
Abstract
Purpose of Review
In this brief review, we will highlight important observational and experimental data in the literature that address the origin of scar-forming cells in lung fibrosis.
Recent Findings
Several cellular sources of activated scar-forming cells (myofibroblasts) have been postulated including alveolar epithelial cells; circulating fibrocytes; and lung stromal cell subpopulations including resident fibroblasts, pericytes, and resident mesenchymal stem cells. Recent advances in lineage-tracing models, however, fail to provide experimental evidence for epithelial and fibrocyte origins of lung myofibroblasts. Resident mesenchymal cells of the lung, which include various cell types including resident fibroblasts, pericytes, and resident mesenchymal stem cells, appear to be important sources of myofibroblasts in murine models of lung injury and fibrosis.
Summary
Lung myofibroblasts likely originate from multiple sources of lung-resident mesenchymal cells. Their relative contributions may vary depending on the type of injury. Although lineage-tracing experiments have failed to show significant contribution from epithelial cells or fibrocytes, they may play important functional roles in myofibroblast activation through paracrine signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myofibroblasts are activated cells responsible for depositing extracellular matrix (ECM) proteins in scar tissue during fibrogenesis. They exhibit enhanced contractility capable of closing open wounds and tissue repair. Myofibroblasts play an integral role in normal wound healing through (ECM) remodeling, hemostasis, and restoration of tissue barrier integrity. Yet, myofibroblasts are also responsible for pathologies encountered in fibrosing diseases where dysregulated activation of myofibroblasts leads to over-exuberant secretion of ECM proteins and replacement of normal organ architecture, ultimately leading to organ failure. Our limited understanding of where these myofibroblasts originate in disease states and the mechanisms by which myofibroblasts become dysregulated has been an important barrier to finding effective therapies. This is especially true in the lung, where fibrosing lung diseases such as idiopathic pulmonary fibrosis (IPF) continue to have limited therapeutic options and poor outcomes. Identifying the origin of myofibroblasts is an important step in understanding mechanisms that are responsible for fibrosis as well as to more therapeutic targets in fibrosing diseases.
Defining the Myofibroblast
Myofibroblasts were first described in ultrastructural examinations of rat wounds [1]. In the first description of myofibroblasts, Gabbiani and colleagues characterized the presence of modified fibroblasts in the granulation tissue of wounds. These modified fibroblasts exhibited cytoplasmic stress fibers similar to those found in contractile smooth muscle cells along with peripheral focal adhesions. Since the original description, molecular and phenotypic features of myofibroblasts have been further detailed.
Normal wound healing is a complex process involving early inflammation, hemostasis, deposition of extracellular matrix and matrix remodeling, and wound closure. Myofibroblasts appear during wound healing and participate in a number of these critical processes. Following wound healing, myofibroblasts disappear, likely through apoptosis. In aberrant wound healing that leads to pathologic fibrosis, however, myofibroblasts persist in the fibrotic scar, obliterating normal tissue architecture and function through continued ECM remodeling and contraction. Thus, myofibroblasts are central to the pathogenesis of fibrotic disease in many organs, and understanding the biology of myofibroblasts is critical to medical therapies in fibrosis.
Although the origin of myofibroblasts remain a topic of some controversy, there are several generally accepted features that define the activated myofibroblast (Table 1). Morphologically, myofibroblasts differ from quiescent interstitial cells or fibroblasts in that they are large cells with ruffled membranes. Actin microfilaments are organized intracellularly and converge at multiple focal points on the cell surface, termed “supermature focal adhesions” in vitro and “fibronexus” in vivo, that connect the intracellular network of stress fibers to points of contact in the ECM [2]. Functionally, myofibroblasts express of α-smooth muscle actin (α-SMA) which distinguishes it from fibroblasts; they upregulate deposition of ECM products such as type I collagen (COL 1), increase release of inflammatory mediators, and display enhanced contractile forces [2]. In particular, many investigators studying myofibroblast precursors have used α-SMA or COL 1 expression as markers that define differentiation into myofibroblasts.
Myofibroblasts in the Lung
The human lung is a structurally complex organ that comprises over 40 cell types [3]. Structurally, the conducting airways include the trachea and subsequent branching segments of bronchi and bronchioles. These terminate in the gas-exchanging alveolar sacs. Throughout the respiratory tree, the airways are lined with epithelial cells, vasculature, and lung-resident mesenchymal cells that are connected to each other through an extensive network of extracellular matrix. Myofibroblasts alter the architecture of local anatomy through enhanced contractility and deposition of excess extracellular matrix, eventually disrupting normal lung physiology. Their involvement in different anatomic regions leads to different pathologies. Myofibroblasts may cause pathology in small conducting airways such as asthma, chronic obstructive pulmonary disease, or bronchiolitis obliterans syndrome. They may affect the pulmonary vasculature and cause pulmonary hypertension. They may affect the pleura in pleural fibrosis and lead to restrictive lung disease. They may expand unchecked in the alveolar interstitium and lead to restriction and impairment in gas exchange, as seen in fibrosing lung diseases. In considering experimental evidence for the origin of myofibroblasts in the lung, it is important to bear in mind that anatomic involvement varies between lung diseases. Myofibroblast involvement around the small conducting airways may be different from their involvement in the alveolar interstitium. While animal models of injury are excellent tools to study the biology of myofibroblasts, they are nevertheless limited in that no one model fully captures the spatial diversity of myofibroblast involvement in lung diseases.
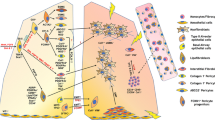
The origin of myofibroblasts in organ fibrosis has been debated. Observations of cellular behavior in vitro and immunohistochemical analyses of fibrotic organs have informed hypotheses about the origin of myofibroblasts. With the advent and widespread adoption of lineage-tracing animal models in the past decade, investigators have been able to follow the fate of candidate precursors of myofibroblasts in vivo and determine whether myofibroblasts truly derive from these candidate populations. In the following sections, we will highlight the main theories of lung myofibroblast precursors (Fig. 1).

Illustration of alveolar interstitium showing multiple sources of myofibroblasts (orange) in the lung. In response to tissue damage (danger signs), various precursor populations have been postulated to contribute to the myofibroblast pool in the lung. These include heterogenous mesenchymal populations such as resident fibroblasts (red and blue), pericytes (purple), and mesenchymal stem cells (MSC, yellow). Other potential sources include circulating fibrocytes (green) and type II alveolar epithelial cells (AECII)
Epithelial Origin
Epithelial-to-mesenchymal transition (EMT) is the process in which differentiated epithelial cells downregulate epithelial markers, lose their adherent phenotype, upregulate mesenchymal markers, and gain fibroblastic functions. Common features observed in EMT include downregulation of E-cadherin and the miRNA200 family, loss of epithelial cell apical–basal polarity, and upregulation of mesenchymal markers including fibroblast-specific protein 1 (FSP1), α-smooth muscle actin (α-SMA), collagen type 1 (Col I), vimentin, and fibronectin [4, 5]. Transcription factors associated with fibroblast proliferation and mesenchymal differentiation such as ZEB1, ZEB2, Twist, Slug, and Snail are also upregulated during EMT [6,7,8,9]. The transitional process is not a binary one, where cells undergoing EMT may exhibit intermediate phenotypes with overlapping features of epithelial and mesenchymal cells. Three types of EMT have been described in different biological contexts: in embryogenesis (type 1), in normal and aberrant wound healing (type 2), and in cancer metastasis (type 3) [4, 10]. Whether epithelial cells serve as progenitors of myofibroblasts in lung fibrosis has been a subject of controversy.
Human lung samples have provided suggestive evidence that cells of epithelial origin contribute to fibrotic scars. For example, primary cultures of human airway epithelial cells (AECs) from control and asthmatic subjects undergo EMT in vitro when exposed to transforming growth factor beta1 (TGF-β1) [11]. In lung transplant recipients who develop bronchiolitis obliterans syndrome (BOS), where terminal respiratory bronchioles in the lung allograft are gradually replaced by granulation tissue in the lumen leading to obstructive lung physiology, airway brushings show increased cell populations with mesenchymal markers by flow cytometry [12]. Furthermore, histologic evaluation of human allograft samples from lung transplant recipients with BOS shows increased mesenchymal markers in bronchioles, and primary bronchiolar epithelial cells undergo EMT when treated with TGF-β1 [13]. Lung biopsies of subjects with IPF showed evidence of cells co-staining type II alveolar epithelial cell marker (pro-SPC) and mesenchymal marker (N-cadherin) [14]. However, another study of human fibrotic lung from subjects with IPF and non-specific interstitial pneumonia (NSIP) reveals no evidence of cells that co-express epithelial and mesenchymal markers [15]. Furthermore, in the bleomycin model of lung fibrosis in mice, cells that are double-positive for epithelial and myofibroblast markers were not detected in the lung by immunohistochemistry [15].
Advances in transgenic mouse models have facilitated the fate-mapping of epithelial cells in experimental models of fibrosis (Table 2). In mouse models of fibrosis, lineage-tracing studies using human Sftpc promoter as driver of β-galactosidase (β-Gal) expression suggest that epithelial cells are precursors of myofibroblasts in experimental models of lung fibrosis [14, 16]. One limitation that has been raised is that histological assessment by β-Gal staining does not provide sufficient detail to differentiate positive staining cells of epithelial origin from adjacent myofibroblasts in fibrotic foci in vivo, and human Sftpc promoter may not adequately represent native epithelial cells in murine models. In contrast, subsequent fate-mapping studies, using endogenous mouse Sftpc promoter to drive fluorescent reporter protein expression in mouse models of fibrosis, failed to show significant contribution to the myofibroblast pool by cells of epithelial origin when examined under confocal microscopy. Using endogenous Sftpc promoter in transgenic mouse models, Rock and colleagues demonstrated that epithelial-derived cells do not contribute significantly to fibrotic regions in the bleomycin lung injury model [17•]. Although kidney fibrosis likely differs from lung fibrosis, a fibrosis study fate-mapping multiple lineages of cell types estimates that EMT contributes to only approximately 5% of myofibroblasts in kidney fibrosis [18].
Thus, evidence for EMT in experimental models for organ fibrosis remains limited. However, it is important to note the differences between animal models of fibrosis and human disease. Whereas many experimental models of lung fibrosis involve single-hit injury (e.g., acid, bleomycin) delivered intratracheally and lead to transient fibrosis with complete resolution, fibrotic disease in the lung represents a heterogeneous category encompassing diverse modes of injury, chronicity, and outcomes. Some are transient and resolve completely after infection or acute respiratory distress syndrome (ARDS), while others may become persistent, such as fibrotic ARDS resulting in chronic respiratory failure. More dramatic still, some are progressive, such as fibrosing interstitial lung diseases (e.g., idiopathic pulmonary fibrosis, non-specific interstitial pneumonitis). The mechanism of tissue injury and the progressive, non-resolving nature of myofibroblast activation in chronic, fibrotic lung diseases are critical features that distinguish them from the animal models of lung fibrosis. Although lineage-tracing experiments in animal models do not support EMT as a major process in lung fibrosis, whether injury mechanisms and chronicity in human fibrosing diseases might account for EMT remains to be addressed.
Bone Marrow–Derived Precursors (Fibrocytes)
Bone marrow–derived mesenchymal precursor cells have been described as progenitors of myofibroblasts in tissue injury. This precursor population comprises a small percentage (0.5%) of circulating hematopoietic cells in the normal host, and they were first characterized in human blood as CD45+ CD34+ CD11b+ fibronectin+ vimentin+ Col I+ Col III+ by flow cytometry (Table 2) [19]. This population immuno-stains for hematopoietic (CD45), stem cell (CD34), and various mesenchymal markers. Infiltrating cells in mouse cutaneous healing wounds further demonstrate the existence of cells co-expressing hematopoietic, stem cell, and mesenchymal markers [19]. Fibrocytes have also been observed in human lung fibrotic disease. For example, confocal microscopy and immunofluorescent imaging of human lungs from IPF subjects and controls reveal the presence of fibrocytes only in fibrotic foci [20]. The level of fibrocytes in circulation in IPF subjects might also correlate with disease activity and progression [21]. Meanwhile, fibrocytes have been observed in lung allografts of transplant patients with bronchiolitis obliterans syndrome, and their circulating levels may also correlate with development and severity of disease [22, 23]. Evidence of bone marrow–derived cells that exhibit mesenchymal features and the demonstration of their relationship with human fibrotic diseases form the basis of interest in fibrocytes as precursors of myofibroblasts.
Circulating fibrocytes are recruited to sites of injury by tissue-derived chemokines [24,25,26]. Cultured fibrocytes respond to TGFβ in vitro, differentiating into cells that exhibit features of myofibroblasts including α-SMA expression and collagen gel contraction [27, 28]. Animal models of injury in lung and skin suggest that fibrocytes contribute significantly to α-SMA+ myofibroblasts or produce collagen in scars following experimentally induced injury [25, 29]. Yet, other groups were not able to replicate these findings. In one study, GFP-labeled bone marrow–derived Col I+ fibrocytes were collected from lung fibrotic foci following bleomycin-induced lung injury [30]. GFP+ fibrocytes isolated from scars failed to express α-SMA even with TGFβ stimulation in vitro. Furthermore, although the process of fibrosis is different between organs, studies of fibrocytes in liver and kidney fibrosis also reveal little evidence of fibrocytes differentiating into α-SMA+ myofibroblasts at sites of tissue injury and repair [31, 32].
While the contribution of fibrocytes to the myofibroblast population remains a topic of controversy, there is evidence that fibrocytes likely indirectly contribute to tissue fibrogenesis. Fibrocytes obtained from burn patients do not express significant collagen I or α-SMA compared to fibrocytes from control subjects. However, conditioned media from fibrocytes collected from burn patients significantly induced myofibroblast features in dermal fibroblasts compared to conditioned media from control subject fibrocytes [33]. Furthermore, fibrocytes may contribute to airway remodeling by promoting myofibroblast differentiation through stem cell factor (SCF) and interleukin-31 [34]. Another mechanism by which fibrocytes may indirectly influence fibroblastic behavior in resident cells is by release of exosomes. Exosomes are extracellular nanoparticles secreted by many cell types. They contain lipid bilayers that envelop various protein and nucleic acid contents that are protected from degradation. Exosomes can be taken up by recipient cells and the contents of exosomes may modulate recipient cell behavior, thus they serve as a mode of intercellular communication. Pro-fibrotic exosomes released by fibrocytes have been described in the literature; however, more studies will need to be conducted to characterize their contribution to human fibrotic disease in the lung [35].
Perivascular Stromal Origin
Pericytes are mural cells in capillary beds that surround the vasculature. They share a common basement membrane with endothelial cells in the microvascular niche and are classically defined by ultrastructure demonstrating their relationship with endothelial cells [36]. In addition to histology, investigators have used common markers to define pericytes in tissues such as platelet-derived growth factor receptor beta (PDGFRβ), neuron-glial antigen 2 (NG2), CD146 (melanoma cell adhesion molecule or MCAM), α-SMA, desmin, vimentin, and regulator of G protein signaling (RGS5) (Table 2) [37]. The biology of pericytes in vascular development and regulation of blood–brain barrier has been studied [36]. Yet, recent studies show that pericytes may have other functional roles following organ injury [32, 38•, 39, 40].
Interest in pericytes as progenitors of myofibroblasts in organ fibrosis began with observations of pericyte function in wound healing in the central nervous system. Pericytes contribute to the generation of scar tissue in the spinal cord [41, 42]. In the kidney, pericytes have been shown to contribute to the myofibroblast pool [39]. In the lung, we have shown that perivascular stromal cells defined by FoxD1-lineage and enriched for PDGFRβ expression are present in fibrotic foci in the bleomycin model of lung fibrosis [38•]. Primary stromal cells of the FoxD1-lineage isolated from transgenic mouse lungs upregulate α-SMA expression in response to TGFβ stimulation. Furthermore, up to approximately half of the α-SMA+ myofibroblasts in fibrotic foci are FoxD1-lineage cells. In another lineage-tracing study, however, Rock and colleagues did not find significant contribution to myofibroblasts in the lung using NG2 as the genetic label for lung pericytes [17•]. Differences in these findings may be explained by the use of different markers to label pericytes and the relatively inefficient labeling of NG2+ cells by the NG2-CreERT2 transgenic model used in the study. It is possible that a subpopulation of NG2− pericytes in the FoxD1-lineage cells in the lung upregulates α-SMA following bleomycin injury, but further studies will need to be conducted to study this possibility. Although pericyte heterogeneity has been described in other tissues, it has not been carefully characterized in the lung [43, 44]. It is important to note that expression of common pericyte markers is not universal and may be tissue-dependent. Additionally, other cell types may express these markers. These challenges in defining pericytes may lead to conflicting results depending on the strategy used to label pericytes, and they highlight the importance of histology and marker expression in the proper context.
Resident Mesenchymal Cells
Resident mesenchymal populations are most widely accepted as major sources of activated myofibroblasts. Of the resident mesenchymal cells, there is general agreement that quiescent resident fibroblasts represent important precursors of myofibroblasts [17•, 32, 38•, 45•, 46]. Resident fibroblasts contribute to ECM production and turnover in lung homeostasis. In mouse lungs, we found that Col1a1+ cells are enriched for PDGFRα expression but are PDGFRβ− [38•]. Using an unbiased approach by single-cell RNASeq to define the fibroblast population in mouse lungs, another group reported that 80% of the defined fibroblast cluster in uninjured mouse lung expresses Col1a1 [47]. These cells likely represent a population of resident fibroblasts that are transcriptionally distinct from pericyte-like cells that express PDGFRβ. We showed that these resident fibroblasts highly express Col3a1 and Itga8 genes in contrast to pericyte-like cells [38•]. Indeed, in humans, integrin α8 seems to label a major subpopulation of lung fibroblasts similar to the murine model [48]. Other common mesenchymal markers used to identify quiescent lung fibroblasts include Thy-1, fibronectin, and desmin (Table 2). However, it is important to emphasize that there is no one marker that defines resident fibroblasts or any one group of mesenchymal lineages.
Efforts have been made to better characterize the lung mesenchymal population and their heterogeneity, although the absence of a single, well-defined marker poses a challenge in quantifying their relative contributions to myofibroblasts [47,48,49]. In a study using signal transduction pathway readouts to define mesenchymal heterogeneity and lineages, Zepp et al. identified several lineages of mesenchymal cells with distinct transcriptional and functional properties during homeostasis and following injury [45•]. In this study, Axin2-lineage mesenchymal cells can be subgrouped into Axin2-lineage, PDGFRα+, and Axin2-lineage, PDGFRα− populations. The former group occupies the alveolar interstitium, whereas the latter occupies the peri-airway niche. Interestingly, approximately 50% of α-SMA+ myofibroblasts derived from Axin2-lineage mesenchymal cells, while PDGFRα-lineage cells contributed to less than 20% of myofibroblasts. Evaluation of Acta2 expression by qPCR in Axin2-lineage, PDGFRα+, and Axin2-lineage, PDGFRα− populations at day 7 after bleomycin-induced lung injury revealed that the latter population significantly upregulated Acta2 expression, whereas the former did not. Thus, there appears to be preferential myofibroblastic potential in Axin2-lineage, PDGFRα− mesenchymal cells.
There is evidence for other mesenchymal cell types that contribute to α-SMA+ myofibroblasts. Lung-resident mesenchymal stem cells (MSCs) defined by ABCG2-lineage (ABCG2+) have been shown to transition to myofibroblasts in vitro upon stimulation with TGFβ and in vivo with bleomycin lung injury [50•]. These cells express usual MSC markers as well as PDGFRβ. They inhabit the perivascular niche, and although they lack NG2 expression, their transcriptional program is closer to NG2+ pericytes than lung fibroblasts. Thus, this population of perivascular MSCs may represent a subpopulation of pericyte-like cells that are NG2−, defined by ABCG2-lineage, that possess myofibroblastic potential.
Conclusion and Future Directions
Myofibroblasts are central to both normal and aberrant healing following tissue injury. The widespread use of lineage-tracing animal models and more detailed molecular markers has led to more sophisticated characterization of myofibroblast precursors in the lung. Epithelial and bone marrow origins of lung myofibroblasts have been supported by in vitro observations and immuno-histopathology in human fibrotic lung. Yet, lineage-tracing models in the past decade have failed to demonstrate significant, direct contribution of either lineage of cells to the myofibroblast pool in animal models. While direct contributions from the epithelium and circulating fibrocytes remain subjects of controversy, contribution by resident mesenchymal populations has been more broadly accepted. Still, understanding the relative contributions by different lung mesenchymal populations continues to be limited. The lung mesenchyme remains poorly defined without any one marker that demarcates a population with unique features. Advances in next-generation sequencing and analysis may add novel dimensions and complexity to how we understand lung mesenchymal subtypes and how they differentially contribute to lung fibrosis and repair [51].
The search for the lung myofibroblast precursor(s) is merely one aspect in understanding lung fibrogenesis. Future studies that elucidate how different microenvironmental cues alter myofibroblast precursors and activation will be equally critical. Biochemical and physical properties of the ECM, paracrine signaling from other cellular populations (e.g., fibrocytes, macrophages), and diverse modes of lung injury may differentially activate precursor populations. Finally, while activation of myofibroblasts may represent the final convergence event, the paradigm that lung myofibroblasts represent a functionally homogeneous population may need to be revisited. There is evidence in skin fibrosis that myofibroblasts derive from heterogeneous precursors, and their precursor lineages inform distinct transcriptional signatures and functions [52]. Whether multiple myofibroblast origins in lung fibrosis similarly lead to discrete functional populations of myofibroblasts remains an area to be explored.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–50.
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–16.
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–55.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8.
Rout-Pitt N, Farrow N, Parsons D, Donnelley M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res. 2018;19:136.
Karicheva O, Rodriguez-Vargas JM, Wadier N, Martin-Hernandez K, Vauchelles R, Magroun N, et al. PARP3 controls TGFbeta and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget. 2016;7:64109–23.
Yang ZC, Yi MJ, Ran N, Wang C, Fu P, Feng XY, et al. Transforming growth factor-beta1 induces bronchial epithelial cells to mesenchymal transition by activating the Snail pathway and promotes airway remodeling in asthma. Mol Med Rep. 2013;8:1663–8.
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45.
Stone RC, Pastar I, Ojeh N, Chen V, Liu S, Garzon KI, et al. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016;365:495–506.
Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am J Respir Crit Care Med. 2009;180:122–33.
Hodge S, Holmes M, Banerjee B, Musk M, Kicic A, Waterer G, et al. Posttransplant bronchiolitis obliterans syndrome is associated with bronchial epithelial to mesenchymal transition. Am J Transplant. 2009;9:727–33.
Borthwick LA, Parker SM, Brougham KA, Johnson GE, Gorowiec MR, Ward C, et al. Epithelial to mesenchymal transition (EMT) and airway remodelling after human lung transplantation. Thorax. 2009;64:770–7.
Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–5.
Yamada M, Kuwano K, Maeyama T, Hamada N, Yoshimi M, Nakanishi Y, et al. Dual-immunohistochemistry provides little evidence for epithelial-mesenchymal transition in pulmonary fibrosis. Histochem Cell Biol. 2008;129:453–62.
Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69:760–5.
• Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–83. Important recent studies utilizing lineage-tracing animal models to define the origins of myofibroblasts from various cell types.
LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53.
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994;1:71–81.
Andersson-Sjoland A, de Alba CG, Nihlberg K, Becerril C, Ramirez R, Pardo A, et al. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int J Biochem Cell Biol. 2008;40:2129–40.
Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179:588–94.
LaPar DJ, Burdick MD, Emaminia A, Harris DA, Strieter BA, Liu L, et al. Circulating fibrocytes correlate with bronchiolitis obliterans syndrome development after lung transplantation: a novel clinical biomarker. Ann Thorac Surg. 2011;92:470–7 discussion 477.
Andersson-Sjoland A, Erjefalt JS, Bjermer L, Eriksson L, Westergren-Thorsson G. Fibrocytes are associated with vascular and parenchymal remodelling in patients with obliterative bronchiolitis. Respir Res. 2009;10:103.
Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35:175–81.
Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, et al. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest. 2004;114:438–46.
Sakai N, Wada T, Yokoyama H, Lipp M, Ueha S, Matsushima K, et al. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc Natl Acad Sci U S A. 2006;103:14098–103.
Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166:7556–62.
Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282:22910–20.
Mori L, Bellini A, Stacey MA, Schmidt M, Mattoli S. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp Cell Res. 2005;304:81–90.
Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone marrow-derived progenitor cells in pulmonary fibrosis. J Clin Invest. 2004;113:243–52.
Kisseleva T, Uchinami H, Feirt N, Quintana-Bustamante O, Segovia JC, Schwabe RF, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45:429–38.
Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–27.
Wang JF, Jiao H, Stewart TL, Shankowsky HA, Scott PG, Tredget EE. Fibrocytes from burn patients regulate the activities of fibroblasts. Wound Repair Regen. 2007;15:113–21.
Dolgachev VA, Ullenbruch MR, Lukacs NW, Phan SH. Role of stem cell factor and bone marrow-derived fibroblasts in airway remodeling. Am J Pathol. 2009;174:390–400.
Chen L, Brenner DA, Kisseleva T. Combatting fibrosis: exosome-based therapies in the regression of liver fibrosis. Hepatol Commun. 2019;3:180–92.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Yamazaki T, Mukouyama YS. Tissue specific origin, development, and pathological perspectives of pericytes. Front Cardiovasc Med. 2018;5:78.
• Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, et al. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188:820–30. Important recent studies utilizing lineage-tracing animal models to define the origins of myofibroblasts from various cell types.
Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97.
Hung CF, Mittelsteadt KL, Brauer R, McKinney BL, Hallstrand TS, Parks WC, et al. Lung pericyte-like cells are functional interstitial immune sentinel cells. Am J Phys Lung Cell Mol Phys. 2017;312:L556–67.
Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J. A pericyte origin of spinal cord scar tissue. Science. 2011;333:238–42.
Dias DO, Goritz C. Fibrotic scarring following lesions to the central nervous system. Matrix Biol. 2018;68-69:561–70.
Hirschi KK, Rohovsky SA, D'Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–14.
Nehls V, Drenckhahn D. Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol. 1991;113:147–54.
• Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, et al. Distinct mesenchymal lineages and niches promote epithelial self-renewal and myofibrogenesis in the lung. Cell. 2017;170:1134–1148 e1110. Important recent studies utilizing lineage-tracing animal models to define the origins of myofibroblasts from various cell types.
Vyalov SL, Gabbiani G, Kapanci Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am J Pathol. 1993;143:1754–65.
Peyser R, MacDonnell S, Gao Y, Cheng L, Kim Y, Kaplan T, et al. Defining the activated fibroblast population in lung fibrosis using single-cell sequencing. Am J Respir Cell Mol Biol. 2019;61:74–85.
Matsushima S, Aoshima Y, Akamatsu T, Enomoto Y, Meguro S, Kosugi I, et al. CD248 and integrin alpha-8 are candidate markers for differentiating lung fibroblast subtypes. BMC Pulm Med. 2020;20:21.
Heinzelmann K, Lehmann M, Gerckens M, Noskovicova N, Frankenberger M, Lindner M, et al. Cell-surface phenotyping identifies CD36 and CD97 as novel markers of fibroblast quiescence in lung fibrosis. Am J Phys Lung Cell Mol Phys. 2018;315:L682–96.
• Marriott S, Baskir RS, Gaskill C, Menon S, Carrier EJ, Williams J, et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am J Phys Cell Phys. 2014;307:C684–98. Important recent studies utilizing lineage-tracing animal models to define the origins of myofibroblasts from various cell types.
Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, et al. The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell. 2020;26:482–502.
Shook BA, Wasko RR, Rivera-Gonzalez GC, Salazar-Gatzimas E, Lopez-Giraldez F, Dash BC, et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science. 2018;362:eaar2971.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares no conflict of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the author were performed in accordance with all applicable ethical standards including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lung Injury & Fibrosis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hung, C. Origin of Myofibroblasts in Lung Fibrosis. Curr. Tissue Microenviron. Rep. 1, 155–162 (2020). https://doi.org/10.1007/s43152-020-00022-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43152-020-00022-9