
Abstract
As a rare disease leading to male infertility, idiopathic hypogonadotropic hypogonadism (IHH) has strong heterogeneity of clinical phenotype and gene mutation. At present, there is no effective diagnosis and treatment method for this disease. This study is to explore the possible new pathogenic gene of idiopathic hypogonadotrophic hypogonadism and the pathological mechanism affecting its occurrence. We performed a whole-exome sequencing on 9 patients with normosmic idiopathic hypogonadotropic hypogonadism (nIHH), 19 varicocele patients with asthenospermia, oligospermia, or azoospermia, 5 patients with simple nonobstructive azoospermia, and 13 normal healthy adult males and carried out comparative analysis, channel analysis, etc. After preliminary sequencing screening, 309–431 genes harbouring variants, including SNPs and indels, were predicted to be harmful per single patient in each group. In genetic variations of nIHH patients’ analysis, variants were detected in 10 loci and nine genes in nine patients. And in co-analysis of the three patient groups, nine nIHH patients, 19 VC patients, and five SN patients shared 116 variants, with 28 variant-harbouring genes detected in five or more patients. We found that the NEFH, CCDC177, and PCLO genes and the Gene Ontology pathways GO:0051301: cell division and GO:0090066: regulation of anatomical structure size may be key factors in the pathogenic mechanism of IHH. Our results suggest that the pathogenic mechanism of IHH is not limited to the central nervous system effects of GnRH but may involve other heterogeneous pathogenic genetic variants that affect peripheral organs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Idiopathic hypogonadotropic hypogonadism (IHH) is a rare disease with clinical manifestations of azoospermia, hypogonadotropism, pubertal retardation or deletion, and infertility. There is a significant difference in incidence between men (approximately 1:30,000) and women (1:125,000) [1, 2]. It is currently thought that insufficient GnRH secretion is the main cause of IHH. This may be due to abnormal GnRH neuron development, differentiation, or activation; failure of GnRH activation; or insufficient GnRH secretion [1, 3].
A type of IHH known as Kallman syndrome (KS) is related to the development of GnRH neurons [1, 3]. Patients with KS show developmental disorders of the reproductive and olfactory systems [4]. Unlike KS, normosmic idiopathic hypogonadotropic hypogonadism (nIHH) shows complex clinical phenotypic diversity, and its pathogenic mechanism cannot be explained. To investigate the pathogenesis of IHH, many whole-genome sequencing analyses have been performed, resulting in the identification of more than 60 genes associated with IHH [5, 6]. More than 40 of these genes have been shown to affect the development of the reproductive system in animal experiments [7].However, clinical data do not support the hypothesis that IHH stems from the pathogenesis of GnRH neurons [1]. First, despite hypogonadotropism in IHH patients, hormone supplementation is not an effective treatment, and more than half of patients fail to regain fertility. Second, there is extensive genetic heterogeneity among IHH patients, and large-scale next-generation sequencing of IHH patients has shown that no single genetic variant or class of genetic variants is widely present in IHH patients. Variants in the most frequently involved gene, FGFR1, are present in no more than 10% of IHH patients. Although variants in more than 60 genes have been found to be associated with the development of IHH, these genetic data are not sufficient to suggest causation. In addition, most IHH patients have other symptoms, such as hearing impairment, affective disorder, or limb coordination disorder, and the pathogenic mechanism in these patients cannot be explained by the GnRH neuronal origin hypothesis. Moreover, like patients with simple nonobstructive azoospermia (SN, NOA with azoospermia phenotype only), patients with nIHH have azoospermia, but they do not have nervous system abnormalities. Many pathogenic mechanisms of diseases such as SN do not involve the hypothalamus or pituitary gland but are characterised by gonadal dysfunction, such as abnormalities of the testes (including abnormal spermatogenesis and Sertoli cells). Although these abnormalities are heterogeneous, hormone therapy is ineffective in some IHH patients, which may lead to common non-central abnormalities that cause nIHH and SN.
Next-generation sequencing technology has facilitated the detection of genetic variants on a large scale, but studies targeting diseases with genetic heterogeneity remain difficult. In case–control studies, the frequencies of variants in many genes have varied greatly in different populations studied, due to factors such as the characteristics of the population, ethnicity, and geographical location, making it difficult to identify the exact genes associated with the disease. Taking the normal population as the control, due to differences in the natural variation rate of many genes in different populations, the prediction of pathogenic genes associated with rare diseases, such as IHH, is often inaccurate (false positives). For monogenic rare diseases, the detection of the causative gene is largely obscured by the population variation rate. Traditionally, case–control studies of IHH have been conducted to screen for variants by next-generation sequencing, which to some extent increases the likelihood that IHH will be defined and interpreted as a single disease. However, it is also possible that the genetic pathology underlying IHH may be shared with other NOA. Under that premise, overlapping pathologies may be overemphasised, and the mutations only associated with IHH may be ignored. CHD7 is one of the pathogenic genes associated with coloboma, heart defects, atresia choanae (CHARGE) syndrome, and IHH, while reproductive system dysplasia, the main clinical manifestation of IHH, is only one of the secondary symptoms of CHARGE syndrome [8, 9]. Studies have shown that variation in the PORKR2 gene is markedly higher in the Maghrib population (23.3%) than in European populations (5.1%), and it has more reference significance for the diagnosis of KS [10].
In this study, we focused on the Han population in Southwest China. In addition to nIHH patients and normal males, varicocele patients with asthenospermia, oligospermia, or azoospermia (VC) and simple nonobstructive azoospermia (SN) (the patient only has a phenotype with sperm count of 0) were included in the study as 2 other types of NOA (here, we include a wide range of nonobstructive sperm with low quantity or abnormal quality in NOA) to investigate the broader genetic pathology of nIHH. Exome sequencing analysis was combined with pathway analysis to investigate the independent genetic pathogenic basis of nIHH.
Materials and Methods
Subjects and Clinical Evaluation
We have included 9 patients with normosmic idiopathic hypogonadotropic hypogonadism (nIHH); 19 varicocele patients with asthenospermia, oligospermia, or azoospermia; 5 patients with simple nonobstructive azoospermia; and 13 normal healthy adult males.
The research included nine male nIHH patients (Table 1). nIHH patients were included in the study if they had (a) low concentrations of follicle-stimulating hormone (FSH) and luteinising hormone (LH); (b) a low serum testosterone concentration; (c) a zero sperm count; (d) retardation or absence of puberty; (e) computed tomography and magnetic resonance imaging results showing no obvious organic injury in the pituitary region; and (f) a normal chromosome karyotype [1].
We included five SN patients and 19 VC patients. SN patients were included if they had the following: (a) sperm count is 0; (b) normal FSH and LH concentrations; (c) no obvious organic damage to the hypothalamus or pituitary gland; (d) a normal chromosome karyotype; (e) normal reproductive tract and urethral structure; (f) no obvious organic damage to the gonads; and (g) no other type of azoospermia [11]. Varicocele patients with asthenospermia, oligospermia, or azoospermia were included if they had the following: (a) a unilateral or bilateral varicocele under colour Doppler ultrasound and (b) sperm count is 0 or less than or equal to 20*106/ml, or progressive sperm motility are less than 60% [12].
For the normal male control (NC) group, we selected 13 healthy young men in Southwest China. The young men were eligible for the NC group if they (a) were aged 21 to 26; (b) had a body mass index of 20–25; (c) had normal sperm morphology under microscopic examination; (d) had a sperm count > 20 × 106/mL; (e) had a sperm motility rate > 80%; (f) had normal sperm motility; (g) had no hereditary disease affecting their growth and development or their reproductive system; and (h) had no serious disease that may affect the results of the study. All patients were diagnosed and treated at the West China Fourth Hospital, Chengdu, China. After being informed of the details and specific risks of the study, they voluntarily joined the study.
Analysis Methods and Strategy
Sequencing and Data Analysis
DNA Extract and Detect
Genomic DNA extracted from peripheral blood for each sample was fragmented to an average size of 180 ~ 280 bp and subjected to DNA library creation using established Illumina paired-end protocols. The Agilent SureSelect Human All ExonV6 Kit (Agilent Technologies, Santa Clara, CA, USA) was used for exome capture according to the manufacturer’s instructions. The Illumina Novaseq 6000 platform (Illumina Inc., San Diego, CA, USA) was utilized for genomic DNA sequencing in Novogene Bioinformatics Technology Co., Ltd (Beijing, China) to generate 150-bp paired-end reads with a minimum coverage of 10 × for ~ 99% of the genome (mean coverage of 100 ×).
Data Analysis
After sequencing, basecall files conversion and demultiplexing were performed with bcl2fastq software (Illumina). The resulting fastq data were submitted to in-house quality control software for removing low quality reads and then were aligned to the reference human genome (hs37d5) using the Burrows-Wheeler Aligner (bwa)13, and duplicate reads were marked using sambamba tools [14].
SNP/INDEL calling
Single nucleotide variants (SNVs) and indels were called with samtools to generate gVCF [15]. The raw calls of SNVs and INDELs were further filtered with the following inclusion thresholds: (1) read depth > 4; (2) root-mean-square mapping quality of covering reads > 30; and (3) the variant quality score > 20.
CNV calling
The copy number variants (CNVs) were detected with software CoNIFER (V0.2.2) [16].
Annotation
Annotation was performed using ANNOVAR (2017June8) [17]. Annotations included minor allele frequencies from public control data sets as well as deleteriousness and conservation scores enabling further filtering and assessment of the likely pathogenicity of variants.
Rare Variant Filtering
Filtering of rare variants was performed as follows: (1) variants with a MAF less than 0.01 in 1000 genomic data (1000g_all) [18], esp6500siv2_all, gnomAD data (gnomAD_ALL and gnomAD_EAS), and in-house Novo-Zhonghua exome database from Novogene; (2) only SNVs occurring in exons or splice sites (splicing junction 10 bp) are further analysed since we are interested in amino acid changes. (3) Then, synonymous SNVs which are not relevant to the amino acid alternation predicted by dbscSNV are discarded; the small fragment non-frameshift (< 10 bp) indel in the repeat region defined by RepeatMasker is discarded. (4) Variations are screened according to scores of SIFT [19], Polyphen [20], MutationTaster [21], and CADD [22] software. The potentially deleterious variations are reserved if the score of more than half of these four software support harmfulness of variations [23]. Sites (> 2 bp) that did not affect alternative splicing were removed.
ACMG Gene Screening and Comparison
In 2015, the ACMG developed standards and guidelines for the interpretation of sequence variants. These standards have become the gold standard for data interpretation after high-throughput sequencing [24]. The variant classification system developed by the ACMG recommends the use of specific standard terms. The system uses the classifications pathogenic, likely pathogenic, uncertain significance, likely benign, or benign to describe variants found in pathogenic genes of Mendelian diseases.
We performed ACMG analysis on the variant-harbouring genes of all patients after preliminary screening, i.e. next-generation sequencing, quality inspection, coverage determination, and in-depth screening, and identified the variants classified as pathogenic and likely pathogenic. The results of the ACMG analysis for the nIHH group were compared with previously identified pathogenic genes of nIHH.
Metascape Pathway and Pathway Enrichment Analysis
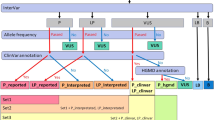
Metascape (metascape.org) is a genome-wide data analysis network platform that integrates more than 40 omics data analyses. The platform provides a series of analysis modes and strategies, including gene pathway and process enrichment analysis, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) biochemical pathway analyses; protein–protein interaction analysis, including string6, biogrid7, and omnipath8; and DisGeNET associated disease analysis. The results are presented in easy-to-understand data tables and images (Fig. 1).

Sequencing and analysis process. Predictive filtering generated from the raw data obtained from the second generation sequencing after routine quality control, basic screening, and hazard prediction. On the one hand, it generates mutation gene analysis and pathway analysis of nIHH, SN, and VC groups after removing NC data; on the other hand, four groups of different mutation site analysis of the same gene were generated
Our data were processed using the pathway enrichment analysis method provided by Metascape. The analysis included KEGG pathways; GO biological processes; reaction group gene sets; standard pathways; and data from the CORUM, Rust, DisGeNET, PaGenBase, transcription factor targets, WikiPathways, and PANTHER Pathway. Enrichment was based on a P value < 0.005, a minimum number of three genes in the same pathway, and an enrichment factor > 1.5. Enrichment values were arranged from small to large. Pathway enrichment was performed for each patient.
Screening Strategy
Analysis of Mutant Genes and Their Biological Functions
First, we eliminated all genetic variants in the three patient groups that were also detected in the NC group. We also compared the data from the three patient groups after preliminary screening. We analysed the data from the nIHH group, after excluding data from the NC group. Previous studies have identified 63 mutated genes affecting the development of nIHH. We compared the nIHH group data with these previously identified genes and then analysed the variants in the nIHH group based on the ACMG criteria. Finally, we filtered the data for genetic variants that were only present in the nIHH group.
We then used a similar method to screen the genetic variants in both the nIHH and NC groups for pathway enrichment analysis and excluded the enriched pathways that appeared in the NC group from the enriched pathways in the nIHH group. Finally, we obtained the screened enriched pathways for nIHH.
Analysis of Common Gene Mutations in nIHH, VC, and SN and Their Biological Functions
After removing the genetic variants identified in the NC group, we compared and screened independent data from the nIHH, VC, and SN groups. We focused on variants that were common to the three groups and analysed those related to spermatogenesis or those with a high level of consistency or large differences between the three groups. Next, we enriched the genes in each group and excluded the enrichment pathway data from the NC group. We combined the enrichment pathway data for the nIHH, VC, and SN groups and identified the enrichment pathways that appeared in all three groups.
We then analysed the data for all four groups without any data excluded. We first counted the common genetic variants present in the four groups, arranged in descending order by the number of co-variants. Of the genes harbouring variants, we selected 18 that most commonly harboured variants and counted their specific variant sites or the exons harbouring the variants (assembly: GRCh37.p13 GCF_000001405.25) to show the distribution of variants located in specific genes in each of the four groups.
Results
Preliminary Screening of Genetic Variants in Each Group
After preliminary sequencing screening, 309–431 genes harbouring variants, including SNPs and indels, were predicted to be harmful per single patient in each group. After eliminating the variants detected in the NC group and comparing the three remaining groups, the number of genes identified per single patient in the IHH, VC, and SN groups was approximately 98–135. Six hundred and forty-five genetic variants were shared between the nIHH and VC groups, and 116 genetic variants were shared by the nIHH, VC, and SN groups (Fig. 2). All analyzed gene mutations occur in the exon region or 2–3 bp away from the exon region and can cause changes in the amino acid sequence of the gene expression protein and are predicted to affect the normal expression of the gene (Supplementary materials).

Intra and inter group genes. Avg: after removing NC data, the average number of gene mutations contained in each patient in the group. Each colour circle represents the mutation of its group after removing the NC group data. The intersection of two circles is the mutation gene shared by the two groups, and the intersection of three circles is the mutation gene shared by the three groups
Genetic Variants in nIHH Patients
We compared the genetic variants in the nIHH group, after excluding those identified in the NC group, with previously identified IHH pathogenic variants (Tables 2 and 3). Variants were detected in 10 loci and nine genes in nine patients. CHD7 gene variants were identified in patients nIHH2 and nIHH4. Variants in the remaining genes, ANOS1, FGFR1, SEMA7A, OTUD4, AMH, KLB, GATA2, and POLR3B, were all detected once.
Subsequently, we performed an analysis based on the ACMG criteria and summarised the results. There were 41 variants identified as pathogenic or likely pathogenic based on the ACMG criteria. Variants in ADAMTS6 and COL12A1 were identified in two patients. The ADAMTS6 variant identified in patients nIHH7 and nIHH8 was 64747447A > C. Only the FGFR1 variant in nIHH patient 3 was a previously identified IHH pathogenic variant (Table 4).
An analysis of variants that were repeated in the nIHH group showed that variants in THSD1, SMPD1, SCL16A3, and KDM2B were each identified in three patients. These genes showed the highest repetition rate in the nIHH group, but no variants in these genes were found in any other group.
In the pathway enrichment analysis of the nIHH group, after excluding variants detected in the NC group, each nIHH patient was found to have 12–41 enriched pathways, and 12 pathways were enriched in more than two patients. Of these, the cell division pathway (GO: 0051301) was enriched in three patients: nIHH3, nIHH4, and nIHH9 (Table 5).
Co-analysis of the Three Patient Groups
Nine nIHH patients, 19 VC patients, and five SN patients shared 116 variants, with 28 variant-harbouring genes detected in five or more patients. PRRC2A was the gene most involved, with variants in this gene detected in one SN patient (SN3) and eight nIHH and VC patients. AKAP13 variants were detected in eight patients; MICAL variants were detected in seven patients; and PBRM1, RECQL5, DOCK8, DCAF13, and SLC26A4 variants were detected in six patients. RECQL5 variants were detected in six patients, five of whom had had splice-site variants at locus 73,626,919 (Table 6). The genetic variants detected in each group are presented in Supplementary Materials.
In the enrichment pathway analysis, only nine pathways were identified in all three groups of patients. “M5880: NABA ECM AFFILIATED”, “R-HSA-9675108: nervous system development”, and “GO:0090066: regulation of anatomical structure size” were identified in five patients; “R-HSA-9716542: signalling by Rho GTPases”, “Miro GTPases”, and “RHOBTB3” were identified in four patients; and the other pathways were identified in three patients (Table 7). Details of enrichment analysis in each group are presented in Supplementary Materials.
Comparison of Different Variant Sites in the Same Gene
We screened 17 variant-harbouring genes with the highest frequency among patients in the nIHH group (TTN, IST1, NEFH, CCDC177, TRIP10, FAM174B, USH2A, PCLO, CASQ2, BPTF, MUC19, ALMS1, PLEC, NT5DC4, MUC17, RIC8A, and OBSCN) and determined the status of these 17 genes in all patients (Table 8). Mutations in these genes occur in the exon region of the corresponding gene and alter the amino acid sequence of the expressed protein, thereby hindering the normal expression of the corresponding gene; the detailed mutation situation is shown in the table (Supplementary Materials). And some of the mutations were positively confirmed by Sanger sequencing on the corresponding patients. Variants in two genes, TTN and IST1, were detected in 83% (38/46) and 98% (45/46) of patients, respectively. Variants in NEFH, CCDC177, TRIP10, FAM174B, and USH2A were detected in more than 50% of patients, and the selected 17 genes accounted for more than 25% of the variants detected.
For specific variant sites and exons, we present the details of some genes for each group (Fig. 3). Of the NEFH variants detected, seven nIHH patients had variants in exon 4; one nIHH patient had a variant in exon 1; and the remaining two SN patients, nine VC patients, and seven NC patients had variants in exon 4. In addition, 21 variants in the NEFH gene were detected in seven nIHH patients, whereas only six NEFH variants were detected in the SN group, 12 in the VC group, and 11 in the NC group.

Specific exon positions of different groups in the same mutant gene. Each gene corresponds to the specific exon of the gene in which the mutations of different groups of patients are located. The percentage shows that more than 100% of the mutations are multiple mutations of the gene in the same patient
Seven nIHH patients had a CCDC177 variant located in exon 1, and all of these were at locus 70,039,793. There were three and four CCDC177 variants detected in the VC and NC groups, respectively. These variants were all located in exon 1 but were not all at the same site as the variant detected in the nIHH patients. PCLO variants were almost exclusively found in nIHH patients, and five of the six PCLO variants were in exon 5.
For OBSCN, two variants were detected in the nIHH group, three were detected in the SN group, seven were detected in the VC group, and four were detected in the NC group. The OBSCN variants were scattered across 16 exons. There were 19 TTN variants in the nIHH group, seven in the SN group, 33 in the VC group, and 23 in the NC group, scattered across 47 exons. Exon 276 was the most frequent site for TTN variants, with four in the nIHH group, one in the SN group, two in the VC group, and four in the NC group. The details of the variant sites are provided in the Supplementary Materials.
Discussion
We selected nIHH, VC, and SN patients and healthy individuals as the experimental subjects. By excluding genetic variants detected in healthy individuals and comparing the variants detected in the three experimental groups, we aimed to identify the specific genetic variants associated with nIHH.
First, we compared the nIHH data with previously identified IHH pathogenic genes. Nine variant-harbouring genes were found in nine patients, with only a CHD7 mutation detected in two patients. This is consistent with previous reports and knowledge of nIHH from the literature, indicating that there is obvious genetic heterogeneity among IHH patients, with no variants in single genes or gene classes cluster in IHH patients on a large scale. It is worth noting that previous studies of CHD7 have focused on KS, CHARGE syndrome, and myocardial function, whereas our two patients with CHD7 variants did not have olfactory dysfunction or other systemic diseases [1, 25,26,27]. Moreover, in the analysis of the nIHH group based on the ACMG criteria, only variants in the FGFR1 gene were identified as pathogenic. ADAMTS6 and COL12A1 variants were detected in two nIHH patients. The site and type of variant were identical in the two patients with ADAMTS6 variants. This is the first report of ADAMTS6 variants associated with male-infertility-related diseases. Only one study has reported an association between this gene and developmental delay, suggesting that ADAMTS6 variants may affect a certain process during growth and development [28]. Of the genes with intragroup duplication, variants in SMPD1 have previously been shown to increase α-synuclein levels and impair acid sphingomyelinase trafficking to lysosomes to induce the development of Alzheimer’s disease [29]. Niemann–Pick disease has also been shown to be related to variants in SMPD1, and type A Niemann–Pick disease manifests with central nervous system abnormalities [30]. Variants in the SMPD1 gene may affect the regulation of the gonad–pituitary axis from the central nervous system and thus affect the development of nIHH. It has also been found that KDM2B inhibits the expression of somatic genes and thus inhibits somatic differentiation during the specification of human primordial germ-cell-like cells, which may be one of the reasons for the azoospermia phenotype in nIHH patients [31].
The results of our pathway analysis showed that only one pathway, cell: division, was enriched in three patients, and 11 pathways were enriched in two patients. None of the 12 enriched pathways has previously been reported to be related to the pathogenesis of nIHH, nor have these pathways been associated with azoospermia. The current hypothesis for the basic aetiology of nIHH, namely, that it originates from damage to GnRH neurons and impaired GnRH synthesis and release, does not appear to be supported by our enrichment analysis results. Our enrichment analysis of a single group of genes with variants in nIHH also showed strong heterogeneity. We did not find any meaningful enrichment of biological pathways involved in GnRH function, that is, the biological function of the hypothalamus or pituitary gland. However, we found many other functional abnormalities, such as cell division, which may be related to spermatogenesis. As mentioned above, variants in KDM2B cause abnormalities in the peripheral reproductive system by affecting the division and proliferation of germ cells. However, our biological function pathway analyses showed great heterogeneity, and therefore, we cannot draw a unified simple conclusion about the pathogenic mechanism of nIHH gene variants. However, our findings show that the main pathogenic basis of nIHH may be in the peripheral, rather than the central and nervous system, and that abnormalities of GnRH function caused by genetic variants is not the only pathogenic mechanism of nIHH.
Many genetic variants were common between nIHH, VC, and SN, but many of them have not been reported to be associated with nIHH or other male infertility disorders. However, some of these variants have been found to be associated with male infertility. The methylation level of PRRC2A has been shown to be significantly correlated with sperm number and sperm motility. Moreover, in a study of NOA in the Han Chinese population, PRRC2A variants were found to lead to abnormal spermatogenesis [32]. A recent study of IHH also suggested that PLXNB1 may induce IHH by affecting changes to GnRH neurons [33]. In addition, some genes have also been found to affect the function of the nervous system. For example, COL11A2 is related to genetic hearing loss and deafness, which is also consistent with some of the phenotypes in our nIHH patients [34, 35]. In the enrichment analysis of three sets of intersecting data, three pathways were enriched in five patients. Of these, the “regulation of the anatomical structure size” pathway, as its name implies, is a collection of genes regulating cellular shape and structure. Most of the genes in this pathway are actin-related genes. Variants in these genes may result in abnormal sperm structure or developmental disruption to a certain extent. Rho-GTPase-related pathways were also enriched in four patients, and these pathways are important in the formation of the actin cytoskeleton, which supports the abovementioned view from another perspective [36]. These results show that patients with IHH, SN, and even some with VC (severe sperm abnormalities) may have common pathogenic genetic variants or enrichment pathways. Moreover, the effects caused by these variants can only be located in the peripheral, rather than the central and nervous system because there were no central nervous system-related symptoms in the SN or VC groups in our study. Although these variants (such as those in PRRC2A) or pathways are not the only cause of IHH, they may be part of the genetic cause and pathogenic mechanism in some IHH patients.
In the initial analysis, many genetic variants shared by the experimental groups and the control group were filtered out so that we only focused on variants that differed in number between the experimental groups and the control group. However, in a subsequent analysis, we found that some variants were frequent in both the nIHH and control groups. These genes have been characterised in the 1000 Genomes Project and found to have a low frequency in the population. We performed an in-depth analysis of the specific exons and sites containing variants for these high consensus genes. In general, these genes could be divided into three categories. The first category included IST1 and NT5DC4. These variants were all located in the same exon, and there was no significant statistical difference in their frequency between groups. IST1 encodes a protein with microtubule-interacting-and-trafficking-interacting motifs that interact with components of endosomal sorting complexes required for transport (ESCRT) [37,38,39]. The IST1 protein regulates the ESCRT-III complex to drive membrane deformation and fission. The role of NT5DC4 is unclear. As the variants detected in this gene occur at a high frequency in the Han Chinese population, they are likely to have no clinical significance. The second category included TTN and OBSCN. Variants in these gene were quite scattered, both in the number of groups and in the exons or specific sites where the variants were located. The third category included CCDC177, NEFH, and PCLO. The number of variants in these genes was significantly greater in the nIHH group than the other groups. The protein encoded by the PCLO gene is part of the presynaptic cytoskeleton matrix, which is involved in the establishment of active synaptic regions and synaptic vesicle trafficking. Some studies have shown that PCLO may be related to human affective disorder, depression, and type 2 diabetes [40, 41]. NEFH encodes a heavy neurofilament protein, which is associated with neuronal damage [42]. Variants in NEFH may be the cause of Charcot–Marie–Tooth neuropathy. However, these variants have not been reported to be related to azoospermia or other related diseases. Our data showed an abnormally high frequency of these variants in IHH, suggesting that they may be related to the pathogenic mechanism of the disease, but this requires further validation and functional research.
The new analysis strategy used in this study provides a new perspective from which to explore the true pathogenic mechanism of nIHH. We conclude that the NEFH, CCDC177, and PCLO genes show an abnormally high variant frequency in nIHH and the pathways GO:0051301: cell division and GO:0090066: regulation of anatomical structure size may be the key difference between nIHH, other types of NOA, and VC. Our results suggest that the pathogenic mechanism of nIHH is not entirely based on GnRH dysfunction. The pathological mechanism of nIHH is not limited to the effects of GnRH on the central nervous system, and other heterogeneous pathogenic genetic variants affecting peripheral organs may also be involved.
Data Availability
All available published data on this study have been included in the main document, tables, figures, and supplementary materials.
Code Availability
Not applicable.
References
Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547–64. https://doi.org/10.1038/nrendo.2015.112.
Mitchell AL, Dwyer A, Pitteloud N, Quinton R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol Metab. 2011;22(7):249–58. https://doi.org/10.1016/j.tem.2011.03.002.
Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci U S A. 1989;86(20):8132–6. https://doi.org/10.1073/pnas.86.20.8132.
Schwanzel-Fukuda M, Bick D, Pfaff DW. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res. 1989;6(4):311–26. https://doi.org/10.1016/0169-328x(89)90076-4.
Dodé C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33(4):463–5. https://doi.org/10.1038/ng1122.
Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5(10):569–76. https://doi.org/10.1038/nrendo.2009.177.
Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. 2021;140(1):77–111. https://doi.org/10.1007/s00439-020-02147-1.
Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48(5):334–42. https://doi.org/10.1136/jmg.2010.087106.
Bajpai R, Chen DA, Rada-Iglesias A, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463(7283):958–62. https://doi.org/10.1038/nature08733.
Sarfati J, Fouveaut C, Leroy C, Jeanpierre M, Hardelin JP, Dodé C. Greater prevalence of PROKR2 mutations in Kallmann syndrome patients from the Maghreb than in European patients. Eur J Endocrinol. 2013;169(6):805–9. https://doi.org/10.1530/eje-13-0419.
Kasak L, Laan M. Monogenic causes of non-obstructive azoospermia: challenges, established knowledge, limitations and perspectives. Hum Genet. 2021;140(1):135–54. https://doi.org/10.1007/s00439-020-02112-y.
Masson P, Brannigan RE. The varicocele. Urol Clin North Am. 2014;41(1):129–44. https://doi.org/10.1016/j.ucl.2013.08.001.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4. https://doi.org/10.1093/bioinformatics/btv098.
Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–32. https://doi.org/10.1101/gr.138115.112.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. https://doi.org/10.1093/nar/gkq603
Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. https://doi.org/10.1038/nature15393.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. https://doi.org/10.1038/nprot.2009.86.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. https://doi.org/10.1038/nmeth0410-248.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6. https://doi.org/10.1038/nmeth0810-575.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. https://doi.org/10.1038/ng.2892.
Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47(1):39–46. https://doi.org/10.1038/ng.3144.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Bergman JE, Janssen N, van der Sloot AM, et al. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. 2012;33(8):1251–60. https://doi.org/10.1002/humu.22106.
Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170A(2):344–54. https://doi.org/10.1002/ajmg.a.37435.
Yan S, Thienthanasit R, Chen D, et al. CHD7 regulates cardiovascular development through ATP-dependent and -independent activities. Proc Natl Acad Sci U S A. 2020;117(46):28847–58. https://doi.org/10.1073/pnas.2005222117.
Malli T, Duba HC, Erdel M, et al. Disruption of the ARID1B and ADAMTS6 loci due to a t(5;6)(q12.3;q25.3) in a patient with developmental delay. Am J Med Genet A. 2014;164a(12):3126–31.
Alcalay RN, Mallett V, Vanderperre B, et al. SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov Disord. 2019;34(4):526–35. https://doi.org/10.1002/mds.27642.
Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab Jan-Feb. 2017;120(1–2):27–33. https://doi.org/10.1016/j.ymgme.2016.12.008.
Yuan W, Yao Z, Veerapandian V, et al. The histone demethylase KDM2B regulates human primordial germ cell-like cells specification. Int J Biol Sci. 2021;17(2):527–38. https://doi.org/10.7150/ijbs.55873.
Bai G, Zhai X, Liu L, et al. The molecular characteristics in different procedures of spermatogenesis. Gene. 2022;826:146405. https://doi.org/10.1016/j.gene.2022.146405.
Welch BA, Cho HJ, Ucakturk SA, et al. PLXNB1 mutations in the etiology of idiopathic hypogonadotropic hypogonadism. J Neuroendocrinol. 2022;34(4):e13103. https://doi.org/10.1111/jne.13103.
Selvam P, Singh S, Jain A, Atwal H, Atwal PS. Novel COL11A2 pathogenic variants in a child with autosomal recessive otospondylomegaepiphyseal dysplasia: a review of the literature. J Pediatr Genet. 2020;9(2):117–20. https://doi.org/10.1055/s-0039-1698446.
Micale L, Morlino S, Schirizzi A, et al. Exon-trapping assay improves clinical interpretation of COL11A1 and COL11A2 intronic variants in stickler syndrome type 2 and otospondylomegaepiphyseal dysplasia. Genes (Basel). 2020;11(12):1513. https://doi.org/10.3390/genes11121513.
Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. https://doi.org/10.1146/annurev.cellbio.21.020604.150721.
Frankel EB, Shankar R, Moresco JJ, Yates JR 3rd, Volkmann N, Audhya A. Ist1 regulates ESCRT-III assembly and function during multivesicular endosome biogenesis in Caenorhabditis elegans embryos. Nat Commun. 2017;8(1):1439. https://doi.org/10.1038/s41467-017-01636-8.
Feng Q, Luo Y, Zhang XN, et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy. 2020;16(4):641–58. https://doi.org/10.1080/15548627.2019.1633862.
Pfitzner AK, Mercier V, Jiang X, et al. An ESCRT-III polymerization sequence drives membrane deformation and fission. Cell. 2020;182(5):1140-1155.e18. https://doi.org/10.1016/j.cell.2020.07.021.
Giniatullina A, Maroteaux G, Geerts CJ, et al. Functional characterization of the PCLO p.Ser4814Ala variant associated with major depressive disorder reveals cellular but not behavioral differences. Neuroscience. 2015;300:518–38. https://doi.org/10.1016/j.neuroscience.2015.05.047.
Ma L, Hanson RL, Que LN, et al. PCLO variants are nominally associated with early-onset type 2 diabetes and insulin resistance in Pima Indians. Diabetes. 2008;57(11):3156–60. https://doi.org/10.2337/db07-1800.
Yan J, Qiao L, Peng H, Liu A, Wu J, Huang J. A novel missense pathogenic variant in NEFH causing rare Charcot-Marie-Tooth neuropathy type 2CC. Neurol Sci. 2021;42(2):757–63. https://doi.org/10.1007/s10072-020-04595-z.
Acknowledgements
The authors sincerely thank the laboratories and personnel of Urology/Pelvic Floor Surgery, West China Fourth Hospital, Sichuan University and Department of Physiology, West China College of Basic Medicine and Forensic Medicine, Sichuan University for their strong support for this research paper.
Funding
1. Sichuan Natural Science Foundation Program (Youth Science Foundation Program), hypothalamic kisspeptins/GPR54 participates in the mechanism study of bisphenol A induced primordial follicle overactivation leading to early ovarian insufficiency, 2022NSFSC12,812,022–01-01 ~ 2023–12-31, 100,000 yuan (四川省自然科学基金项目 (青年科学基金项目), 下丘脑 kisspeptins/GPR54 参与双酚A诱发原始卵泡过度激活致早发性卵巢功能不全的机制研究, 2022NSFSC1281, 2022–01-01 ~ 2023–12-31, 10万元).
2. This research was supported by grants from Sichuan Medical Association (Q20055) and Science and Technology Department of Sichuan, China (Grant no. 2022NSFSC1508).
Author information
Authors and Affiliations
Contributions
Ziyang Ma: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing-original draft, writing-review and editing, visualization.
Yi Dai: validation, formal analysis, writing-original draft, writing-review and editing, visualization.
Lei Jin: methodology, validation, investigation, data curation, writing-original draft, writing-review and editing.
Yi Luo: resources, data curation, supervision, project administration.
Chen Guo: investigation, resources, writing-original draft.
Rui Qu: investigation, resources, writing-original draft, supervision.
Shengyin He: investigation, resources, writing-original draft.
Yugao Liu: investigation, resources, writing-original draft.
Yu Xia: validation, writing-original raft, visualization.
Huan Liu: data curation, visualization.
Lingnan Kong: data curation, visualization.
Miaomiao Xu: data curation, visualization.
Lanlan Zhang: data curation, visualization.
Yue Zhao: data curation, visualization.
Suliya Yushanjiang: data curation, visualization.
Dongzhi Yuan: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing-original draft, writing-review and editing, visualization, supervision, project administration, funding acquisition.
Yang Luo: conceptualization, methodology, writing-review and editing, visualization, supervision, project administration, funding acquisition.
Corresponding author
Ethics declarations
Ethics Approval
This study has passed the review of the Ethics Committee of West China Fourth Hospital, Sichuan University and the project number is HXSY-EC-2021063.
Consent to Participate
All participating patients voluntarily participate in the research of this project after knowing the relevant risks and benefits and agree to provide personal relevant data to this institution for relevant research and thesis publication. Each patient has signed an informed consent form.
Consent for Publication
All data provided in this article does not contain any personal information, and all personal data has been anonymized. All subjects, participants, and researchers gave their consent for publication.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ziyang MaandYi Dai are co-first authors
Attestation Statement
1. The subjects in this research have not concomitantly been involved in other randomized research.
2. Data regarding any of the subjects in the study has not been previously published unless specified.
3. Data will be made available to the editors of the journal for review or query upon request.
Capsule
We found potential pathogenic genes of nIHH that acts on the peripheral.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, Z., Dai, Y., Jin, L. et al. Whole-Exome Sequencing Analysis of Idiopathic Hypogonadotropic Hypogonadism: Comparison of Varicocele and Nonobstructive Azoospermia. Reprod. Sci. 31, 222–238 (2024). https://doi.org/10.1007/s43032-023-01337-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-023-01337-2