
Abstract
Carbon dioxide (CO2) fixation into value-added chemicals has attracted growing attention and one promising atom-efficient pathway is via the cycloaddition with three member-ring compounds like epoxides. Herein, we demonstrated that encapsulation of linear poly(ionic liquid)s (PILs) on ordered mesoporous carbon materials provides a facile and feasible approach towards environmental-friendly heterogeneous catalysts with high performance in CO2 cycloaddition with epoxides under mild conditions. A series of novel linear phenolic hydroxyl group functional imidazolium-based PILs synthesized from hydroxymethylation reaction between 4-(imidazol-1-yl)phenol-1-butyl-imidazolium iodide and formaldehyde was loaded on ordered mesoporous carbon FDU-15–600 derived from mesoporous phenolic resin. By virtue of controlling the initial polymerization temperature, the molecular weight of PILs was facilely modulated, reaching strong host–guest interaction during the PIL immobilization. Highly stable immobilized PIL species with spatial satisfaction of ionic moieties and surface groups were thus realized to enable a synergic CO2 conversion via cycloaddition with epoxides. The optimal catalyst exhibited high yield and stable recyclability by using atmospheric CO2 under metal-additive-solvent-free conditions and the activity surprisingly exceeded the corresponding homogeneous parent IL and PIL. Excellent substrate compatibility was found by extending the transformation of more than ten epoxides including the inert ones such as disubstituted cyclohexene oxide. The significantly enhanced activity is attributed to the synergistic effect of the surface hydrogen groups and ionic moieties to accelerate the rate-determining ring-opening process.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon dioxide (CO2) is one of the chief greenhouse gases causing growing global warming [1,2,3,4], and also an abundant inexpensive and nontoxic C1 feedstock [5, 6]. In this regard, chemically fixing CO2 into high-value-added chemicals has attracted great attention in the mitigation of CO2 emission [7,8,9,10,11]. One of the most promising approaches is the cycloaddition reaction between CO2 and three-member ring organic compounds such as aziridines and epoxides [12,13,14]. This pathway has the features of environmentally friendly and 100% atom-efficiency [5, 8, 15], producing a series of valuable chemicals like cyclic carbonates as solvents, polymer monomers, and organic intermediates [16,17,18,19,20]. To convert CO2 molecules with inherent thermodynamic stability and kinetic inertness under mild condition [12, 21,22,23] encouraged the development of various homogeneous and heterogeneous catalysts including metal-porphyrin [6, 24], metal complexes [25, 26], metal–organic frameworks (MOFs) [27,28,29], covalent organic frameworks (COFs) [30, 31], ionic liquids (ILs) [32, 33], and porous organic polymers (POPs) [34,35,36], etc. The homogeneous catalysts normally exhibited high activity while the heterogeneous one allows facile catalyst separation and recycling. One of the dreaming ways to combine their advantages is the heterogenization of high-performance homogeneous catalysts but remains extremely challenging due to the potential activity decline and active sites leaching during this process.
Cycloaddition of CO2 with epoxides usually undergoes ring opening, CO2 insertion, and ring closure [37,38,39]. The first step is normally recognized as the rate-determining step and is triggered by an attack by a nucleophilic agent. The epoxide activation can be greatly promoted by Lewis acids (metal sites) [24,25,26, 28, 29] or hydrogen bond donors (HBDs) such as the hydroxyl group [5]. The anions of ILs were the efficient nucleophilic agents and the ILs’ structure associating with the corresponding catalytic function can be facilely modulated at a molecular level by integrating versatile functional groups to promote CO2 affinity and epoxide activation. Therefore, ILs and their derivatives have been extensively investigated as a large family of environmental-friendly metal-free catalysts for CO2 cycloaddition [21, 40]. For example, phenolic hydroxyl group functional IL showed excellent activity in the CO2 cycloaddition and achieved a high yield of 96% in the conversion of epichlorohydrin (ECH) at room temperature and atmospheric conditions [33]. To facilitate catalyst recycling, great efforts have been made to develop IL-related heterogeneous catalysts mainly comprising two following strategies [41,42,43]. One is the synthesis of ionic moieties containing polymers through the polymerization of IL monomer or with other linkers through various polymerization pathways such as free radical polymerization, hypercrosslinkage, and condensation [44,45,46,47]. For example, free radial self-polymerization of 2-(dimethylamino) ethyl methacrylate derived IL monomers afforded a series of PILs that can effectively catalyze the CO2 cycloaddition with multiple epoxides with the yields of 70–98% at 110 °C and 20 bar [46]. Bifunctional ionic polymers were directly synthesized through co-polymerization of tris (4-vinylphenyl) phosphine and functional dibromides and the resulting carboxyl-containing one exhibited the yield above 99% at 140 °C in the transformation of atmospheric CO2 via cycloaddition with ECH [47]. Alternatively, anchoring ILs and PILs on the porous materials provided a promising approach toward IL-derived solid materials that can inherit the abundant porosity of parent supports [8, 45]. More importantly, the combination of surface groups in the supports and immobilized moieties favors reaching the highly effective synergistic conversion of CO2 via cycloaddition with epoxide. Particularly, encapsulation of PILs on the ordered porous materials benefits to balance the stability and activity thanks to the confinement of the ordered porous channels and the spatial satisfaction of different reactive sites [48, 49]. This strategy is feasible and versatile for IL solidification because it does not rely on the designation of polymerizable IL monomer for the pore formation during the polymerization process. Many ordered porous materials with the large surface area such as zeolites, MOFs, COFs, and mesoporous silica have been explored to immobilize ILs and PILs for the preparation of heterogeneous IL-derived catalysts [16, 27, 28, 50]. Carbon materials with the features such as large surface area, tunable porosity, and abundant surface functional groups have been explored as the supports and catalysts in the CO2 cycloadditions [51,52,53,54,55,56,57]. For instance, as a metal-free heterogeneous catalyst, graphene oxide (GOs) achieved 90.3% conversion and 98.6% selectivity in converting styrene oxide to styrene cyclic carbonate at 140 °C and 1 bar by using N,N-Dimethylformamide (DMF) as the solvent [55]. A PIL/graphene composite was prepared via an in-situ surface construction strategy and gave a yield of 99% in the coupling of 15 bar CO2 with propylene oxide at 100 °C [56]. Carbon supported single Zn atom catalyst was constructed from the straightforward carbonization of carbon supported phenanthroline-ligated Zn(OAc)2 complex and exhibited the yields of 93–98% in the conversion of seven epoxides via coupling with 5 bar CO2 at 100 °C in the presence of tetrabutyl ammonium bromide (TBAB) as an additive [57]. The abundant surface oxygen groups such as hydrogen groups on the carbon materials can serve as effective HBD to promote the epoxide activation to accelerate the corresponding transformation into the target cyclic carbonates [3, 41]. In addition to the disordered carbon materials mentioned above, ordered porous carbon materials such as CMK, FDU, and CGM-Cs (carbon-based Cornell Graded Materials) series [58,59,60]. Nonetheless, the encapsulation of PILs into the ordered porous carbon materials is still to be explored for the preparation of metal-free heterogeneous catalysts for CO2-epoxide coupling.
Herein, we triggered the application of ordered mesoporous carbon materials in the encapsulation of linear PILs and constructed a family of facilely adjustable PIL-carbon hybrid heterogeneous catalysts for CO2 fixation via cycloaddition with epoxide. The novel linear PILs were synthesized via hydroxymethylation reaction between phenolic hydroxyl functional imidazolium-based IL with formaldehyde solution under alkaline conditions [61], with molecular weight modulation via controlling the polymerization temperature. The target ordered mesoporous carbon material FDU-15–600 was prepared through the carbonization of mesoporous phenolic resin that was synthesized in a soft-template route [62]. The linear PILs were confined in the mesochannels of FDU-15–600, reaching strong host–guest interaction that depended on the initial molecular weight of PILs. Herein, an effective synergy between the surface hydrogen groups and the ionic moieties with the nucleophilic agent was achieved and thus resulted in high efficiency in the cycloaddition of atmospheric CO2 with epoxides under the mild condition without any additive and solvent. High yield, stable recycling performance and broad scope were observed in the catalysis evaluation. The activity of the champion heterogeneous PIL of the present work even outperformed its parent homogeneous IL and PIL, showing the great potential of this strategy.
1.1 Experimental section
1.1.1 Materials
The commercial chemicals and reagents were used as received without extra purification unless otherwise stated. 4-(Imidazol-1-yl) phenol (97%) was provided by Meyer (Shanghai) Chemical Technology Co. LTD. Iodobutane was provided by Xilong Science Co. LTD. Triblock copolymer (PEO-PPO-PEO, pluronic F127) was purchased from Sigma-Aldrich Co. LTD. Phenol was purchased from Shanghai Lingfeng Chemical Reagent Co. LTD. Activated carbon (AC) was purchased from Shanghai Macklin Biochemical Co. LTD.
1.1.2 Characterization
1H nuclear magnetic resonance (NMR) spectra were collected on a Bruker DPX 500 spectrometer. C, H, and N combustion chemical analyses were performed on the Vario EL cube. Brunauer–Emmett–Teller (BET) surface areas and pore volumes were measured at the temperature of liquid nitrogen (-196 °C) by using a BELSORP-MINI analyzer. Prior to the measurement, all the samples were degassed at 150 °C for 3 h to a vacuum of 10−3 Torr. Scanning electron microscopy (SEM) images were acquired with a Hitachi S-4800 scanning electron microscope (10 kV). Transmission electron microscope (TEM) images were recorded on a JEOL JEM-2010 (200 kV) instrument. Small-angle X-ray diffraction (XRD) patterns were collected by using a Smart Lab X-ray diffractometer from Rigaku. Solid-state 13C cross-polarization (CP)/magic-angle-spinning (MAS) NMR (13C MAS NMR) analysis was carried out on a Bruker AVANCE-III spectrometer. X-ray photoelectron spectroscopy (XPS) spectra were collected on a PHI 5000 Versa Probe X-ray photoelectron spectrometer equipped with Al Kα radiation (1486.6 eV). Thermal gravimetric analysis (TGA) experiments were performed on an STA409 instrument under N2 flow. The molecular weight of linear PILs was analyzed by gel permeation chromatography (GPC, 1515, Waters, USA). Raman spectra were collected on a Horiba HR 800 spectrometer with a Spectra-Physics 2018 Argon/Krypton Ion Laser system (excitation line: 514 nm).
1.1.3 Synthesis of soluble phenolic resol
Soluble phenolic resol was prepared under basic conditions according to a reported procedure [61]. Phenol (4.0 g, 42.5 mmol) was melted at 50 °C and then mixed with a sodium hydroxide solution (20 wt%, 0.85 g, 4.25 mmol) under stirring, followed by dropwise addition of formalin (37 wt%, 6.9 g, 85 mmol). The mixture was stirred at 75 °C for 1 h and then cooled to room temperature. The pH value of the solution above was adjusted to be 7.0 by using an aqueous hydrochloric acid solution with a concentration of 0.6 M. Subsequently, water was removed by rotary evaporation under vacuum and the resulting soluble phenolic resol was re-dissolved in ethanol for further use.
1.1.4 Synthesis of FDU-15–600
Triblock copolymer (PEO-PPO-PEO, pluronic F127; 0.6 g) was dissolved in ethanol (10.0 g) at 55 °C and then mixed with 3.0 g of ethanol solution containing soluble phenolic resol precursors (20 wt%). The mixture was stirred for 30 min to give a homogeneous solution, which was transferred to a 150 mm culture dish. Ethanol was evaporated at room temperature for 5–8 h. The resulting transparent membrane was then heated in an oven at 100 °C for 24 h. The obtained solids were calcined at 350 °C under nitrogen for 2 h with a heating rate of 1 °C min−1 and then calcined at 600 °C for 3 h to afford FDU-15–600.
1.1.5 Synthesis of [p-ArOH-IM]I
The IL monomer [p-ArOH-IM]I was synthesized according to the reported procedure [33]. 4-(1-Imidazolyl)phenol (0.32 g, 2 mmol, 97%), n-Iodobutane (0.368 g, 2 mmol, 99%), and anhydrous alcohol (10 ml) were stirred at room temperature for 1 h and then heated at 100 °C for 24 h. The mixture was cooled to room temperature and washed with ethyl acetate (3 × 20 mL). The residue solvent was removed by rotary evaporation and the resulting solid was dried in a vacuum at 70 °C. 1H NMR (400 MHz, Deuterium Oxide, Fig. S1): δ 9.08 (t, J = 1.7 Hz, 1H), 7.71 (t, J = 1.8 Hz, 1H), 7.60 – 7.51 (m, 1H), 7.47 – 7.35 (m, 2H), 7.05 – 6.93 (m, 2H), 4.23 (t, J = 7.2 Hz, 2H), 1.91 – 1.77 (m, 2H), 1.36 – 1.24 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H).
1.1.6 Synthesis of linear PILs
In a typical synthesis, [p-ArOH-IM] I (0.162 g) and NaOH solution (20 wt%, 10 μL) were mixed in a tube, followed by the addition of formaldehyde solution (37 wt%, 100 μL). The mixture was sonicated for 30 min and then heated at 90 °C for 60 min. After that, the solution was neutralized by an aqueous HCl solution (0.6 M) and washed subsequently with ethanol and ethyl acetate to remove the residue [p-ArOH-IM]I and NaCl. The gel-like product was dried at 90 °C under vacuum to offer a linear PIL PIL-90. Varying the polymerization temperature to room temperature (RT), 55 °C, and 110 °C afforded PIL-RT, PIL-55, and PIL-110, respectively.
1.1.7 Preparation of FDU-15–600 supported linear PILs
Linear PIL was immobilized on FDU-15–600 by a wet-impregnation method. Typically, PIL-90 (0.089 g) was dissolved in ethanol (5 mL) to give a homogeneous solution, which was mixed with FDU-15–600 (0.504 g, pre-activated at 120 °C under vacuum for 3 h) in a 100 mL beaker. The mixture was stirred at room temperature for 12 h, with the auto-evaporation of ethanol, and then dried in a vacuum oven at 100 °C for 12 h to give the product 15%PIL-90@FDU-15–600. By varying the PIL precursor and the loading amount, various other hybrids were prepared.
1.1.8 Preparation of control samples
For comparison, several control samples, including 15%S-PIL-90@FDU-15–600, 15%PIL90@LOMC, 15%PIL-90@AC, and 15%IL@FDU-15–600 were prepared as follows.
15%S-PIL-90@FDU-15–600
Pre-activated FDU-15–600 (0.504 g), [p-ArOH-IM]I (0.086 g), and ethanol (5 mL) were mixed in a 50 mL flask and sonicated for 30 min to give the mixture A. An aqueous NaOH solution (20 wt%, 5 μL) was diluted with formalin (37 wt%, 50 μL) in a centrifuge tube and sonicated for 30 min to give the mixture B. The mixtures A and B were mixed in a 50 mL flask and stirred at 90 °C for 60 min under a nitrogen atmosphere. The resulting solid was dried in a vacuum oven at 100 °C for 12 h to give 15%S-PIL-90@FDU-15–600.
15%PIL-90@LOMC
Disordered carbon (LOMC) was synthesized with the same procedure as that of FDU-15–600 except that more F127 (1 g) was involved. PIL-90 was loaded on LOMC with the same loading amount and procedure as that of 15%PIL-90@FDU-15–600 to give 15%PIL-90@LOMC.
15%PIL-90@AC
15%PIL-90@AC was prepared by loading PIL-90 with the same loading amount and procedure as that of 15%PIL-90@FDU-15–600.
15%IL@FDU-15–600
15%IL@FDU-15–600 was prepared by loading IL monomer [p-ArOH-IM]I on FDU-15–600 with the same loading amount and procedure to that of 15%PIL-90@FDU-15–600.
1.1.9 CO2 cycloaddition with epoxide
The cycloaddition of atmospheric CO2 with epoxides was carried out in a Schleck tube reactor (25 mL) equipped with a CO2 balloon. Epoxide (5 mmol) and 15%PIL-90@FDU-15–600 (50 mg, 0.4 mol%) were stirred at the desired temperature for the preset time. After the reaction, the solid was separated by centrifugation and the internal standard n-dodecane (0.5 g) was added into the liquid phase, which was further diluted with ethyl acetate. The resulting solution was qualitatively analyzed by a gas chromatography-mass spectrometer (Bruker Scion 436 GC–MS) and quantitatively analyzed by a gas chromatograph (Agilent 7890B) equipped with a flame ionization detector (FID) and a capillary column (HP-5, 30 m × 0.25 mm × 0.25 μm). The cycloaddition of high-pressure CO2 with epoxide was conducted in a stainless-steel autoclave (25 mL) and the product was analyzed by GC and GC–MS as mentioned above.
The catalyst was isolated after the cycloaddition reaction, washed with ethyl acetate several times and dried at 90 °C for 12 h in a vacuum. Afterward, the catalyst was reused for the next catalytic run under the same reaction conditions. This procedure was repeated five times to examine the recyclability of the catalyst PIL-90@FDU-15–600.
2 Results and discussion
2.1 Structure of FDU-15–600 supported linear PIL
Scheme 1 depicts the preparation procedure of ordered mesoporous carbon confined linear PILs for CO2 cycloaddition with epoxide. The ordered mesoporous carbon FDU-15–600 was synthesized by using F127 as the soft template and calcined at 350 °C to remove the template, with further carbonization at 600 °C. The linear PILs were synthesized via hydroxymethylation reaction, where [p-ArOH-IM]I bearing phenolic hydroxyl group and formaldehyde respectively served as the α-H resource component and a cross-linking agent [63, 64]. The linear PILs were immobilized on FDU-15–600 via the wet-impregnation process. By varying the polymerization temperature from room temperature (RT) to 110 °C, a series of linear PILs were synthesized and named PIL-T, where T = RT, 55, 90, and 110, represent the polymerization temperature. As shown in Fig. S2, the color of the PIL-T series gradually become shallower with increasing polymerization temperature. CHN elemental analyses indicated that the PIL-T series had similar chemical composition to the IL precursor [p-ArOH-IM]I while slightly lower H content (Table S1 entries1-5), attributable to the α-H elimination during the polymerization [40]. The polymerization degree of PIL-T was analyzed by gel permeation chromatography (GPC) (Fig. S3). The molecular weights of PIL-T were listed in Table 1. Polymerization at RT caused a low molecular weight that was about three times that of IL precursor (Table 1, entries 1 and 2), suggesting that PIL-RT was composed of about three [p-ArOH-IM]I molecules. With elevating polymerization temperature, the molecular weights of PIL-T series increased to 23,041, 30,494, and 48,084 at 55 °C, 90 °C, and 110 °C, respectively (Table 1, entries 3–5). This result implies that the polymerization degree of PIL-T was facilely modulated by controlling the polymerization temperature.

Schematical illustration of the preparation of ordered mesoporous carbon (FDU-15-600) encapsulated linear poly(ionic liquid) for CO2 cycloaddition with epoxides
A family of carbon encapsulated linear PIL samples, termed X%PIL-T@FDU-15–600 (X% index the loading amount), were prepared by using PIL-T with different loading amounts, including 15%PIL-T@FDU-15–600, 10%PIL-90-FDU-15–600, and 20%PIL-90@FDU-15–600. There was no N element in FDU-15–600 and the detectable N species on X%PIL-T@FDU-15–600 samples reflected the immobilization of PIL-T (Table S1, entries 6–12). The PIL content linearly increased with the loading amount, while close to each other by fixing the loading amount. The small-angle XRD patterns of the parent FDU-15–600 and X%PIL-T@FDU-15–600 were displayed in Fig. 1a and Fig. S4a. For FDU-15–600, a strong diffraction peak was observed at the 2θ of 0.86, corresponding to the 100 plane [61]. Two weak peaks were found at the 2θ of 1.36 and 1.67, corresponding to the 110 and 200 planes [62]. The observation of these diffraction peaks revealed the formation of a well-ordered 2D hexagonal structure with a space group of p6mm [65]. The remaining of these peaks in the XRD patterns of X%PIL-T@FDU-15–600 indicated the preservation of the parent highly ordered hexagonal mesostructure after PIL-T loading. The weakening of the peak intensity came from the occupation of the guest PIL-T inside the meso-channels. The solid-state 13C MAS NMR spectra of FDU-15–600 and the typical PIL loaded sample 15%PIL-90@FDU-15–600 were shown in Fig. 1b. Both of them exhibited similar and distinct peaks at ~ 28.1, ~ 127, and 153.4 ppm, which are respectively corresponded to the C atom of the methylene group linking the two benzene rings, the C atoms of the benzene ring adjacent the phenolic hydroxyl groups, and the other C atoms of the benzene ring [61, 63]. Comparing the spectrum of 15%PIL-90@FDU-15–600 with FDU-15–600, it was found that there existed an extra shoulder band at 138 ppm, attributable to the C2 atom of the imidazole ring [63]. This phenomenon is in line with the encapsulation of imidazolium based PIL in the FDU-15–600. Notably, a slight shift of the strong peak at 126.6 ppm to 127.4 ppm, which may be caused by the coincidence of the imaged C4 and C5 carbon atoms with the benzene ring carbon atoms, resulting in the change of the chemical shift of the characteristic peak of the aromatic carbon and imidazolium ring (C4 and C5 atoms) not obvious there [63].

a Small-angle XRD patterns. b Solid-state 13C CP/MAS NMR spectra. c Nitrogen sorption isotherms and d pore size distribution curves. The sorption isotherms for samples FDU-15-600, 15%PILRT@FDU-15-600, 15%PIL-55@FDU-15-600, and 15%PIL-90@FDU-15-600 are shifted by 200, 250, 160,and 70 cm3 g-1. The pore size distribution curves for samples FDU-15-600, 15%PIL-RT@FDU-15-600, 15%PIL-55@FDU-15-600, and 15%PIL-90@FDU-15-600 are shifted by 0.7, 0.55, 0.4, and 0.15 cm3 g-1, respectively
Nitrogen sorption isotherms of FDU-15–600 and X%PIL-T@FDU-15–600 were investigated to provide the pore information (Fig. 1c). All of them displayed the typical IV type isotherm with an apparent H1-type hysteresis loop, characteristic of classical 2D-hexagonal mesopores [61, 64]. The pore size distribution curves calculated by using Barret-Joyner-Halenda (BJH) method further visualized the existence of mesoporous structure (Fig. 1d). The Brunauer–Emmett–Teller (BET) surface areas and pore volumes were summarized in Table 1. FDU-15–600 had a high surface area of 798 m2 g−1 and a large pore volume of 0.62 cm3 g−1 (Table 1, entry 6). Decreased surface area and pore volume were observed after loading of PIL-T, and the higher loading, the less porosity (Table 1, entries 7–12). For example, the typical sample 15%PIL-90@FDU-15–600 has a surface area of 283 m2 g−1 and a pore volume of 0.29 cm3 g−1 (Table 1, entry 9). These results indicate that PIL-T occupies inside the mesopores.
X-ray photoelectron spectroscopy (XPS) analyses were performed to discern the interactions between the guests' PIL-X and the supports in 15%PIL-X@FDU-15–600 (X = RT, 55, 90, and 110). For comparison, FDU-15–600, [p-ArOH-IM]I, and 15%IL@FDU-15–600 were measured in parallel. The survey scan XPS spectrum of FDU-15–600 showed the signals at 284 and 532 eV for C1 and O1s species, respectively (Fig. S5a) [66]. Additional signals at 402 eV and 618 eV respectively for N1s and I3d species (Fig. S5a) were observed in the IL or PIL containing samples, reflecting the loading of these guests [67, 68]. The high-resolution N1s and I3d XPS (Figs. 2a and b) spectra indicated that [p-ArOH-IM]I exhibited one signal at 402.5 eV for N1s (NI species) and a set of double peaks at 630.8 eV for I3d(3/2) and 619.3 eV for I3d(5/2) (II species) [33, 69]. Loading [p-ArOH-IM]I on FDU-15–600 caused the emergence of one small N1s signal at 400.0 eV and a set of double I3d signals at 632.7/620.6 eV in the N1s and I3d XPS spectra of 15%IL@FDU-15–600, respectively. These singles came from the formation of less positively charged N species (NII) and less negatively charged I species (III), attributable to the strong host–guest interaction that varied the cation–anion interaction in the immobilized [p-ArOH-IM]I on FDU-15–600. A similar phenomenon was observed in the N1s and I3d XPS spectra of 15%PIL-X@FDU-15–600. The proportion of different N and I species were calculated from their peak area and summarized in Table S2 and Table S3. More NII and III species formed in 15%PIL-X@FDU-15–600 relative to 15%IL@FDU-15–600. With increasing polymerization temperature in the preparation of PIL precursor, the content of III species varied slightly, while the content of NII species firstly increased and then decreased, reaching the maximum value at T = 90 for 15%PIL-90@FDU-15–600. This result indicated that modulating the polymerization temperature can facilely adjust the interaction between PIL and FDU-15–600, reaching a strong interaction at moderate polymerization temperature. The reason can be assigned that linear PIL with moderate molecular weight was synthesized under this condition, providing more binding sites to strengthen the host–guest interaction while avoiding the potential agglomeration by using the PIL precursor with excessive molecular weight. Besides, the O1s XPS spectra of all these samples were deconvoluted into two signals at 532 eV and 533 eV, respectively for C = O and C–OH species, proving the existence of hydroxyl species (Fig. S5b).

a N1s and b I3d XPS spectra
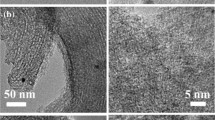
Additional characterizations were performed by taking the typical sample 15%PIL-90@FDU-15–600 as an example and the parent FDU-15–600 was tested in parallel to demonstrate the variation before and after immobilization of linear PIL. The morphology of FDU-15–600 and 15%PIL-90@FDU-15–600 were monitored by scanning electron microscopy (SEM) images (Fig. 3a and b). FDU-15–600 was composed of irregular particles on the micrometer scale, while 15%PIL-90@FDU-15–600 demonstrated the almost same morphology. No spongy-like particles of the amorphous linear polymer were found, suggesting the formation of linear PIL inside FDU-15–600’s mesopores. The transmission electron microscope (TEM) image of 15%PIL-90@FDU-15–600 shows the highly ordered mesoporous structure (Fig. 3c) [62, 63]. The corresponding elemental mapping images displayed the relatively homogeneous dispersion of C, N, O, and I elements, representative of the highly dispersive ionic moieties (Fig. 3d-g).

SEM images of a FDU-15-600 and b 15%PIL-90@FDU-15-600. c TEM image of 15%PIL-90@FDU-15-600 and d-g corresponding elemental mapping images of C, O, N, and I, respectively
Thermogravimetric (TG) profiles of FDU-15–600, 15%PIL-90@FDU-15–600, and [p-ArOH-IM]I were presented in Fig. 4a and Fig. S6, showing negligible weight loss below 240 °C. Dramatical weight loss of 15%PIL-90@FDU-15–600 happened from 240 °C to 380 °C, resembling the TG curve of [p-ArOH-IM]I (Fig. S6). This result suggests that the weight loss above came from the decomposition of the framework ionic moieties. What’s more, the weight loss difference between FDU-15–600 and 15%PIL-90@FDU-15–600 was around 15%, close to the theoretical loading amount of PIL-90 and in line with the elemental analysis result (Table S1, entry 4). Raman spectra of FDU-15–600 and 15%PIL-90@FDU-15–600 were presented in Fig. 4b. Each Raman spectrum demonstrated two signals at 1336 cm−1 (D band) and 1587 cm−1 (G band) [61] respectively for the disordered carbon structure and graphitic domains [70]. The defect density is reflected by the ratio of the peak intensity of the D band to that of the G band (ID/IG). 15%PIL-90@FDU-15–600 exhibited slightly higher ID/IG values of 1.52 than FDU-15–600 (1.46), which is assignable to the existence of an amorphous linear polymer in the meso-channels.
CO2 adsorption isotherms of 15%PIL-X@FDU-15–600 were measured up to 1 bar at 298 K and compared with the ones of FDU-15–600 and 15%IL@FDU-15–600 (Fig. S7 and Fig. S8). These adsorption isotherms were fitted by using the Langmuir model [30] and the fitting parameters qc and kc of these samples were listed in Table S4, respectively corresponding to the saturated adsorption capacity of the adsorption site and the adsorption equilibrium constant. The CO2 uptake at 1 bar was 2.16 mmol.g−1 for FDU-15–600 and decreased after loading [p-ArOH-IM]I or PIL-T due to the decline of the surface area (Fig. 4c). The surface area normalized CO2 uptakes at 1 bar was thus calculated to be 0.0027, 0.0030, 0.0021, 0.0028, 0.0040, and 0.0024 mmol.m−2 for FDU-15–600, 15%IL@FDU-15–600, 15%PIL-X@FDU-15–600 (X = RT, 55, 90, and 110), respectively (Fig. 4d). 15%PIL-90@FDU-15–600 exhibited the highest surface area normalized CO2 uptake, reflecting the favorable CO2 trapping ability capacity.

a TG curves of FDU-15-600 and 15%PIL-90-60@FDU-15-600. b Raman spectra of FDU-15-600 and 15%PIL-90@FDU-15-600. c CO2 adsorption isotherms at 298 K and d Unit adsorption capacity
2.2 CO2 fixation through cycloaddition with epoxide
The catalytic performance of the above [p-ArOH-IM]I, PIL-T, and X%PIL-T@FDU-15–600 samples were evaluated in the CO2 cycloaddition with epoxides to produce cyclic carbonates (Table 2). The investigation started with CO2 coupling with styrene oxide (SO) under a relatively mild condition of 110 °C and atmospheric pressure without any additive or solvent. For comparison, various control catalysts were tested in parallel, including the parent support FDU-15–600, IL precursor [p-ArOH-IM]I, and PIL-T series. FDU-15–600 was inactive in the reaction, giving a low yield of 2.6% (Table 2, entry 1). Feeding 0.4 mol% [p-ArOH-IM]I relative to SO led to a moderate yield of 80.0% and a turnover number (TON) of 200 (Table 2, entry 2). In parallel, the catalytic performance of PIL-T series and other solid catalysts below unless otherwise mentioned were tested by using the same dosage of ionic moieties to [p-ArOH-IM]I. The yield was 61.7% over PIL-RT and continuously increased along with the elevated polymerization temperature (Table 2, entry 3). Notably, the yield of PIL-90 (71.7%) was close to that of PIL-110 (72.6%), suggesting that excessive polymerization temperature caused only slight activity variation. Despite this, loading PIL-T on FDU-15–600 presented a different picture, with a maximum yield of 94.5% over 15%PIL-90@FDU-15–600. The influence of the reaction temperature and time was presented in Fig. S9, demonstrating a rapid increase in the yield with the temperature or time. Compared with previous encapsulated PILs catalysts (Table 3) [3, 27, 71], it is the first time to reach effective CO2 cycloaddition with epoxides under atmospheric conditions over an encapsulated PIL catalyst. Considering the substrate difference, Table 3 listed the activity involving various substrates. The comparison indicated that previous carbon supported IL or PIL catalysts can only catalyze the transformation of robust and moderate active substrates under high pressure [27, 42, 72,73,74]. By contrast, the scope surveying indicated the great substrate tolerance of 15%PIL-90@FDU-15–600, offering high yields even for those inert ones such as long-alkyl chain bearing terminal epoxide and disubstituted cyclohexene oxide under atmospheric condition, further demonstrating the superiority of this catalyst.
All the 15%PIL-T@FDU-15–600 catalysts showed a higher yield than the corresponding PIL-T sample and the activity of 15%PIL-90@FDU-15–600 is even superior to that of [p-ArOH-IM]I (Table 2, entries 2–10), revealing a synergistically catalytic process by combining the PIL-T and carbon support. Immobilization of PIL-110 on FDU-15–600 caused slight activity enhancement, assignable to the limited dispersion of the bulky linear polymer in a confined nano-space. In addition, the loading amount of PIL-90 on FDU-15–600 was explored by preparing two control samples with either lower (10%) or higher (20%) loading. The nitrogen sorption result indicated a negative relationship between the loading amount and the porosity, i.e. the higher loading, the lower surface area and pore volume (Fig. S10 and Table 1, entries 9, 11, and 12). Varying the loading amount always provided a higher yield than the parent PIL-90 and moderate loading of 15% gave the highest activity when charging the same dosage of ionic moieties (Table 2, entries 9, 11, and 12), further reflecting the cooperation of PIL-90 and FDU-15–600. This is further supported by the result that 15%PIL-90@FDU-15–600 was more active than 15%PIL-90 + FDU-15–600, the physical mixture of FDU-15–600 and PIL-90 (yield: 94.5% vs 78.9%) (Table 2, entries 9 and 13).
For additional comparison, PIL-90 was impregnated on commercial activated carbon (AC) and a disordered carbon (LOMC) (Table S1, entries 13–16). AC is an amorphous carbon with a large surface area of 1526 m2 g−1 and a pore volume of 0.52 cm3 g−1 (Fig. S11 and Table 1, entry 13). After loading with 15%PIL-90, the resulting sample 15%PIL-90@AC had a surface area of 942 m2 g−1 and a pore volume of 0.40 cm3 g−1 (Table 1, entry 14). CO2 cycloaddition with SO catalyzed by 15%PIL-90@AC gave a similar yield to PIL-90 (72.3% vs 71.7%) (Table 2, entry 14), suggesting the lack of a synergistic effect between PIL-90 and AC. LOMC was derived from the carbonization of a disordered phenolic resin that was synthesized with the same procedure as that of FDU-15–600 except for the involvement of excessive soft-template F127. Small-angle XRD patterns of LOMC revealed a disordered structure, which was also observable on the sample 15%PIL-90@LOMC after loading with PIL-90 (Fig. S4b). Nitrogen sorption isotherm and pore size distribution curve revealed the mesoporous structure of LOMC, with a similar surface area and pore volume to FDU-15–600 (Fig. S11 and Table 1, entry 15). Loaded sample 15%PIL-90@LOMC had a surface area of 227 m2 g−1 and a pore volume of 0.19 cm3 g−1 (Table 1, entry 16). Under the identical reaction conditions, 15%PIL-90@LOMC exhibited a yield of 80.4% (Table 2, entry 15), close to that over [p-ArOH-IM]I but lower than the one over 15%PIL-90@FDU-15–600. These comparisons further reflect that PIL-90 confined in an ordered meso-channels of FDU-15–600 resulted in the synergistic CO2 cycloaddition with epoxide. The reason can be assigned to that the ordered meso-channels afforded the isolated nano-space to achieve better dispersion of these linear PILs, allowing the suitable distance between the surface groups of FDU-15–600 (for example, the hydroxyl groups) and ionic moieties. Besides, the in situ condensations of [p-ArOH-IM]I and formaldehyde on FDU-15–600 was conducted to provide a control sample 15%S-PIL-90@FDU-15–600, with a yield of 75.5% (Table 2, entry 16 and Table S1, entry 17). At the same time, it has 15% PIL-90@FDU-15–600 The similar specific surface area proves that the differ rence in activity comes from the different loading modes (Table 1, entry 17). This phenomenon indicates that the immobilization of pre-polymerized PIL led to better dispersion for higher activity. Directly loading IL precursor [p-ArOH-IM]I on FDU-15–600 was conducted and the resulting sample 15%IL@FDU-15–600 (Figs. S12, S13, and Table S1, entry 18) offered a lower yield of 89.2% (Table 2, entry 17) than that of 15%PIL-90@FDU-15–600 and their specific surface areas are similar (Table 1, entry 18), further revealing the superiority of immobilization of PIL on this carbon support.
During the CO2 cycloaddition with epoxide, the ring-opening is usually the rate-determining step and the epoxide activation can be promoted by the HBD through H-bond interaction [8, 21, 81,82,83]. For the supported ILs or PILs, the HBD species can come from the functional groups in ionic moieties and the support, but requiring spatial satisfactory [3, 14, 73, 84]. Previously, we have carefully investigated the role of phenolic hydroxyl group of imidazolium-based ILs in the cycloaddition with epoxide and afforded a highly active IL [p-ArOH-IM]I [33]. Herein, the heterogenization of this homogeneous IL was reached by first polymerization and successive loading on ordered mesoporous carbon FDU-15–600. The delocalized I− anions of ionic moieties have well leaving ability, allowing superior spatial facilitation to complete the ring-opening with the assistance of HBD groups in the ionic moieties and surface groups. Based on the result above and previous mechanism investigation [19, 64, 85], the most probable reaction pathway is proposed for 15%PIL-90@FDU-15–600 catalyzed CO2 cycloaddition with epoxide (Scheme 2), in which the I– anions served as the nucleophilic reagent [40], while the abundant phenolic hydroxyl groups in the ionic moieties and inside the wall of mesochannels of FDU-15–600 acted as efficient HBD [5, 63, 86]. In the beginning, epoxide was adsorbed into the mesochannels of 15%PIL-90@FDU-15–600 and activated by the hydroxyl groups with the polarization of C–O bonds of epoxide through hydrogen bonding interaction [33, 40]. The nucleophilic attack of I– anions to the C atom of activated epoxide led to the ring-opening, generating iodine-alkoxide followed by CO2 insertion. After that, ring closure produced cyclic carbonate, with the left of I– anions to regenerate the catalyst. The ring-opening is normally recognized as the rate-determining step [21], and the synergy of nucleophilic reagent and HBD is crucial for the epoxide activation and transition state stabilization [8, 33]. Modulating the molecular weight of PIL precursors can finely adjust the host–guest interaction to reach stable immobilization and favorable dispersion. Owing to this balance, the optimal sample 15%PIL-90@FDU-15–600 allowed the I– anions to near the surface hydroxyl groups, satisfying the requirable synergistic effect. By contrast, the loading of IL precursor resulted in unstable active sites, showing apparent deactivation during the recycling test (Table 2, entry 18) due to the leaching of loaded IL molecules as demonstrated by the decline of the N content in the elemental analysis of the spent catalyst (Table S1, entry 19).

Proposed reaction route for CO2 cycloaddition with epoxide catalyzed by 15%PIL-90@FDU-15-600
2.3 Reusability and scope
The recycling stability of a heterogeneous catalyst is of great importance for practical application. Delightedly, the separation of 15%PIL-90@FDU-15–600 from the reaction mixture was readily realized by facile centrifugation. Recycling investigation showed that the catalytic activity of 15%PIL-90@FDU-15–600 was well retained during the 5 runs test in CO2 cycloaddition with SO (Fig. 5a and Table 2, entry 19). The ordered mesopores and structural integrity was observed by recording the small angle XRD and specific surface area (Table 1, entry 19) pattern of the spent 15%PIL-90@FDU-15–600 recovered after the 5th run (Fig. 5b). The preservation of the chemical composition and porosity during the recycling cycloaddition with epoxide was evidenced by the almost same elemental analysis result (Table S1, entry 20), N2 sorption isotherm and the pore size distribution curve (Figs. 5c and 5d) of the spent 15%PIL-90@FDU-15–600 as the fresh one. All of these are responsive to excellent recyclability and stability.

a Catalytic reusability of 15%PIL-90@FDU-15-600. Reaction conditions: styrene oxide (5 mmol), catalyst (0.4 mol%), 110 oC, 1 bar CO2 (balloon), 24 h. b Small-angle PXRD pattern. c N2 sorption isotherm and d pore size distribution curve of spent 15%PIL-90@FDU-15-600 recovered after the 5th run
The scope 15%PIL-90@FDU-15–600 was extended to the CO2 fixation with various epoxides (Table 4). Good to excellent yields were achieved in these terminal epoxides comprising the ones containing long alky chains, the conversion of which was more difficult than those active epoxides like epichlorohydrin (Table 4, entries 1–10) [3, 41]. Notably, the effective cycloaddition of atmospheric CO2 with inert disubstituted cyclohexene oxide over the present catalyst afforded a high yield of 91.7% (Table 4, entry 11). This is rarely reached by heterogeneous catalysts before [3, 40]. The scope surveying indicated the great substrate tolerance of this catalyst. It is worth mentioning that a high TON of 2645 and turnover frequency (TOF) of 7935 (Table 4, entry 12) was observed over 15%PIL-90@FDU-15–600 by using high pressure CO2 (3 MPa), greatly outperforming those carbon encapsulated ILs and PILs listed in Table 3. Though various effective heterogeneous catalysts with high TON and TOF have been reported before, it is still scarcely for metal-free heterogeneous catalysts under additive and solve free conditions. A comprehensive comparison with previous catalysts under similar conditions [3, 45, 81,82,83] indicated that the above TON and TOF over 15%PIL-90@FDU-15–600 under harsh conditions are superior to majority of those state-of-art ones, and particularly, a record-high TOF was reached over this metal-free catalyst. All of these reveal the high performance of 15%PIL-90@FDU-15–600 in the heterogeneous CO2 cycloaddition with epoxides.
3 Conclusion
A family of linear PILs with tunable molecular weight was synthesized through a hydroxy methylation reaction between a phenolic hydroxyl group functional imidazolium-based IL and formaldehyde by controlling the polymerization temperature. The resulting PILs were encapsulated on the mesoporous phenolic resin derived ordered mesoporous carbon FDU-15–600 to give the corresponding heterogeneous PIL-carbon hybrid catalysts. The interaction between PILs and carbon was strengthened by involving PIL with moderate molecular weight synthesized using moderate polymerization temperature. Not only the stability was improved, but also the suitable spatial adjacency of the ionic moieties and surface groups in the wall of mesochannels was reached, affording synergistically catalytic CO2 cycloaddition with epoxide. The best-performing catalyst was highly active in the transformation of a series of epoxides through coupling with CO2 under the atmospheric condition without any metal species, additive or solvent, and even more active than the parent IL and PIL precursors. The catalyst was facilely recovered and reused, and stable activity was observable during the recycling test. This work provides a facile and feasible perspective toward IL-derived heterogeneous catalysts and highlights the great potential of ordered mesoporous carbon materials in the rational designation of highly effective metal-free catalysts for CO2 fixation.
Availability of data and materials
Not applicable.
Abbreviations
- AC:
-
Activated carbon
- LOMC:
-
Disordered carbon
- Triblock copolymer:
-
PEO-PPO-PEO, pluronic F127
References
Zhu X, Xie W, Wu J, Miao Y, Xiang C, Chen C, Ge B, Gan Z, Yang F, Zhang M, O’hare D, Li J, Ge T, Wang R (2022) Recent advances in direct air capture by adsorption. Chem Soc Rev 51(15):6574–6651. https://doi.org/10.1039/d1cs00970b
Singh G, Lee J, Karakoti A, Bahadur R, Yi J, Zhao D, Albahily K, Vinu A (2020) Emerging trends in porous materials for CO2 capture and conversion. Chem Soc Rev 49(13):4360–4404. https://doi.org/10.1039/d0cs00075b
Li G, Dong S, Fu P, Yue Q, Zhou Y, Wang J (2022) Synthesis of porous poly(ionic liquid)s for chemical CO2 fixation with epoxides. Green Chem 24(9):3433–3460. https://doi.org/10.1039/d2gc00324d
Wang S, Wang Y, Kuang Y, Xu S, Gao S, Liu L, Niu H, Xiao P, Huang B (2022) Adsorption behaviour of molecular sieve and activated carbon for CO2 adsorption at cold temperatures. Carbon Neutrality 1(1):16. https://doi.org/10.1007/s43979-022-00017-5
Guo Z, Jiang Q, Shi Y, Li J, Yang X, Hou W, Zhou Y, Wang J (2017) Tethering Dual Hydroxyls into Mesoporous Poly(ionic liquid)s for Chemical Fixation of CO2 at Ambient Conditions: A Combined Experimental and Theoretical Study. ACS Catal 7(10):6770–6780. https://doi.org/10.1021/acscatal.7b02399
Deacy AC, Kilpatrick AFR, Regoutz A, Williams CK (2020) Understanding metal synergy in heterodinuclear catalysts for the copolymerization of CO2 and epoxides. Nat Chem 12(4):372–380. https://doi.org/10.1038/s41557-020-0450-3
Zhou Y, Zhang J, Wang L, Cui X, Liu X, Wong SS, An H, Yan N, Xie J, Yu C, Zhang P, Du Y, Xi S, Zheng L, Cao X, Wu Y, Wang Y, Wang C, Wen H, Chen L, Xing H, Wang J (2021) Self-assembled iron-containing mordenite monolith for carbon dioxide sieving. Science 373:315–320. https://doi.org/10.1126/science.aax5776
Jia D, Ma L, Wang Y, Zhang W, Li J, Zhou Y, Wang J (2020) Efficient CO2 enrichment and fixation by engineering micropores of multifunctional hypercrosslinked ionic polymers. Chem Eng J 390:124652. https://doi.org/10.1016/j.cej.2020.124652
Zhai G, Liu Y, Mao Y, Zhang H, Lin L, Li Y, Wang Z, Cheng H, Wang P, Zheng Z, Dai Y, Huang B (2022) Improved photocatalytic CO2 and epoxides cycloaddition via the synergistic effect of Lewis acidity and charge separation over Zn modified UiO-bpydc. Appl Catal B 301:120793. https://doi.org/10.1016/j.apcatb.2021.120793
Cheng J, Cen K (2022) Mechanisms of strengthening energy and mass transfer in microbial conversion of flue-gas-derived CO2 to biodiesel and biogas fuels. Carbon Neutrality 1(1):11. https://doi.org/10.1007/s43979-022-00004-w
Chernyak SA, Corda M, Dath JP, Ordomsky VV, Khodakov AY (2022) Light olefin synthesis from a diversity of renewable and fossil feedstocks: state-of the-art and outlook. Chem Soc Rev 51(18):7994–8044. https://doi.org/10.1039/d1cs01036k
Xu G-Q, Ma X-N, Jia X-B, Dong Y-H, Jiang Y-Q, Li X-J (2022) Synthesis of nucleoside-substituted carbonate and diol derivatives through the carbon dioxide reaction using polyionic liquid catalysts. Green Chem 24(11):4573–4580. https://doi.org/10.1039/d2gc00566b
Chen SJ, Li JS, Haddad R, Sadeghzadeh SM (2022) Cycloaddition of allylic chlorides, aryl alkynes, and carbon dioxide using nanoclusters of polyoxomolybdate buckyball supported by ionic liquid on dendritic fibrous nanosilica. J CO2 Util 61:10235. https://doi.org/10.1016/j.jcou.2022.102035
Zhou Y, Zhang W, Ma L, Zhou Y, Wang J (2019) Amino Acid Anion Paired Mesoporous Poly(ionic liquids) as Metal-/Halogen-Free Heterogeneous Catalysts for Carbon Dioxide Fixation. ACS Sustainable Chem Eng 7(10):9387–9398. https://doi.org/10.1021/acssuschemeng.9b00591
Li J, Jia D, Guo Z, Liu Y, Lyu Y, Zhou Y, Wang J (2017) Imidazolinium based porous hypercrosslinked ionic polymers for efficient CO2 capture and fixation with epoxides. Green Chem 19(11):2675–2686. https://doi.org/10.1039/c7gc00105c
Dan M, Zhong R, Hu S, Wu H, Zhou Y, Liu Z-Q (2022) Strategies and challenges on selective electrochemical hydrogen peroxide production: Catalyst and reaction medium design. Chem Catal 2(8):1919–1960. https://doi.org/10.1016/j.checat.2022.06.002
Tong H, Qu Y, Li Z, He J, Zou X, Zhou Y, Duan T, Liu B, Sun J, Guo K (2022) Halide-free pyridinium saccharinate binary organocatalyst for the cycloaddition of CO2 into epoxides. Chem Eng J 444:135478. https://doi.org/10.1016/j.cej.2022.135478
Luo R, Yang Y, Chen K, Liu X, Chen M, Xu W, Liu B, Ji H, Fang Y (2021) Tailored covalent organic frameworks for simultaneously capturing and converting CO2 into cyclic carbonates. J Mater Chem A 9(37):20941–20956. https://doi.org/10.1039/d1ta05428g
Guo L, Lamb KJ, North M (2021) Recent developments in organocatalysed transformations of epoxides and carbon dioxide into cyclic carbonates. Green Chem 23(1):77–118. https://doi.org/10.1039/d0gc03465g
Guo YC, Chen WJ, Feng L, Fan YC, Liang JS, Wang XM, Zhang X (2022) Greenery-inspired nanoengineering of bamboo-like hierarchical porous nanotubes with spatially organized bifunctionalities for synergistic photothermal catalytic CO2 fixation. J Mater Chem A 10(23):12418–12428. https://doi.org/10.1039/d2ta02885a
Yang GW, Xu CK, Xie R, Zhang YY, Zhu XF, Wu GP (2021) Pinwheel-Shaped Tetranuclear Organoboron Catalysts for Perfectly Alternating Copolymerization of CO2 and Epichlorohydrin. J Am Chem Soc 143(9):3455–3465. https://doi.org/10.1021/jacs.0c12425
Gong L, Sun J, Liu Y, Yang G (2021) Photoinduced synergistic catalysis on Zn single-atom-loaded hierarchical porous carbon for highly efficient CO2 cycloaddition conversion. J Mater Chem A 9(38):21689–21694. https://doi.org/10.1039/d1ta06159c
Meng X, Ju Z, Zhang S, Liang X, Von Solms N, Zhang X, Zhang X (2019) Efficient transformation of CO2 to cyclic carbonates using bifunctional protic ionic liquids under mild conditions. Green Chem 21(12):3456–3463. https://doi.org/10.1039/c9gc01165j
Luo R, Chen M, Zhou F, Zhan J, Deng Q, Yu Y, Zhang Y, Xu W, Fang Y (2021) Synthesis of metalloporphyrin-based porous organic polymers and their functionalization for conversion of CO2 into cyclic carbonates: recent advances, opportunities and challenges. J Mater Chem A 9(46):25731–25749. https://doi.org/10.1039/d1ta08146b
Yang Q, Yang CC, Lin CH, Jiang HL (2019) Metal-Organic-Framework-Derived Hollow N-Doped Porous Carbon with Ultrahigh Concentrations of Single Zn Atoms for Efficient Carbon Dioxide Conversion. Angew Chem Int Ed 58(11):3511–3515. https://doi.org/10.1002/anie.201813494
Sinha I, Lee Y, Bae C, Tussupbayev S, Lee Y, Seo M-S, Kim J, Baik M-H, Lee Y, Kim H (2017) Computer-aided rational design of Fe(iii)-catalysts for the selective formation of cyclic carbonates from CO2 and internal epoxides. Catal Sci Technol 7(19):4375–4387. https://doi.org/10.1039/c7cy01435j
Ding M, Jiang H-L (2018) Incorporation of Imidazolium-Based Poly(ionic liquid)s into a Metal-Organic Framework for CO2 Capture and Conversion. ACS Catal 8(4):3194–3201. https://doi.org/10.1021/acscatal.7b03404
Sun Y, Huang H, Vardhan H, Aguila B, Zhong C, Perman JA, Al-Enizi AM, Nafady A, Ma S (2018) Facile Approach to Graft Ionic Liquid into MOF for Improving the Efficiency of CO2 Chemical Fixation. ACS Appl Mater Interfaces 10(32):27124–27130. https://doi.org/10.1021/acsami.8b08914
Yuan Y, Li J, Sun X, Li G, Liu Y, Verma G, Ma S (2019) Indium-Organic Frameworks Based on Dual Secondary Building Units Featuring Halogen-Decorated Channels for Highly Effective CO2 Fixation. Chem Mater 31(3):1084–1091. https://doi.org/10.1021/acs.chemmater.8b04792
Cao JJ, Shan WJ, Wang Q, Ling XC, Li G, Lyu Y, Zhou Y, Wang J (2019) Ordered Porous Poly(ionic liquid) Crystallines: Spacing Confined Ionic Surface Enhancing Selective CO2 Capture and Fixation. ACS Appl Mater Interfaces 11(6):6031–6041. https://doi.org/10.1021/acsami.8b19420
Zhi Y, Shao P, Feng X, Xia H, Zhang Y, Shi Z, Mu Y, Liu X (2018) Covalent organic frameworks: efficient, metal-free, heterogeneous organocatalysts for chemical fixation of CO2 under mild conditions. J Mater Chem A 6(2):374–382. https://doi.org/10.1039/c7ta08629f
Liu J, Yang G, Liu Y, Zhang D, Hu X, Zhang Z (2020) Efficient conversion of CO2 into cyclic carbonates at room temperature catalyzed by Al-salen and imidazolium hydrogen carbonate ionic liquids. Green Chem 22(14):4509–4515. https://doi.org/10.1039/d0gc00458h
Guo Z, Hu Y, Dong S, Chen L, Ma L, Zhou Y, Wang L, Wang J (2022) “Spring-loaded” mechanism for chemical fixation of carbon dioxide with epoxides. Chem Catal 2(3):519–530. https://doi.org/10.1016/j.checat.2021.12.023
Kong L, Han S, Zhang T, He L, Zhou L (2021) Developing hierarchical porous organic polymers with tunable nitrogen base sites via theoretical calculation-directed monomers selection for efficient capture and catalytic utilization of CO2. Chem Eng J 420:127621. https://doi.org/10.1016/j.cej.2020.127621
Wang X, Dong Q, Xu Z, Wu Y, Gao D, Xu Y, Ye C, Wen Y, Liu A, Long Z, Chen G (2021) Hierarchically nanoporous copolymer with built-in carbene-CO2 adducts as halogen-free heterogeneous organocatalyst towards cycloaddition of carbon dioxide into carbonates. Chem Eng J 403:126460. https://doi.org/10.1016/j.cej.2020.126460
Cui C, Sa R, Hong Z, Zhong H, Wang R (2020) Ionic-Liquid-Modified Click-Based Porous Organic Polymers for Controlling Capture and Catalytic Conversion of CO2. Chemsuschem 13(1):180–187. https://doi.org/10.1002/cssc.201902715
Qu Y, Chen Y, Sun J (2022) Conversion of CO2 with epoxides to cyclic carbonates catalyzed by amino acid ionic liquids at room temperature. J CO2 Util 56:101840. https://doi.org/10.1016/j.jcou.2021.101840
Park S, Morales-Collazo O, Freeman B, Brennecke JF (2022) Ionic Liquid Stabilizes Olefin Facilitated Transport Membranes Against Reduction. Angew Chem Int Ed 61(25):e202202895. https://doi.org/10.1002/anie.202202895
Xin X, Shan H, Tian T, Wang Y, Yuan D, You H, Yao Y (2020) Conversion of CO2 into Cyclic Carbonates under Ambient Conditions Catalyzed by Rare-Earth Metal Complexes Bearing Poly(phenolato) Ligand. ACS Sustainable Chem Eng 8(35):13185–13194. https://doi.org/10.1021/acssuschemeng.0c01736
Bobbink FD, Vasilyev D, Hulla M, Chamam S, Menoud F, Laurenczy G, Katsyuba S, Dyson PJ (2018) Intricacies of Cation-Anion Combinations in Imidazolium Salt-Catalyzed Cycloaddition of CO2 Into Epoxides. ACS Catal 8(3):2589–2594. https://doi.org/10.1021/acscatal.7b04389
Wang X, Zhou Y, Guo Z, Chen G, Li J, Shi Y, Liu Y, Wang J (2015) Heterogeneous conversion of CO2 into cyclic carbonates at ambient pressure catalyzed by ionothermal-derived meso-macroporous hierarchical poly(ionic liquid)s. Chem Sci 6(12):6916–6924. https://doi.org/10.1039/c5sc02050f
Kojčinović A, Likozar B, Grilc M (2022) Heterogeneous catalytic materials for carboxylation reactions with CO2 as reactant. J CO2 Util 66:102250. https://doi.org/10.1016/j.jcou.2022.102250
Song H, Wang Y, Liu Y, Chen L, Feng B, Jin X, Zhou Y, Huang T, Xiao M, Huang F, Gai H (2021) Conferring Poly(ionic liquid)s with High Surface Areas for Enhanced Catalytic Activity. ACS Sustainable Chem Eng 9(5):2115–2128. https://doi.org/10.1021/acssuschemeng.0c07399
Singh SK, Savoy AW (2020) Ionic liquids synthesis and applications: An overview. J Mol Liq 297:112038. https://doi.org/10.1016/j.molliq.2019.112038
Liu M, Wang X, Jiang Y, Sun J, Arai M (2018) Hydrogen bond activation strategy for cyclic carbonates synthesis from epoxides and CO2: current state-of-the art of catalyst development and reaction analysis. Catal Rev 61(2):214–269. https://doi.org/10.1080/01614940.2018.1550243
Ying T, Tan X, Su Q, Cheng W, Dong L, Zhang S (2019) Polymeric ionic liquids tailored by different chain groups for the efficient conversion of CO2 into cyclic carbonates. Green Chem 21(9):2352–2361. https://doi.org/10.1039/c9gc00010k
Wan YL, Zhang ZM, Ding C, Wen LL (2021) Facile construction of bifunctional porous ionic polymers for efficient and metal-free catalytic conversion of CO2 into cyclic carbonates. J CO2 Util 52:101673. https://doi.org/10.1016/j.jcou.2021.101673
Du YR, Xu BH, Xia SP, Ding GR, Zhang SJ (2021) Dehydrative Formation of Isosorbide from Sorbitol over Poly(ionic liquid)-Covalent Organic Framework Hybrids. ACS Appl Mater Interfaces 13(1):552–562. https://doi.org/10.1021/acsami.0c18105
Sun Q, Aguila B, Perman J, Nguyen N, Ma S (2016) Flexibility Matters: Cooperative Active Sites in Covalent Organic Framework and Threaded Ionic Polymer. J Am Chem Soc 138(48):15790–15796. https://doi.org/10.1021/jacs.6b10629
Aguila B, Sun Q, Wang X, O’rourke E, Al-Enizi AM, Nafady A, Ma S (2018) Lower Activation Energy for Catalytic Reactions through Host-Guest Cooperation within Metal-Organic Frameworks. Angew Chem Int Ed 57(32):10107–10111. https://doi.org/10.1002/anie.201803081
Buaki-Sogó M, Vivian A, Bivona LA, García H, Gruttadauria M, Aprile C (2016) Imidazolium functionalized carbon nanotubes for the synthesis of cyclic carbonates: reducing the gap between homogeneous and heterogeneous catalysis. Catal Sci Technol 6(24):8418–8427. https://doi.org/10.1039/c6cy01068g
Polidoro D, Perosa A, Rodríguez-Castellón E, Canton P, Castoldi L, Rodríguez-Padrón D, Selva M (2022) Metal-Free N-Doped Carbons for Solvent-Less CO2 Fixation Reactions: A Shrimp Shell Valorization Opportunity. ACS Sustainable Chem Eng 10(41):13835–13848. https://doi.org/10.1021/acssuschemeng.2c04443
Gbe J-LK, Ravi K, Singh M, Neogi S, Grafouté M, Biradar AV (2022) Hierarchical porous nitrogen-doped carbon supported MgO as an excellent composite for CO2 capture at atmospheric pressure and conversion to value-added products. J CO2 Util 65:102222. https://doi.org/10.1016/j.jcou.2022.102222
Zhong H, Yang C, Fan L, Fu Z, Yang X, Wang X, Wang R (2019) Dyadic promotion of photocatalytic aerobic oxidation via the Mott-Schottky effect enabled by nitrogen-doped carbon from imidazolium-based ionic polymers. Energy Environ Sci 12(1):418–426. https://doi.org/10.1039/c8ee02727g
Zhang S, Zhang H, Cao F, Ma Y, Qu Y (2018) Catalytic Behavior of Graphene Oxides for Converting CO2 into Cyclic Carbonates at One Atmospheric Pressure. ACS Sustainable Chem Eng 6(3):4204–4211. https://doi.org/10.1021/acssuschemeng.7b04600
Guo XX, Zhang FL, Muhammad Y, Hu DL, Cai ZT, Xiao GM (2022) Enhancement in the active site exposure in a porphyrin-based PIL/graphene composite catalyst for the highly efficient conversion of CO2. Dalton Trans 51(8):3331–3340. https://doi.org/10.1039/d1dt04338b
Cui X, Dai X, Surkus A-E, Junge K, Kreyenschulte C, Agostini G, Rockstroh N, Beller M (2019) Zinc single atoms on N-doped carbon: An efficient and stable catalyst for CO2 fixation and conversion. Chin J Catal 40(11):1679–1685. https://doi.org/10.1016/s1872-2067(19)63316-4
Hesse SA, Beaucage PA, Smilgies DM, Wiesner U (2021) Structurally Asymmetric Porous Carbon Materials with Ordered Top Surface Layers from Nonequilibrium Block Copolymer Self-Assembly. Macromolecules 54(6):2979–2991. https://doi.org/10.1021/acs.macromol.0c02720
Jeong U, Kim H, Ramesh S, Dogan NA, Wongwilawan S, Kang S, Park J, Cho ES, Yavuz CT (2021) Rapid Access to Ordered Mesoporous Carbons for Chemical Hydrogen Storage. Angew Chem Int Ed 60(41):22478–22486. https://doi.org/10.1002/anie.202109215
Jeong Y, Cui M, Choi J, Lee Y, Kim J, Son Y, Khim J (2020) Development of modified mesoporous carbon (CMK-3) for improved adsorption of bisphenol-A. Chemosphere 238:124559. https://doi.org/10.1016/j.chemosphere.2019.124559
Meng Y, Gu D, Zhang F, Shi Y, Yang H, Li Z, Yu C, Tu B, Zhao D (2005) Ordered Mesoporous Polymers and Homologous Carbon Frameworks: Amphiphilic Surfactant Templating and Direct Transformation. Angew Chem Int Ed 117(43):7215–7221. https://doi.org/10.1002/ange.200501561
Liu F, Huang K, Wu Q, Dai S (2017) Solvent-Free Self-Assembly to the Synthesis of Nitrogen-Doped Ordered Mesoporous Polymers for Highly Selective Capture and Conversion of CO2. Adv Mater 29(27):2932–2941. https://doi.org/10.1002/adma.201700445
Zhang W, Wang Q, Wu H, Wu P, He M (2014) A highly ordered mesoporous polymer supported imidazolium-based ionic liquid: an efficient catalyst for cycloaddition of CO2 with epoxides to produce cyclic carbonates. Green Chem 16(11):4767–4774. https://doi.org/10.1039/c4gc01245c
Zhang W, Mei Y, Wu P, Wu H-H, He M-Y (2019) Highly tunable periodic imidazole-based mesoporous polymers as cooperative catalysts for efficient carbon dioxide fixation. Catal Sci Technol 9(4):1030–1038. https://doi.org/10.1039/c8cy02595a
Nie D, Liang Y, Zhou T, Sun Q, Shi G, Jin L (2012) FDU-15-Pt composites with different Pt loading and their electrocatalytic reduction to ultratrace nitroaromatic compounds. Int J Environ Anal Chem 92(7):832–843. https://doi.org/10.1080/03067319.2010.520124
Yang C, Chen Y, Wang X, Sun J (2022) Polymeric ionic liquid with carboxyl anchored on mesoporous silica for efficient fixation of carbon dioxide. J Colloid Interface Sci 618:44–55. https://doi.org/10.1016/j.jcis.2022.03.066
Demirci S, Yurddaskal M, Dikici T, Sarioglu C (2018) Fabrication and characterization of novel iodine doped hollow and mesoporous hematite (Fe2O3) particles derived from sol-gel method and their photocatalytic performances. J Hazard Mater 345:27–37. https://doi.org/10.1016/j.jhazmat.2017.11.009
Muniandy L, Adam F, Rahman NRA, Ng E-P (2019) Highly selective synthesis of cyclic carbonates via solvent free cycloaddition of CO2 and epoxides using ionic liquid grafted on rice husk derived MCM-41. Inorg Chem Commun 104:1–7. https://doi.org/10.1016/j.inoche.2019.03.012
Li K, Zhao Y, Zhang P, He C, Deng J, Ding S, Shi W (2016) Combined DFT and XPS investigation of iodine anions adsorption on the sulfur terminated (001) chalcopyrite surface. Appl Surf Sci 390:412–421. https://doi.org/10.1016/j.apsusc.2016.08.095
Shan W, Li S, Cai X, Zhu J, Zhou Y, Wang J (2018) Carbon catalyzed hydroxylation of benzene with dioxygen to phenol over surface carbonyl groups. ChemCatChem 11:1076–1085. https://doi.org/10.1002/cctc.201801668
Lan D-H, Gong Y-X, Tan N-Y, Wu S-S, Shen J, Yao K-C, Yi B, Au C-T, Yin S-F (2018) Multi-functionalization of GO with multi-cationic ILs as high efficient metal-free catalyst for CO2 cycloaddition under mild conditions. Carbon 127:245–254. https://doi.org/10.1016/j.carbon.2017.11.007
Du Y-R, Yang X, Wang Y-F, Guan P-X, Wang R, Xu B-H (2022) Immobilization poly(ionic liquid)s into hierarchical porous covalent organic frameworks as heterogeneous catalyst for cycloaddition of CO2 with epoxides. Mol Catal 520:112164. https://doi.org/10.1016/j.mcat.2022.112164
Lan DH, Fan N, Wang Y, Gao X, Zhang P, Chen L, Au CT, Yin SF (2016) Recent advances in metal-free catalysts for the synthesis of cyclic carbonates from CO2 and epoxides. Chin J Catal 37(6):826–845. https://doi.org/10.1016/S1872-2067(15)61085-3
Lian S, Song C, Liu Q, Duan E, Ren H, Kitamura Y (2021) Recent advances in ionic liquids-based hybrid processes for CO2 capture and utilization. J Environ Sci 99:281–295. https://doi.org/10.1016/j.jes.2020.06.034
Yang CK, Chen YL, Qu Y, Zhang JX, Sun JM (2021) Phase-controllable polymerized ionic liquids for CO2 fixation into cyclic carbonates. Sustain Energ Fuels 5(4):1026–1033. https://doi.org/10.1039/d0se01293a
Du Y-R, Ding G-R, Wang Y-F, Xu B-H, Zhang S-J (2021) Construction of a PPIL@COF core–shell composite with enhanced catalytic activity for CO2 conversion. Green Chem 23(6):2411–2419. https://doi.org/10.1039/d1gc00267h
Ye Y, Chen Y, Huang J, Sun J (2022) In-situ Synthesis of Ionic Liquids on B-doped Mesoporous SiO2 Catalyst for Epoxide-CO2 Cycloaddition. Asian J Org Chem 11(8):e202200234. https://doi.org/10.1002/ajoc.202200234
Jiang B, Liu J, Yang G, Zhang Z (2022) Efficient conversion of CO2 into cyclic carbonates under atmospheric by halogen and metal-free Poly (ionic liquid)s. Chin J Chem Eng. https://doi.org/10.1016/j.cjche.2022.05.018
Červenková Št’astná L, Krupková A, Petrickovic R, Müllerová M, Matoušek J, Koštejn M, Cuřínová P, Jandová V, Šabata S, Strašák T (2020) Multivalent Bifunctional Carbosilane Dendrimer-Supported Ammonium and Phosphonium Organocatalysts for the Coupling of CO2 and Epoxides. ACS Sustainable Chem Eng 8(31):11692–11703. https://doi.org/10.1021/acssuschemeng.0c03367
Dokhaee Z, Ghiaci M, Farrokhpour H, Buntkowsky G, Breitzke H (2020) SBA-15-Supported Imidazolium Ionic Liquid through Different Linkers as a Sustainable Catalyst for the Synthesis of Cyclic Carbonates: A Kinetic Study and Theoretical DFT Calculations. Ind Eng Chem Res 59(28):12632–12644. https://doi.org/10.1021/acs.iecr.0c01050
Shaikh RR, Pornpraprom S, D’elia V (2017) Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions. ACS Catal 8(1):419–450. https://doi.org/10.1021/acscatal.7b03580
Xu B-H, Wang J-Q, Sun J, Huang Y, Zhang J-P, Zhang X-P, Zhang S-J (2015) Fixation of CO2 into cyclic carbonates catalyzed by ionic liquids: a multi-scale approach. Green Chem 17(1):108–122. https://doi.org/10.1039/c4gc01754d
Luo R, Liu X, Chen M, Liu B, Fang Y (2020) Recent Advances on Imidazolium-Functionalized Organic Cationic Polymers for CO2 Adsorption and Simultaneous Conversion into Cyclic Carbonates. Chemsuschem 13:3945–3966. https://doi.org/10.1002/cssc.202001079
Zhou X, Weber J, Yuan J (2019) Poly(ionic liquid)s: Platform for CO2 capture and catalysis. Curr Opin Green Sustainable Chem 16:39–46. https://doi.org/10.1016/j.cogsc.2018.11.014
Wang JQ, Dong K, Cheng WG, Sun J, Zhang SJ (2012) Insights into quaternary ammonium salts-catalyzed fixation carbon dioxide with epoxides. Catal Sci Technol 2(7):1480–1484. https://doi.org/10.1039/c2cy20103h
Toda Y, Komiyama Y, Kikuchi A, Suga H (2016) Tetraarylphosphonium Salt-Catalyzed Carbon Dioxide Fixation at Atmospheric Pressure for the Synthesis of Cyclic Carbonates. ACS Catal 6(10):6906–6910. https://doi.org/10.1021/acscatal.6b02265
Acknowledgements
The computational resources generously provided by the High-Performance Computing Center of Nanjing Tech University are greatly appreciated.
Funding
Open access funding provided by Shanghai Jiao Tong University. This work was supported by the National Natural Science Foundation of China (grants 22072065, 22178162, and 22222806), the Distinguished Youth Foundation of Jiangsu Province (BK20220053), and the Six Talent Peaks Project in Jiangsu Province (grant JNHB-035).
Author information
Authors and Affiliations
Contributions
YW conducted the investigation and wrote the original draft. LM contributed on the experiment design, test and written of original draft. ZS, SD and ZG conducted partial experiments and data analysis. JW and YZ conceived and supervised the project and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Additional file 1: Fig. S1.
1H NMR spectrum of [p-ArOH-IM]. Fig. S2. Optical images of PIL-RT, PIL-55, PIL-90, and PIL-110. Fig. S3. GPC spectra of PIL-RT, PIL-55, PIL-90, and PIL-110. Fig. S4. Small-angle XRD patterns of (a) FDU-15-600 encapsulated PILs and (b) various control samples. Fig. S5. (a) Survey scan and (b) O1s XPS spectra.Fig. S6. TG curve of [p-ArOH-IM]I under N2 atmosphere. Fig. S7.CO2 adsorption isotherms of 15%IL@FDU-15-600 at 298 K. Fig. S8. CO2 adsorption isotherms at 298 K of (a) FDU-15-600, (b) 15%IL@FDU-15-600, (c) 15%PIL-RT@FDU-15-600, (d)15%PIL-55@ FDU-15-600, (e) 15%PIL-90@FDU-15-600, and (f) 15%PIL-110@ FDU-15-600. Fig. S9. Yield as a function of (a) reaction temperature and (b) reaction time in 15%PIL-90@FDU-15-600 catalyzed CO2 cycloaddition with styrene oxide. Reaction conditions: styrene oxide (5 mmol), 15%PIL-90@FDU-15-600 (0.4 mol %), 1 bar CO2 (balloon). Fig.S10.(a) N2 sorption isotherms and (b) pore size distribution curves of 10%PIL-90@FDU-15-600 and 20%PIL-90@FDU-15-600.Fig. S11.(a) Nitrogen sorption isotherms and (b) pore size distribution curves. The sorption isotherms for samples AC, 15%PIL-90@AC, LOMC, and 15%PIL-90@LOMC are shifted by 220, 230, 200, and 210 cm3g-1. The pore size distribution curves for samples AC, 15%PIL-90@AC, LOMC, and 15%PIL-90@LOMC are shifted by 1.2, 0.9, 0.5, and 0.3 cm3 g-1, respectively. Fig. S12. Small-angle XRD pattern of 15%IL@FDU-15-600. Fig. S13. (a) N2 sorption isotherm and (b) pore size distribution curve of 15%IL@FDU-15-600. Table S1. Elemental analysisa. Table S2. Types and content of N in the samples. Table S3. Types and content of I- in the samples. Table S4. Fitting parameters for the CO2sorption isothermsa.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Ma, L., Song, Z. et al. Ordered mesoporous carbon encapsulated linear poly(ionic liquid)s enabling synergy effect of surface groups and ionic moieties for CO2 fixation under mild conditions. Carb Neutrality 2, 1 (2023). https://doi.org/10.1007/s43979-022-00041-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43979-022-00041-5