 94%, hence providing cost effective targeted PCR analysis of these specific alleles as a convenient diagnostic tool. Failure to detect mutations in two unrelated probands using the Sanger screening approach alone may be explained by the heterogeneity of the ARPKD disease where mutations may lay in other exons or indeed in other recessive cystogenes that phenocopy ARPKD. None of the unsolved patients had have family history of kidney disease, therefore mutations in autosomal recessive genes that lead to ARPKD-like phenotypes such as the DZIP1L [8] or even in dominant cystic kidney disease genes such as HNF1B, PKD1 and PKD2 that often occur de novo are possible [25]. Whole exome sequencing approaches that would allow inclusion of genes such as DZIP1L are the suggested option for these unsolved patients. With the current improvements of high throughput sequencing of different renal ciliopathy genes, it is anticipated that the majority of patients with cystic kidney disease phenotypes can receive a precise molecular genetic diagnosis.
To date, 748 unique PKHD1 variants have been recorded in the Human ARPKD/PKHD1 Mutation Database (http://www.humgen.rwth-aachen.de/index.php). Approximately 45% of these variants are missense alterations resulting in substitution of conserved amino acids, which usually leads to partial or complete dysfunction of fibrocystin. All of the PKHD1 variants in this study are missense alterations of highly conserved amino acids. Although three of these variants are reported in the ARPKD / PKHD1 Mutation Database, p. (Thr36Met) is the most persistent mutation found in PKHD1 in ARPKD patients to date. Structurally, fibrocystin is an integral membrane protein consisting of a large amino terminal extracellular domain (about 3860 aa) containing various glycosylation sites, a single transmembrane segment and a short cytoplasmic C-terminal tail (about 195 aa) comprising four potential protein kinase A phosphorylation sites [3] (Supplementary Figure S2B). The localization of fibrocystin to primary cilia and its integral structure predicted a sensory role at which fibrocystin acts as receptor transducing the extracellular information into the cell through stimulation of signal cascades, thus controlling cell-cell adhesion and proliferation [3]. The 4 missense variants identified in this study are located in exons encoding the extracellular domain (Supplementary Figure S2C) and are either very rare or not observed in the reference databases of healthy controls (including ExAC, gnomAD, 1000G project (Table 3)).
In the Omani population our previous NGS studies did not find PKHD1 truncating mutations that have been described in patients with perinatal lethal phenotypes [22, 26]. The most frequent change identified in Omani families was p.(Thr36Met) located in exon 3, detected homozygously in 16 families and heterozygously in 8 families, accounting for almost 75% of the families. Patients with homozygous p.(Thr36Met) change (n = 18) had an earlier age of onset and an increased severity of disease (6 had a severe perinatal presentation and died before 1 year of life, while the remaining had either infantile or early childhood presentation leading to ESKD) (Table 2). Consistent with observations made by Bergman et al. [26], the p.(Thr36Met) variant may lead to variability in the age of onset and severity of disease. Additionally, p.(Thr36Met) in combination with the missense changes p.(Thr136Ala) and p.(Arg1624Trp) in some of our cases caused a relatively severe form of ARPKD, which is in agreement with previous reported studies [21, 27, 28].
The relatively common pathogenic PKHD1 allele p.(Thr36Met) has been described in many populations and ethnicities. Whether this allele is a highly conserved ancestral change that is frequent in some populations such as the Central European population [15, 27], or caused by recurrent mutational events is uncertain [22]. The p.(Thr36Met) allele appears to be common in European genomes, with an expected carrier frequency of 1:412 [29]. However, detections of this change in patients from different ethnicities and origins are suggestive that p.(Thr36Met) is a PKHD1 ‘hotspot’ mutation caused by the frequent methylation events of cytosine to thymine in the CpG sites [17, 27]. It is also assumed that the substitution of the amino acid Threonine to Methionine creates a potential alternative translation start codon that may be even stronger than the original start codon [21]. The protein product initiating from position c.107 would be predicted to lead to complete loss of protein function due to improper protein folding [21].
The missense change, p.(Arg1624Trp), was previously reported in patients from different ethnicities, including Caucasian Americans [17, 19], Dutch [18], Czech Republicans [28], Slovenians [30], Saudi Arabians [17, 31, 32] and Kuwaitis [33]. The p.(Arg1624Trp) mutation has been described with late onset or older ARPKD presentations when present homozygously [17, 31] and heterozygously in trans with other truncating or missense change [17, 30]. In contrast, 6 of our patients with the p.(Arg1624Trp) mutation developed clinical features of ARPKD in infancy, 5 presented during childhood period and 1 at 11 years of age. The p.(His3124Tyr) combined with p.(Arg1624Trp) was found in a 26 year old patient with stage 4 CKD, who was initially diagnosed with polycystic kidney disease in early childhood (Table 2). These findings are in contrast to those made by Bergmann et al. [16] and Gunay-Aygun [19] that correlated p.(His3124Tyr) with a severe perinatal-fatal phenotype.
These results therefore demonstrate that establishing genotype-phenotype correlations in ARPKD is challenging. Any correlation is complicated by the large number of missense variants distributed over the entire length of the coding exons of PKDH1 and its complex splicing pattern [3]. It was believed that two truncating mutations are associated with severe perinatal lethality and at least the presence of one missense is required for survival beyond the neonatal period. However, evidence is accumulating on the increased pathogenicity of some missense mutations that may cause complete loss of function effects [22]. The wide variability in ARPKD severity among patients may in part be explained by differences in PKHD1 mutations, influences of modifiers genes and environmental factors [25].
ARPKD is generally a severe form of pediatric ciliopathy with recognized phenotypic variability. While a significant number of ARPKD patients surviving the neonatal period reaches adulthood, some patients have an adulthood presentation and their kidney function ranges from normal to moderate kidney insufficiency to ESKD [34]. Although bilateral kidney enlargement with multiple cysts is the major clinical characteristic, liver manifestations may lead to symptomatic disease complications in ARPKD patients. Liver disease tends to manifest later than kidney disease typically with progressive hepatic fibrosis and portal hypertension [34]. Hypersplenism, portal hypertension, and variceal bleeding are major liver involvements that may develop as a result of progressive liver fibrosis. In rare cases, both kidney and liver disease may present in late adolescence or in adulthood [35]. The low prevalence, limited clinical information and atypical sonographic pattern of adult ARPKD patients can challenge the clinical diagnosis and management, hence genetic testing may be demanded for the establishment of definite diagnosis [36]. In this study, the absence of late presenting ARPKD in our cohort may be due to recruiting predominantly cystic kidney disease phenotypes.
The Omani population is characterized by a unique structure of tribal communities occupying definite geographical regions. This structure is conserved over many generations and has created genetic isolates [37]. The custom of consanguineous marriages as well as within-tribe (endogamous) marriages are extremely conserved in Oman, accounting for 56.3% [38] and 20.4% of total marriages, respectively [39]. Over 300 genetic diseases have been identified in the Omani population [40]. The high frequency of recessive disorders in this population is probably related to a combination of genetic drift, consanguinity, and geographical isolation. The detection of only 4 pathogenic variants in different geographical regions of the country may be explained by the presence of PKHD1 founder alleles and reveals a high degree of homogeneity in this population. Similarly, genetic studies of population isolates such as Finnish, French, Ashkenazi Jews and Africans represent a powerful method of finding founder mutations in PKHD1, which can be utilized for efficient diagnostic testing of at-risk individuals and pregnancies in these populations (Supplementary Table S3) [27, 41,42,43].
Currently there is no clinical cure for ARPKD other than managing the clinical complications [34]. Together translational research and clinical trials in patients may facilitate successful drug development in coming future. With the absence of clinical biomarkers and lack of comprehensive assessment of the available therapeutic options for ARPKD patients on one hand and great morbidity and mortality of disease on the other hand, there is a serious need for prospective and retrospective population studies and construction of an international clinical database. Such effort can elaborate the current understanding of ARPKD and deliver more information on extrarenal manifestations and treatment options. Recently, the German Society for Pediatric Nephrology (GPN) and the European Study Consortium for Chronic Kidney Disorders Affecting Pediatric Patients (ESCAPE) collaborated to initiate an international multicenter registry of ARPKD (ARegPKD) [44]. The continued identification of PKHD1 variants and their associated phenotypes is to be promoted and inclusion of cohorts from different ethnicities is valuable and should be encouraged.
Conclusions
In conclusion, this study shows that prior NGS identification of PKHD1 mutations and subsequent screening of only 4 exons of the PKHD1 gene was sufficient to identify the expected causative alleles in 94% of the studied families and was suggestive of founder effects in this gene. There are many advantages for identifying high frequency limited disease associated mutations in a population, including simplifying the diagnostic testing, providing genetic counseling for individuals at risk and allowing rapid detection of mutations in other family members.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information file]. The raw data are available from the corresponding author on reasonable request.
Abbreviations
- ARPKD:
-
Autosomal recessive polycystic kidney disease
- CHF:
-
Congenital hepatic fibrosis
- CKD:
-
Chronic kidney disease
- DZIP1L :
-
DAZ-interacting zinc finger protein 1-like
- ExAc:
-
Exome Aggregation Consortium
- ESCAPE:
-
European Study Consortium for Chronic Kidney Disorders Affecting Pediatric Patients
- ESKD:
-
End stage kidney disease
- gnomAD:
-
Genome Aggregation Database
- GPN:
-
German Society for Pediatric Nephrology
- KDOQI:
-
Kidney Disease Outcome Quality Initiative
- NGS:
-
Next generation sequencing
- OMIM:
-
Online Mendelian Inheritance in Man
- PKHD1 :
-
Polycystic kidney and hepatic disease 1
- PolyPhen-2 :
-
Polymorphism Phenotyping v2
- SIFT:
-
Sorting Intolerant From Tolerant
- UTI:
-
Urinary tract infections
References
Lonergan GJ, Rice RR, Suarez ES. Autosomal recessive polycystic kidney disease: radiologic-pathologic correlation. Radiographics. 2000;20(3):837–55.
Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014;165(3):611–7.
Bergmann C. Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front Pediatr. 2017;5:221.
Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, Liang D, Zhao P, Ma J, Chen XZ, et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol. 2008;19(3):455–68.
Denamur E, Delezoide AL, Alberti C, Bourillon A, Gubler MC, Bouvier R, Pascaud O, Elion J, Grandchamp B, Michel-Calemard L, et al. Genotype-phenotype correlations in fetuses and neonates with autosomal recessive polycystic kidney disease. Kidney Int. 2010;77(4):350–8.
Ebner K, Dafinger C, Ortiz-Bruechle N, Koerber F, Schermer B, Benzing T, Dotsch J, Zerres K, Weber LT, Beck BB, et al. Challenges in establishing genotype-phenotype correlations in ARPKD: case report on a toddler with two severe PKHD1 mutations. Pediatr Nephrol. 2017;32(7):1269–73.
Al Alawi I, Al Salmi I, Al Mawali A, Al Maimani Y, Sayer JA. End-stage kidney failure in Oman: an analysis of registry data with an emphasis on congenital and inherited renal diseases. Int J Nephrol. 2017;2017:6403985.
Lu H, Galeano MCR, Ott E, Kaeslin G, Kausalya PJ, Kramer C, Ortiz-Bruchle N, Hilger N, Metzis V, Hiersche M, et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat Genet. 2017;49(7):1025–34.
Rajab A, Bappal B, Al-Shaikh H, Al-Khusaibi S, Mohammed AJ. Common autosomal recessive diseases in Oman derived from a hospital-based registry. Community Genet. 2005;8(1):27–30.
Al Riyami MS, Al Shehhi M, Al Sulaimi T, Al Mamary L, Al Maskari A, Al Ghaithi B, Al Riyami M, Al Kalbani N, Al Saidi S. Epidemiology and outcome of CKD in Omani children. Kidney Int Rep. 2019;4(5):727–32.
Hogg RJ, Furth S, Lemley KV, Portman R, Schwartz GJ, Coresh J, Balk E, Lau J, Levin A, Kausz AT, et al. National Kidney Foundation's kidney disease outcomes quality initiative clinical practice guidelines for chronic kidney disease in children and adolescents: evaluation, classification, and stratification. Pediatrics. 2003;111(6 Pt 1):1416–21.
Al Alawi I, Al Salmi I, Al Rahbi F, Al Riyami M, Al Kalbani N, Al Ghaithi B, Al Mawali A, Sayer JA. Molecular genetic diagnosis of omani patients with inherited cystic kidney disease. Kidney Int Rep. 2019;4(12):1751–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30(3):259–69.
Rossetti S, Torra R, Coto E, Consugar M, Kubly V, Malaga S, Navarro M, El-Youssef M, Torres VE, Harris PC. A complete mutation screen of PKHD1 in autosomal-recessive polycystic kidney disease (ARPKD) pedigrees. Kidney Int. 2003;64(2):391–403.
Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Furu L, et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004;23(5):453–63.
Sharp AM, Messiaen LM, Page G, Antignac C, Gubler MC, Onuchic LF, Somlo S, Germino GG, Guay-Woodford LM. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J Med Genet. 2005;42(4):336–49.
Losekoot M, Haarloo C, Ruivenkamp C, White SJ, Breuning MH, Peters DJ. Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum Genet. 2005;118(2):185–206.
Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C(4):296–306.
Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70(5):1305–17.
Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, et al. Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol. 2003;14(8):2004–14.
Bergmann C, Senderek J, Windelen E, Kupper F, Middeldorf I, Schneider F, Dornia C, Rudnik-Schoneborn S, Konrad M, Schmitt CP, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005;67(3):829–48.
Al-Lawati TT. Fibropolycystic disease of the liver and kidney in Oman. Arab J Gastroenterol. 2013;14(4):173–5.
Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30(1):15–30.
Bergmann C. Early and severe polycystic kidney disease and related Ciliopathies: an emerging field of interest. Nephron. 2019;141(1):50–60.
Bergmann C, Senderek J, Schneider F, Dornia C, Kupper F, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Moser M, Buttner R, et al. PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2004;23(5):487–95.
Bergmann C, Senderek J, Sedlacek B, Pegiazoglou I, Puglia P, Eggermann T, Rudnik-Schoneborn S, Furu L, Onuchic LF, De Baca M, et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J Am Soc Nephrol. 2003;14(1):76–89.
Obeidova L, Seeman T, Elisakova V, Reiterova J, Puchmajerova A, Stekrova J. Molecular genetic analysis of PKHD1 by next-generation sequencing in Czech families with autosomal recessive polycystic kidney disease. BMC Med Genet. 2015;16:116.
Ward CJ, Wu Y, Johnson RA, Woollard JR, Bergstralh EJ, Cicek MS, Bakeberg J, Rossetti S, Heyer CM, Petersen GM, et al. Germline PKHD1 mutations are protective against colorectal cancer. Hum Genet. 2011;129(3):345–9.
Smolović B, Muhović D, Hodžić A, Bergant G, Peterlin B. The role of next generation sequencing in the differential diagnosis of Caroli's syndrome. Balkan J Med Genet. 2018;21(2):49–53.
Al-Hamed MH, Kurdi W, Alsahan N, Alabdullah Z, Abudraz R, Tulbah M, Alnemer M, Khan R, Al-Jurayb H, Alahmed A, et al. Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. J Med Genet. 2016;53(5):338–47.
Edrees BM, Athar M, Al-Allaf FA, Taher MM, Khan W, Bouazzaoui A, Al-Harbi N, Safar R, Al-Edressi H, Alansary K, et al. Next-generation sequencing for molecular diagnosis of autosomal recessive polycystic kidney disease. Gene. 2016;591(1):214–26.
Vivante A, Hwang DY, Kohl S, Chen J, Shril S, Schulz J, van der Ven A, Daouk G, Soliman NA, Kumar AS, et al. Exome sequencing discerns syndromes in patients from consanguineous families with congenital anomalies of the kidneys and urinary tract. J Am Soc Nephrol. 2017;28(1):69–75.
Buscher R, Buscher AK, Weber S, Mohr J, Hegen B, Vester U, Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease (ARPKD): kidney-related and non-kidney-related phenotypes. Pediatr Nephrol. 2014;29(10):1915–25.
Fonck C, Chauveau D, Gagnadoux MF, Pirson Y, Grunfeld JP. Autosomal recessive polycystic kidney disease in adulthood. Nephrol Dial Transplant. 2001;16(8):1648–52.
Burgmaier K, Kilian S, Bammens B, Benzing T, Billing H, Buscher A, Galiano M, Grundmann F, Klaus G, Mekahli D, et al. Clinical courses and complications of young adults with autosomal recessive polycystic kidney disease (ARPKD). Sci Rep. 2019;9(1):7919.
Rajab A, Al Rashdi I, Al Salmi Q. Genetic services and testing in the Sultanate of Oman. Sultanate of Oman steps into modern genetics. J Community Genet. 2013;4(3):391–7.
Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6:17.
Rajab A, Patton MA. A study of consanguinity in the Sultanate of Oman. Ann Hum Biol. 2000;27(3):321–6.
Rajab A, Hamza N, Al Harasi S, Al Lawati F, Gibbons U, Al Alawi I, Kobus K, Hassan S, Mahir G, Al Salmi Q, et al. Repository of mutations from Oman: The entry point to a national mutation database. F1000Res. 2015;4:891.
Michel-Calemard L, Dijoud F, Till M, Lambert JC, Vercherat M, Tardy V, Coubes C, Morel Y. Pseudoexon activation in the PKHD1 gene: a French founder intronic mutation IVS46+653A>G causing severe autosomal recessive polycystic kidney disease. Clin Genet. 2009;75(2):203–6.
Quint A, Sagi M, Carmi S, Daum H, Macarov M, Ben Neriah Z, Meiner V, Elpeleg O, Lerer I. An Ashkenazi founder mutation in the PKHD1 gene. Eur J Med Genet. 2016;59(2):86–90.
Lambie L, Amin R, Essop F, Cnaan A, Krause A, Guay-Woodford LM. Clinical and genetic characterization of a founder PKHD1 mutation in Afrikaners with ARPKD. Pediatr Nephrol. 2015;30(2):273–9.
Ebner K, Feldkoetter M, Ariceta G, Bergmann C, Buettner R, Doyon A, Duzova A, Goebel H, Haffner D, Hero B, et al. Rationale, design and objectives of ARegPKD, a European ARPKD registry study. BMC Nephrol. 2015;16(1):22.
Acknowledgements
The authors thank the supportive nephrologists and nurses from the Royal Hospital and all referral hospitals across Oman for their help in gathering the families and their clinical information. The authors would like also to thank Mozah Al-Busaidi, a genetic counselor at Sohar Hospital for her help in collecting the clinical information of patients as well as their history of disease and pedigrees. The cooperation of the tested families is appreciated.
Funding
This study was supported by the Ministry of Health and The Research Council Grant for Issa Al Salmi, (ORG/HSS/14/015), Oman. EM is supported by Kidney Research UK. JAS is funded by Kidney Research UK and the Northern Counties Kidney Research Fund.
Funders have no role in the study.
Author information
Authors and Affiliations
Contributions
IAA: molecular genetic study, data analysis and wrote the manuscript, EM: data analysis and manuscript review, IAS: organized samples collections, clinical data evaluation and manuscript review, FAR: organized samples collections, clinical data collection, AAM: manuscript review and JAS: conceived the study, data analysis and supervision and manuscript review. All authors have read and approved the manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was ethically approved by the Research and Ethical Review and Approval Committee, Ministry of Health (MOH) in Oman and by the Ethics Committee, College of Medicine and Health Sciences, Sultan Qaboos University (MREC#1096) in Oman.
All study participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1
: Figure S1. Pedigrees of the 32 analysed families. Figure S2. Representation of the missense variants of the PKHD1 gene detected in ARPKD patients in relation to the gene exon structure and protein domains. Table S1. Disease categories and genes selected for targeted NGS panel for cystic kidney disease. Table S2. Primers used for PCR amplification and sequencing of PKHD1 gene. Table S3. Different PKHD1 founder mutations associated with different ethnicities.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Al Alawi, I., Molinari, E., Al Salmi, I. et al. Clinical and genetic characteristics of autosomal recessive polycystic kidney disease in Oman. BMC Nephrol 21, 347 (2020). https://doi.org/10.1186/s12882-020-02013-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-020-02013-2