
Abstract
This paper reports the pressureless sintering behavior and the activation energy of the powdered MAX phases Ti3SiC2 and Ti2AlC. A non-isothermal technique was used to determine the sintering kinetic parameter. The Ti3SiC2 and Ti2AlC MAX phases showed the maximum sintering rate at 1723 K, 0.14 and 0.10 µm/s, respectively. The sintering rate of the sample at different temperatures followed a cubic equation which was determined. The sintering activation energy (Ea) for the Ti3SiC2 and Ti2AlC samples was 362.1 kJ/mol and 640.3 kJ/mol, respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Mn+1AXn phases also known as MAX phases, where n ≥ 1, M is an early transition metal, A is a group A element (mainly IIIA, IVA) and X is a carbon or nitrogen combination of properties. Characteristic properties of ceramics, such as high melting point, high temperature oxidation resistance and resistance to harsh environments, with resin-like behavior quite unusual for non-metallic materials grade [1, 2]. The MAX phase is a great material for use in a wide range of disciplines (structural and high temperature applications, protection against wear and corrosion, etc.) [3, 4] because of its favorable features as ceramic composites or monoliths [5, 6].
Ti3SiC2 and Ti2AlC, two of the most studied compounds of the MAX phase family, are of particular interest for this work. Ti2AlC has been reported to have excellent machinability at room temperature and metallics properties (i.e.: high electrical conductivity) and remarkable high temperature ductility [7, 8] and Ti3SiC2 exhibits many outstanding ceramic properties, such as elastic hardness, low density, strong chemical corrosion resistance, high melting point and at the same time have metallic properties such as resistance to thermal shock, relatively high electrical and thermal conductivity and good damage tolerance [1, 8].
Ti3SiC2, pure Ti2AlC, and composites based on these compounds have been synthesized utilizing various starting materials and procedures. Powder synthesis techniques include self-propagating high-temperature synthesis (SHS), mechanically activated annealing, conventional heat treatment of unpacked powder mixtures, and self-propagating temperature sintering laser-induced transmission. Additionally, a great deal of study has concentrated on producing bulk samples through sintering utilizing various treatment techniques, such as pulse discharge sintering, spark plasma sintering, isostatic hot pressing, or alternative non-pressure methods [9,10,11,12,13,14,15,16,17,18,19,20,21,22]. The thickening tendency of powders has, however, received little attention in research to date.
Sintering is a thermal energy-based manufacturing technique used to create materials and parts with controlled density from metal, ceramic, or composite powders. Sintering is becoming more essential as a material handling technology since material synthesis and processing have become more important in recent years for materials development [23,24,25,26,27,28,29,30].
A fundamental and complete understanding of the fundamentals of materials science is required for understanding the sintering process and tackle the challenges involved. As a result, we present the non-pressure sintering behavior of cold-fused Ti3SiC2 and Ti2AlC powders in Ar in this study. A non-isothermal kinetic approach was used to calculate the sintering parameters.
2 Experimental
The Ti3SiC2 and Ti2AlC MAX phases powders were previously synthesized following the procedure described elsewhere [22, 31]. To yield compacted MAX phase cylinders (diameter x height, 3 × 15 mm) a two steps procedure was used. At first a uniaxial pressing at 2 ton for 5 min (Evacuable KBr Die, Perkin Elmer) was used to shape the cylinder morphology of the powdered phases Ti3SiC2 and Ti2AlC, and subsequently a cold isostatic pressing second step at 200 MPa for 10 min to compact the mentioned cylinders.
The green bodies shrinkage behavior was measured by a horizontal dilatometer (DIL 402 Expedis Classic, Netzsch). With a linear variable differential transducer (sensitivity: 2 nm; force resolution: 0.001 mN, sample pressure applied 0.2 N), the shrinkage was axially measured.
To gauge the temperature, a calibrated thermocouple was placed directly above the sample. Using an established program, a stepwise isothermal dilatometry measurement was taken. Automatically recorded information included time (s), temperature (K), and length shrinkage (μm). The temperature holding steps for the stepwise isothermal dilatometry program were 1373, 1473, 1523, 1573, 1623, 1673, and 1723 K for 60 min each, with a 10 K/min ramping rate between the isothermal holdings. Each experiment was performed twice. Green bodies were heated up to 1773 K at various constant heating rates (2.5, 5, and 10 K/min) for the kinetic field analysis. All of the runs were carried out using an Argon gas flow of 100 ml/min (Air Liquide España S. A., Madrid, Spain; H2O 3 ppm, O2 2 ppm, and CnHm 0.5 ppm). Through a heated transfer capillary, in situ, gas measurement was carried out using a mass spectrometer (Netzsch: QMS 403 C Aëolos). The shrinkage rate as a function of temperature fitting was performed working with the OriginPro 8.5 software (OriginLab Corporation, 167 Northampton, MA, USA).
The kinetic parameters were derived from the product evolution profile using a non-isothermal approach (at a constant heating rate) [32,33,34,35,36]. The reaction is assumed to follow an Arrhenius expression:
where, Tm (K) is the temperature for the onset, m is the heating rate (K/min), Ea is the activation energy (J/mol), k is the Boltzmann constant (1.380(6)⋅10−23 JK−1), NA is the Avogadro constant (6.022(1)⋅1023 mol−1), and A0 is the frequency factor.
The water immersion technique was used to measure the bulk densities of the bodies (Archimedes method (ISO-3369)). The rule of the mixture was used to determine the theoretical density of the compositions studied (theoretical density [1] of Ti3SiC2 = 4.52 g/cm3; Ti2AlC = 4.11 g/cm3; Ti3AlC2 = 4.24 g/cm3; TiC = 4.90 g/cm3).
The thermogravimetric (TG) and differential scanning calorimetric (DSC) methods were employed to assess the thermal stability of the powdered raw sample mixtures. They were carried out on a Netzsch Jupiter STA 449C. In sintered Al2O3 crucibles, 20 mg of the substance was retained, and the temperature was raised from ambient temperature to 1773 K at a rate of 10 K/min. These tests were carried out with an argon atmosphere flow of 100 cc/min.
The MAX phase Ti3SiC2 and Ti2AlC raw powders were analyzed by Rietveld analysis of the high-resolution XRD and a description is showed below in the results and discussion.
Due to the layered structure of Ti3SiC2 and Ti2AlC, additional care was taken during the assembly of diffractometric specimens. It was found that slurry mounting in acetone was useful in reducing the adverse effects of preferred orientations. A Panalytical X'Pert Pro instrument outfitted with a Θ/Θ goniometer, a Cu Kα radiation source (40 kV, 40 mA), a secondary Kβ filter, and an X'Celerator detector was used to obtain X-ray diffraction patterns. Step-scan mode was used to scan the diffraction patterns from 20° to 80° (2Θ) with 0.05° steps and a counting time of 600 s/step.
According to the Scherrer equation, the full-width at half-maximum (FWHM) of the Ti3SiC2 (104) and Ti2AlC (103) diffraction peaks were used to determine the average length of coherently diffracting domains [37, 38]. The instrumental broadening was corrected using LaB6 standard from NIST (National Institute of Standards and Technology, Gaithersburg, Maryland, EE.UU.).
3 Results and discussion
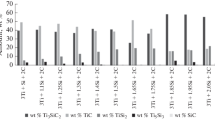
XRD patterns of the powdered MAX phases are shown in Fig. 1a and b, which are the profiles for samples 1 (Ti3SiC2) and 2 (Ti2AlC), respectively. The relative intensities of Ti3SiC2 and Ti2AlC XRD peaks supported a random orientation (Ti3SiC2, JCPDS file 74-0310 and Ti2AlC, JCPDS file 29-0095). The phase quantification, the initial density of the green body and the size of the coherently diffracting domain of the samples studied are summarized in Table 1. Sample 1 is constituted by 90% of Ti3SiC2 and a 10% of TiC and Ti3SiC2 phase crystallite size was 48 nm. The sample 2 was constituted by 89% of Ti2AlC, 3% of TiC and 8% of Ti3AlC2 and the crystallite size of the Ti2AlC phase was 20 nm.

X-ray diffractograms of a sample 1 (Ti3SiC2) and, b sample 2 (Ti2AlC)
The shrinkage evolution for the samples was determined by obtained shrinkage data from each sample at various hold temperatures during the sintering process, as shown in Fig. 2a and b. Every studied temperature was held during 60 min and each experiment was carried out two times. Any contraction was observed below 1473 K. An increment of the total shrinkage registered at every temperature studied was observed up to 1673 K, after that, at 1723 K, the shrinkage was lower compared with the previous temperature studied. Sample 1 (Ti3SiC2) and sample 2 (Ti2AlC) showed a total shrinkage of ~ 1.93 ± 0.12 mm (11.98%) and ~ 1.41 ± 0.09 mm (8.40%), respectively.

Characteristic stepwise temperature variation for shrinkage evolution of: a sample 1 (Ti3SiC2) and, b sample 2 (Ti2AlC). Every step was hold 60 min
The shrinkage rate is considered to gain a better understanding of the changes in densification during thermal treatment. The relative linear shrinkage rate, as shown in Fig. 3, is a measure of the densification rate. Only one maximum in densification rate was noticed in Ti3SiC2 and Ti2AlC, showing normal densification behavior throughout the sintering process.

Shrinkage rate as a function of temperature [K] of: a sample 1 (Ti3SiC2) and, b sample 2 (Ti2AlC)
In Fig. 3a and b, an analysis of the shrinkage rate at the different temperatures studied for samples 1 (Ti3SiC2) (Fig. 3a) and 2 (Ti2AlC) (Fig. 3b) is presented. In both samples there was an shrinkage rate increment with the temperature. The maximum shrinkage rate was reached at 1673 K (1400 °C). The values observed were 0.14 and 0.10 µm/s for samples 1 and 2, respectively. The shrinkage rate at each temperature defined a profile with the following cubic equation (goodness of fitness (χ2)):
where y and x are the shrinkage rate and the temperature [K], respectively.
The maximum of this curve represents the temperature at which the shrinkage rate is the largest and, again, the temperature at which grain growth starts. This method makes it simple to set up the sintering conditions needed to create materials with fine grains. Given that the properties of sintered compacts are significantly influenced by the grain size, this is particularly advantageous in the production of ceramics for various applications.
Samples 1 and 2 were heated up to 1773 K (1500 °C) at different heating rates and the profile dL/Lo versus temperature are shown in Figs. 4 and 5, respectively. At 2.5 K/min sample 1 (Ti3SiC2, (Fig. 4) showed a slight expansion from room temperature till 1406 K. The value of the rate of thermal expansion (RTE: it represents the velocity, in terms of temperature (K) of the material expansion (first derivative, the slope of the tangent line to the function at the point x) was 1.8 ⋅ 10–6 K−1. From this temperature to the sintering temperature (1570 K) a little contraction was observed. When the heating rate was increased to 5 K/min and 10 K/min, the samples studied showed four different states before sintering temperature. For the 5 K/min run, a first slight expansion from room temperature to 730 K with a RTE of 1.09 ⋅ 10–5 K−1 was observed, an increment in the expansion rate from 730 to 1145 K (RTE = 6.5 ⋅ 10–5 K−1), a plateau from 1145 to 1431 K and a slight contraction from 1431 K to the sintering temperature (1613 K). The sample heated at 10 K/min showed similar behavior, but the expansion was from room temperature to 765 K (RTE = 1.1 ⋅ 10–5 K−1), a higher expansion rate from 765 to 1188 K (RTE = 1.2 ⋅ 10–4 K−1), the plateau from 1188 to 1457 K and a slight shrinkage (but higher than 5 K/min run) from this temperature to 1646 K (sintering temperature).

Expansion–shrinkage evolution of sample 1 (Ti3SiC2) at different constant heating rates

Expansion–shrinkage evolution of sample 2 (Ti2AlC) at different constant heating rates
In Fig. 5 the expansion–shrinkage evolution as the function of the temperature at the different heating rates is shown in the sample 2 (Ti2AlC). Independently of the heating rate Ti2AlC exhibited similar trends, an expansion from room temperature to the sintering transition state. When the sample was heated at 2.5 K/min showed a RTE of 1.0 ⋅ 10–5 K−1, and an onset sintering temperature of 1612 K. At 5 K/min and 10 K/min, the RTE and onset sintering temperature were 9.5 ⋅ 10–6 K−1 and 8.8 ⋅ 10–6 K−1 and 1637 K and 1658 K, respectively.
The relative density of the sintered specimen (ρs) was estimated using the equation given by Ran et al. [39], assuming isotropy in the densification of the specimen. Based on a theoretical density of 4.56 g/cm3 for sample 1, calculated by the rule of mixtures, the green density of the powder compact after cold pressing was determined to be 62.1% of the theoretical. To correct the thermal expansion, the thermal expansion coefficient determined Manoun B. et al. (Ti3SiC2 CTE, 2.095 × 10–5 + 7.7 × 10−9 T; temperature is in degrees Celsius) [40] was used. The final maximum density obtained by this equation was, 73.6%, 71.4% and 69.0%, for 2.5 K/min, 5 K/min, and 10 K/min, respectively.
A similar procedure was done for the Ti2AlC [41, 42] with similar results. The theoretical density for sample 2, calculated by the rule of mixtures, was 4.14 g/cm3, and for the compact body after cold pressing was determined to be 54.8% of the theoretical. The final maximum density obtained by this equation was, 65.2%, 63.4% and 60.5%, for 2.5 K/min, 5 K/min and 10 K/min, respectively.
The computed final densities following the dilatometer experiments were in acceptable agreement with those obtained by the Archimedes method (Sample 1, 74.0%, 71.1%, and 69.4%, and Sample 2, 65.0%, 63.2%, and 60.9% of the theoretical, for 2.5 K/min, 5 K/min and 10 K/min, respectively). The calculated and determined by Archimedes method density of Ti3SiC2 and Ti2AlC samples are shown in Fig. 6 for the three different heating rates of 2.5, 5 and 10 K/min. It was discovered that the maximum density was dependent on the heating rate, with higher heating rates resulting in lower sintered densities. It is widely known that when the heating rate is lower, the compact is exposed to sintering for a longer period of time; as a result, the relative density is higher.

Calculated and Archimedes method relative density (% of the theoretical) after sintering as a function of the heating rate of sample 1 (Ti3SiC2) and sample 2 (Ti2AlC)
In Fig. 7a and b ln(Tm2/m) as function of 1/RTm based on Eq. (1) for samples 1 (Ti3SiC2) and 2 (Ti2AlC) is plotted, respectively. The activation energy (Ea) and frequency factor (A0) were calculated using linear regression. The rate of reaction was determined by these two parameters. The extracted data was listed in Table 2. The activation energy of the Ti3SiC2 (sample 1) and Ti2AlC (sample 2) were 362.1 and 640.3 kJ/mol, respectively. The consistent slope at each density indicated that the activation energy remained constant throughout the sintering process. The activation energy is conformed to the Arrhenius curve, indicating that the diffusion mechanism is the primary material migration mode.

ln(Tm2/m) as a function of 1/RTm of: a sample 1 (Ti3SiC2) and b sample 2 (Ti2AlC)
However, due to the diversity of the systems, the activation energy levels vary from one system to another, precisely like the data in Table 2. The Ti–Al bond's cleavage energy is lower (1.23 eV) [43] than the Ti–Si bond's (2.88 eV) [44] when comparing the Ti3SiC2 and Ti2AlC systems. The variances in their electronegativity make this clear. Ti, Al, and Si atoms have electronegativities of 1.54, 1.61, and 1.90, respectively. Ti–Si (0.36) has a significantly higher electronegativity difference than Ti–Al (0.07). In this way, the Ti-Si link creates an ionic bond, making it stronger than the Ti–Al bond. If both samples are compared, from a point of view of their components bond energy cleavage and electronegativity, we are coming into a contradiction with their sintering activation energies showed in Table 2. However, according to the sintering theory, the activation energy of sintering should be consistent with that of the apparent diffusion coefficient of the rate-limiting species of sintering, so taking into account the structure and characteristics of Mn+1AXn phases [45,46,47], (i) the M–A bonds are relatively weaker than the M–X bonds, and, (ii) and when n = 1, the A-layers are separated by two M-layers (M2AX, Ti2AlC), and when n = 2, they are separated by three layers (M3AX2, Ti3SiC2), is possible to understand these activation energy values.
Since the data of Si or Al diffusion on MAX phase is not available, diffusion data in other materials available in the literature were analyzed. It was found that Si activation energy is in the range of 400 kJ/mol [48], and for Al is in the range of 690 kJ/mol [49]. Both values match closely with the Ea determined for their respective studied MAX phases (362.1 and 640.3 kJ/mol, for Ti3SiC2 and Ti2AlC, respectively). These results suggest the possibility that the A element diffusion is playing a key role during sintering of MAX phase powder through the diffusion mechanism.
In his study of the crystal-chemical connections in the sintering of various materials, Bron et al. [50] discovered that sintering was facilitated by excess disordering, which was caused by lattice elongation and bonding loosing. Because of its greater lattice size, this fact favors a lower Ea for M3AX2 phases (Samples 1, Ti3SiC2) over M2AX (Sample 2, Ti2AlC).
And finally, another sample characteristic to be analyzed during sintering is the crystallite size (determined by XRD methods) [51,52,53,54]. The efficiency of grain boundaries as vacancy sinks is significantly influenced by the size of the crystallites. It has been proven [55] that powder derived from single-crystal particles has slight grain growth at high sintering temperatures, while polycrystalline particles show strong grain growth, which is consistent with their increased activity. In our study, a larger crystallite size (Table 1) is showed by sample 1 (Ti3SiC2), which is in contradiction with the expected Ea trend. This fact shows us that the effect of the crystallite size on the sintering Ea is negligible compared to the chemical and structural characteristics of the two studied samples.
Few reports can be found about MAX phase sintering kinetic analysis. In this way, the work carried out by Rajkumar et al. [56] on Cr2AlC phase (a kind of M2AX, similar to Ti2AlC), showed an activation energy of about 490 kJ/mol, and the research carried out by Perevislov et al. [57] on Zr3AlC2 (a kind of M3AX2, similar to Ti3SiC2) showed an activation energy of 392 kJ/mol, showing a similar trend as that presented in this research, a greater activation energy value for Ti2AlC phase (M2AX type) than for Ti3SiC2 phase (M3AX2).
Finally, concerning other kinds of materials (alloys, ceramics, high entropy ceramics, composites, etc.), or ways to sintering (pressureless, Hot Isostatic Pressing (HIP), Spark Plasma Sintering (SPS), etc.), the activation energy (Ea) obtained in this work is in the range of those described in the literature. For example, the pressureless sintering Ea for the Fe(25%)-Co(15%)-Cu(60%) alloy powder was 453.11 kJ/mol [58], for the (TiZrHfVNbTa)C high entropy ceramics was 839 kJ/mol [59], and for the Yttrium Aluminium Garnet (YAG) was 522 kJ/mol [60], and the Ea for the sintering by Hot Isostatic Press of the MoSi2-WSi2- (1%)Si3N4 composite was in the order of 396 kJ/mol [61].
For the initial green bodies, concurrent TG/DSC and gas analyses were performed to determine the stability during thermal processing. The weight evolution and heat flow (DSC) as a function of temperature under an argon environment at a fixed heating rate of 10 K/min are depicted in Fig. 8a and b. At around 600 K, sample 1 (Ti3SiC2, Fig. 8a) experienced a weight loss of − 1.4%. Gas analysis at the same temperature reveals the presence of CO and H2O as weight loss by products (Fig. 8a inset). After that, a slight weight increment was observed (+ 0.6%), probably due to oxidation. Any characteristic reaction was observed in the DSC profile. The onset temperature for the weight increment was fixed at ~ 1350 K. This temperature matched with the decomposition model described by Emmerlich et al. [62], in which one, the presence of residual oxygen in the argon atmosphere used, helped by the void formation (stage III, 1375 K) and an increment of the pore size (stage IV, 1575 K), described this slight weight increment.

TG/DSC curves of: a sample 1 (Ti3SiC2) (gas analysis is in the inset) and, b sample 2 (Ti2AlC) measured in argon
Sample 2 (Ti2AlC, Fig. 8b) showed an increment on the weight of + 2.5% from ~ 750 K up to 1773 K. This almost lineal increment was associated an oxidation due to the presence of the residual oxygen in argon atmosphere and the non-formation of the protective α-Al2O3 external layer which would protect against further oxidation of the underlying Ti2AlC [63]. In contrast to oxidation at high temperature, oxidation at lower temperature (750 K) typically produces porous phase-mixed oxide coatings that lack protective properties [64].
4 Conclusions
A phenomenological equation was defined and was successfully applied to an analysis of the sintering behavior and kinetics of Ti3SiC2 and Ti2AlC MAX phases. The sintering activation energy (Ea) for the Ti3SiC2 and Ti2AlC samples was 362.1 kJ/mol and 640.3 kJ/mol, respectively.
The maximum shrinkage rate was reached at 1673 K for both phases. The values observed were 0.14 and 0.10 µm/s for the Ti3SiC2 and Ti2AlC phases, respectively. The shrinkage rate at each temperature was defined by cubic equation.
It was discovered that the maximum density was dependent on the heating rate, with higher heating rates resulting in lower sintered densities. When a slower heating rate is utilized to achieve the same temperature, a lengthy time scale for mass transport and diffusion has been to be claimed for this.
Data availability
No/Not applicable (this manuscript does not report data generation or analysis).
References
Barsoum MW. The MN+1AXN phases: a new class of solids. Progr Solid State Chem. 2000;28:201–81.
Eklund P, Beckers M, Jansson U, Hogberg H, Hultman L. The Mn + 1AXn phases: materials science and thin-film processing. Thin Solid Films. 2010;518:1851–78.
Cover MF, Warschkow O, Bilek MMM, McKenize DR. A comprehensive survey of M2AX phase elastic properties. J Phys Condensed Matter. 2009;21: 305403.
Wang XH, Zhou YC. Layered machinable and electrically conductive Ti2AlC and Ti3AlC2 ceramics: a review. J Mater Sci Techn. 2010;26:385–416.
Amini S, Cordoba JM, Daemen L, McGhie AR, Ni CY, Hultman L, Oden M, Barsoum MW. On the stability of mg nanograins to coarsening after repeated melting. Nano Lett. 2009;9:3082–6.
Tzenov NV, Barsoum MW. Synthesis and characterization of Ti3AlC2. J Am Ceram Soc. 2000;83:825–32.
Barsoum MW, Ali M, El-Raghy T. Processing and characterization of Ti2AlC, Ti2AlN, and Ti2AlC0.5N0.5. Metall Mater Trans A. 2000;31:1857–65.
Barsoum MW, El-Raghy T. A progress report on Ti3SiC2, Ti3GeC2, and the H-phase, M2BX. J Mater Synth Process. 1997;5(3):197–216.
Liang BY, Wang MZ, Li XP, Sun SB, Zou Q, Mu YC, Li X. Synthesis of Ti2AlC by laser-induced self-propagating high-temperature sintering. J Alloys Compd. 2010;501:L1–3.
He XD, Bai YL, Li YB, Zhu CC, Kong XH. In situ synthesis and mechanical properties of bulk Ti3SiC2/TiC composites by SHS/PHIP. Mater Sci Eng A. 2010;527:4554–9.
Kim CS, Hwang SI, Ha JS, Kang SM, Cheong DS. Factors affecting surface roughness of Al2O3 films deposited on Cu substrates by an aerosol deposition method. J Ceram Process Res. 2010;11:82–5.
Shi L, Zhang JF, Wang LJ, Jiang W, Fabrication SPS. Microstructure and electric properties of TiC/Ti2AlC/TixAly in-situ composites. J Inorg Mater. 2009;24:1168–72.
Chen YL, Yan M, Sun YM, Mei BC, Zhu JQ. The phase transformation and microstructure of TiAl/Ti2AlC composites caused by hot pressing. Ceram Int. 2009;35:1807–12.
Zhou WB, Mei BC, Zhua JQ. Rapid reactive synthesis of Ti2AlC-TiB2 composites by spark plasma sintering. J Ceram Process Res. 2009;10:102–4.
Gauthier-Brunet V, Cabioc’h I, Chartier P, Jaouen M, Dubois S. Reaction synthesis of layered ternary Ti2AlC ceramic. J Eur Ceram Soc. 2009;29:187–94.
Zhu JF, Gao JQ, Yang JF, Wang F, Niihara K. Synthesis and microstructure of layered-ternary Ti2AlC ceramic by high energy milling and hot pressing. Mater Sci Eng A. 2008;490:62–5.
Rosen J, Ryves L, Persson POA, Bilek MMM. Deposition of epitaxial Ti2AlC thin films by pulsed cathodic arc. J App Phys. 2007;10: 056101.
Riley DP, Kisi EH, Wu E, McCallum A. Self-propagating high-temperature synthesis of Ti3SiC2 from 3Ti + SiC + C reactants. J Mater Sci Letters. 2003;22:1101–4.
Orthner HR, Tomasi R, Botta WJ. Reaction sintering of titanium carbide and titanium silicide prepared by high-energy milling. Mater Sci Eng A. 2002;336:202–8.
El-Raghy T, Barsoum MW. Processing and mechanical properties of Ti3SiC2: I, reaction path and microstructure evolution. J Am Ceram Soc. 1999;82:2849–54.
Arunajatesan S, Carim AH. Synthesis of titanium silicon carbide. J Am Ceram Soc. 1995;78:667–72.
Cordoba JM, Sayagues MJ, Alcala MD, Gotor FJ. Monophasic nanostructured powders of niobium, tantalum, and hafnium carbonitrides synthesized by a mechanically induced self-propagating reaction. J Am Ceram Soc. 2007;90:825–30.
Oghbaei M, Mirzaee O. Microwave versus conventional sintering: a review of fundamentals, advantages and applications. J Alloys Compd. 2010;494:175–89.
Ragulya AV. Consolidation of ceramic nanopowders. Adv Appl Ceram. 2008;107:118–34.
Olevsky EA. Theory of sintering: from discrete to continuum. Mater Sci Eng R Rep. 1998;23:41–100.
Kuang X, Carotenuto G, Nicolais L. A review of ceramic sintering and suggestions on reducing sintering temperatures. Adv Perform Mater. 1997;4:257–74.
Hlavacek V, Puszynski JA. Chemical engineering aspects of advanced ceramic materials. Industrial Eng Chem Res. 1996;35:349–77.
Suttor D, Fischman GS. Densification and sintering kinetics in sintered silicon nitride. J Am Ceram Soc. 1992;75:1063–7.
Cordoba JM, Alcala MD, Aviles MA, Sayagues MJ, Gotor FJ. New production of TiCxN1−x-based cermets by one step mechanically induced self-sustaining reaction: powder synthesis and pressureless sintering. J Eur Ceram Soc. 2008;28:2085–98.
Cordoba JM, Sanchez-Lopez JC, Aviles MA, Alcala MD, Gotor FJ. Properties of Ti(C, N) cermets synthesized by mechanically induced self-sustaining reaction. J Eur Ceram Soc. 2009;29:1173–82.
Vershinnikova VI, Kovaleva DYu. Synthesis of the Ti 2 AlC MAX phase with a reduction step via combustion of a TiO 2+ Mg+ Al+ C mixture. Inorg Mater. 2018;54:949–52.
Dunkerton LV, et al. Migration of sulphur from organic to the inorganic phase under hydrodesulphurization conditions. Fuel. 1988;67:967–72.
Juntgen H, Vanheek KH. An update of german non-isothermal coal pyrolysis work. Fuel Process Technol. 1979;2:261–93.
Koch E. Non-isothermal reaction analysis. New York: Academic Press; 1977.
Brukh R, Mitra S. Kinetics of carbon nanotubeoxidation. J Mater Chem. 2007;17:619–23.
Cordoba JM, et al. Morphology influence of the oxidation kinetics of carbon nanofibers. Corros Sci. 2009;51:926–30.
Patterson AL. The Scherrer formula for X-Ray particle size determination. Phys Rev. 1939;56:978–82.
Scherrer P. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen. Mathematisch-Physikalische Klasse. 1918;2:394.
Ran S, Winnubst L, Wiratha W, Blank DHA. Sintering behavior of 0.8 mol%-CuO-doped 3Y-TZP ceramics. J Am Ceram Soc. 2006;89(1):151–5.
Manoun B, Saxena SK, Liermann H-P. Thermal expansion of polycrystalline Ti3SiC2 in the 25–1400 ºC temperature range. J Am Ceram Soc. 2005;88(12):3489–91.
Wang J, Wang J, Li A, Li J, Zhou Y. Theoretical study on the mechanism of anisotropic thermal properties of Ti2AlC and Cr2AlC. J Am Ceram Soc. 2014;97(4):1202–8.
Barsoum MW, Ali M, El-Raghy T. Processing and characterization of Ti2AlC, Ti2AlN, and Ti2AlC0.5N0.5. Metal Mater Trans A. 2000;31:1857–65.
Wang J, Wang J, Zhou Y. Stable M2AlC (0001) surfaces (M= Ti, V and Cr) by first-principles investigation. J Phys Condens Matter. 2008;20: 225006.
Zhang HZ, Wang SQ. First-principles study of Ti3AC2 (A = Si, Al) (0 0 1) surfaces. Acta Mater. 2007;55:4645–55.
Hug G, Fries E. Full-potential electronic structure of Ti2AlC and Ti2AlN. Phys Rev B. 2002;65: 113104.
Hug G, Jaouen M, Barsoum MW. XAS, EELS, and full-potential augmented plane wave study of the electronic structures of Ti2AlC, Ti2AlN, Nb2AlC, and (Ti 0.5Nb0.5)2AlC. Phys Rev B. 2005;71:24105.
Sun ZM, Zhou YC. Ab initio calculation of Ti3SiC2. Phys Rev B. 1999;60:1441.
Jana DC, et al. Effective activation energy for the solid-state sintering of silicon carbide ceramics. Metall Mater Trans A. 2018;49A:5599–606.
Bae SI, Baik S. Sintering and grain growth of ultrapure alumina. J Mater Sci. 1993;28:4197–204.
Thürnrrüer F, Thornrna W. The sintering process. Metall Rev. 1967;115:69–108.
Oel HJ. Agglomeration. New York and London: Interscience Publishers; 1962. p. 271.
Modern developments in powder metallurgy. Vol. I: Fundamentals and methods. New York: H. H. Hausner; 1966
Oel HJ. Berichte über die II Internat. Pulvermet. Tagung, 1961, p. 85. 1962: Berlin, ed. by Akademie-Verlag.
Moorthy VK, Rao SVK. Plansee Proc., 1962, 181.
Yamaguchi T. Effect of powder parameters on grain growth in manganese-zinc ferrite. J Amer Ceram Soc. 1964;47:131.
Rajkumar Y, Panigrahi BB. Sintering mechanisms of ultrafine Cr2AlC MAX phase powder. Mater Today Comm. 2016;8:46–52.
Perevislov SN, Arlashkin IE, Stolyarova VL. Synthesis and sintering of MAX phases in the Zr–Al–C system. J Am Ceram Soc. 2024;107:488–500.
Xie D, et al. Pressureless sintering curve and sintering activation energy of Fe-Co-Cu pre-alloyed powders. Mater Design. 2015;87:482–7.
Zhang W, et al. Grain growth kinetics and densification mechanism of (TiZrHfVNbTa)C high-entropy ceramic under pressureless sintering. J Mater Sci Technol. 2022;110:57–64.
Huang Y. Sintering kinetics of YAG ceramics. J Rare Earths. 2014;32(5):416–21.
Titov DD et al., IOP Conf. Ser.: Mater. Sci. Eng. 2018;347: 1–7
Emmerlich J, et al. Thermal stability of Ti3SiC2 thin films. Acta Mater. 2007;55:1479.
Byeon J, et al. Microstructure and residual stress of alumina scale formed on Ti2AlC at high temperature in air. Oxid Met. 2007;68:97–111.
Wang XH, Zhou YC. Intermediate-temperature oxidation behaviour of Ti2AlC in air. J Mater Res. 2002;17:2974–81.
Acknowledgements
I would like to thank María Córdoba Catalán for her unwavering strength and support, for always putting a smile on my face even in the most difficult and hazardous situations.
Author information
Authors and Affiliations
Contributions
José Manuel Córdoba Gallego developed the research from the beginning till the end.
Corresponding author
Ethics declarations
Competing interests
No, I declare that the authors have no competing interests as defined by Springer, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Córdoba Gallego, J.M. Pressureless sintering kinetics analysis of Ti3SiC2 and Ti2AlC powdered MAX phases. Discov Mater 4, 26 (2024). https://doi.org/10.1007/s43939-024-00098-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43939-024-00098-2