
Abstract
Genomes are incredibly dynamic within diverse eukaryotes and programmed genome rearrangements (PGR) play important roles in generating genomic diversity. However, genomes and chromosomes in metazoans are usually large in size which prevents our understanding of the origin and evolution of PGR. To expand our knowledge of genomic diversity and the evolutionary origin of complex genome rearrangements, we focus on ciliated protists (ciliates). Ciliates are single-celled eukaryotes with highly fragmented somatic chromosomes and massively scrambled germline genomes. PGR in ciliates occurs extensively by removing massive amounts of repetitive and selfish DNA elements found in the silent germline genome during development of the somatic genome. We report the partial germline genomes of two spirotrich ciliate species, namely Strombidium cf. sulcatum and Halteria grandinella, along with the most compact and highly fragmented somatic genome for S. cf. sulcatum. We provide the first insights into the genome rearrangements of these two species and compare these features with those of other ciliates. Our analyses reveal: (1) DNA sequence loss through evolution and during PGR in S. cf. sulcatum has combined to produce the most compact and efficient nanochromosomes observed to date; (2) the compact, transcriptome-like somatic genome in both species results from extensive removal of a relatively large number of shorter germline-specific DNA sequences; (3) long chromosome breakage site motifs are duplicated and retained in the somatic genome, revealing a complex model of chromosome fragmentation in spirotrichs; (4) gene scrambling and alternative processing are found throughout the core spirotrichs, offering unique opportunities to increase genetic diversity and regulation in this group.
Similar content being viewed by others


Introduction
For most eukaryotes, the genetic sequences received from parents are used directly by the offspring. However, in some species the germline and somatic genomes differ dramatically in their composition. For example, the germ cells of sea lampreys possess the full complement of genomic DNA, whereas the somatic cells possess a smaller fraction of the germline genome, which is the result of programmed genome rearrangement (PGR) (Smith et al. 2018; Timoshevskiy et al. 2016). PGR occurs from vertebrates to protists. Examples of PGR at a genome-wide level, where specific regions throughout the germline chromosomes are eliminated during development of the somatic genome, have been described in nematodes (Müller and Tobler 2000), copepods (Kloc and Zagrodzinska 2001), hagfish (Kubota et al. 1997), foraminiferans (Lee et al. 1991), and ciliates (Jahn and Klobutcher 2002).
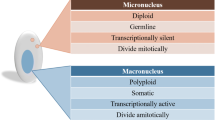
Although PGR is widespread among distinct groups of eukaryotes, it appears most pronounced in ciliates, a highly divergent group of single-celled eukaryotes that is estimated to be about one billion years old (Arnaiz et al. 2012; Chen et al. 2014; Hamilton et al. 2016; Maurer-Alcalá et al. 2018a; Wang et al. 2022; Zhao et al. 2021). Ciliates display nuclear dimorphism, where separate nuclei contain the diploid germline genome (the micronucleus, or MIC) and the polyploid somatic genome (macronucleus, or MAC) within the same cell. The MAC is transcriptionally active throughout the entire life cycle and undergoes amitotic division during asexual reproduction (Katz 2001; Prescott 1994; Seyfert and Cleffmann 1982). In contrast, the transposon-rich MIC is transcriptionally silent in vegetative cells (Chen et al. 2014; Tian et al. 2022) and divides mitotically during vegetative (or asexual) growth (Prescott 1994). During the sexual process (conjugation), the MIC becomes transcriptionally active, undergoing meiotic and mitotic events to produce both migratory and stationary haploid gametic nuclei. Conjugating pairs exchange their migratory gametic nuclei which then fuse with the stationary gametic nucleus found in the partner cell to form a zygotic nucleus (Gong et al. 2020; Jiang et al. 2019; Zhang et al. 2022). The zygotic nucleus undergoes successive mitotic divisions followed by the development of new MACs and MICs. PGR happens specifically during development of the MAC, a series of events that results in chromosomal fragmentation, DNA elimination, and DNA amplification (Gao et al. 2023; Prescott 1994; Rzeszutek et al. 2020).
The extent and pattern of PGR vary dramatically among ciliates. Tetrahymena thermophila (class Oligohymenophorea) generates 181 MAC chromosomes from five zygotic chromosomes (Sheng et al. 2020; Wang et al. 2021), eliminating ~ 12,000 germline-limited sequences (called internal eliminated sequences, IES), which account for over 30% of its germline genome sequences (Fass et al. 2011; Hamilton et al. 2016). In comparison, ciliates in the class Spirotrichea (spirotrichs, the focus of this study) process their genomes much more extensively. Oxytricha trifallax, for example, generates ~ 19,000 “gene-sized” nanochromosomes from ~ 100 zygotic chromosomes, eliminating over 200,000 IESs (~ 90% of the germline genome) (Chen et al. 2014; Lindblad et al. 2019). The mean length of the resulting nanochromosomes is only 3.2 kb, with each containing a minimal amount of non-coding DNA outside the coding domains, untranslated regions, and telomeres. PGR in this species is also more complicated, with 20–30% of the macronucleus-destined sequences (MDS, the sequences retained in the somatic genome) “scrambled” in a non-canonical order in the germline genome, requiring complex genome rearrangements to form a functional MAC genome. Even for species in the same class, the patterns of PGR can be quite different. For example, the somatic genomes of the spirotrich genus Euplotes (e.g., E. vannus and E. woodruffi) have similar size and structure to, but larger MDSs than, O. trifallax (Chen et al. 2014, 2019; Feng et al. 2022). Furthermore, only about 4–7% of the genes in the germline genome are scrambled in Euplotes, far lower than the 20–30% seen in Oxytricha.
To expand our knowledge of the diversity of genome structures and the evolution of complex genome rearrangements, we focus on spirotrichs which are characterized by having germline genomes and highly fragmented somatic genomes with gene-sized ‘‘nanochromosomes”. In the present study, we provide the first insights into the structure of the germline genome of Strombidium cf. sulcatum and Halteria grandinella. Strombidium cf. sulcatum belongs to the subclass Oligotrichia and is one of the best known marine microzooplankton species as it plays important roles in food webs and energy flow in marine pelagic waters (Song et al. 2020; Wang et al. 2021). Halteria grandinella is a cosmopolitan species that lives predominantly in freshwater habitats and is typically planktonic. It was recently shown to be closely related to hypotrichs based on both phylogenetic and phylogenomic analyses (Gao et al. 2016; Wang et al. 2019), although it shares similar morphological features with oligotrichs which might result from convergent evolution to adapt to a planktonic lifestyle. We also report the first somatic genome of S. cf. sulcatum and compare its features with H. grandinella and two other model ciliates in the same class, namely E. vannus (subclass Euplotia) and O. trifallax (subclass Hypotrichia). Specifically, comparing the somatic and germline genome sequences in S. cf. sulcatum and H. grandinella allows us to compare the features of PGR in these species with those in E. vannus and O. trifallax. This comparison offers detailed insights into the diversity and architecture of PGR in ciliates with gene-sized somatic chromosomes.
Materials and methods
Cell isolation and nucleic acid extraction
Halteria grandinella was isolated from a freshwater pond in Baihuayuan Park (36°04ʹ N, 120°22ʹ E) in Qingdao, northern China. Strombidium cf. sulcatum was collected from surface seawater in Daya Bay (22°37ʹ N, 114°38ʹ E) in Huizhou, southern China. Species were identified by morphological features and small subunit ribosomal RNA gene sequences. Single cells of each species were isolated, washed, and cultivated at room temperature (ca. 25 °C) in flasks containing autoclaved habitat water. Escherichia coli strain DH5α was used as the food source. All downstream experiments were based on clones produced by asexual reproduction of a single cell. Cells were starved for 48 h before DNA or RNA extraction to reduce contamination by E. coli and other bacteria. Cells were harvested by centrifugation at 300 g for 5 min. DNA was extracted using phenol chloroform extraction followed by ethanol precipitation. RNA extraction was performed with the Rneasy Plus Mini Kit (Qiagen, Germany, Cat No. 74134). QIAGEN REPLI-g Single Cell Kit (Cat No. 150345) was used for MIC genome amplification from each single cell with Multiple Displacement Amplification (MDA). All operations followed the manufacturer’s instructions.
High-throughput sequencing
DNA libraries were generated using Truseq Nano DNA HT Sample Preparation Kits (cat. FC-121-2003, Illumina USA) following the manufacturer’s recommendations. Briefly, the DNA sample was fragmented by sonication to a size of 350 bp. The DNA fragments were end-polished, A-tailed, and ligated with the full-length adapter for Illumina sequencing with further PCR amplification. PCR products were purified (AMPure XP system) and libraries were analyzed for size distribution by Agilent2100 Bioanalyzer and quantified using real-time PCR. The RNA libraries were generated using NEBNext Ultra RNA library prep kits for Illumina (cat. E7530, NEB, USA) following the manufacturer’s instructions. All the samples were sequenced based on the Illumina Hiseq PE150 platform. In total, we sequenced at least 20 Gb raw data for each DNA sample and 10 Gb raw data for each RNA sample. The reads produced by the high-throughput sequencing machine (Illumina) were evaluated and processed by FastP (v0.20.0, -c -detect_adapter_for_pe -l 75) to discard low-quality reads (Chen et al. 2018).
Genome assembly and cleanup
DNA reads were assembled by SPAdes (v3.13.0) with “-k 21,33,55,77--careful”. “--sc” was additionally enabled for Multiple Displacement Amplification (MDA) data to deal with non-uniform coverage and to remove potential chimeras (Bankevich et al. 2012; Nurk et al. 2013; Papudeshi et al. 2017; Prjibelski et al. 2020; Xu and Zhao 2018). GapCloser (v1.1.2, a module of SOAPdenovo2) was employed to fill gaps in assemblies (Luo et al. 2012). QUAST (v4.6.3) was used to obtain N50, GC content and other genomic statistics (Gurevich et al. 2013). CD-HIT (v4.6.1, cd-hit-est -c 0.98 -r 1 -n 10) was applied for removing redundant sequences. The telomeres were identified using a custom Perl script (Code S1) that recognized the telomeric repeats at the ends of contigs (regular expression pattern of Perl: /[AC]*CCCCAA[AC]*CCAAAA/ and /GGGGTT[GT]*TTGGGG[GT]*/). Poorly supported contigs (coverage < 1) in the assemblies were also discarded. Mitochondrial genomic sequences of ciliates and the dataset of bacterial sequences were downloaded from GenBank as a BLAST database to identify contamination caused by mitochondria or bacteria (BLAST v2.10.0 + , cutoff: E-value <1 e–10) (Camacho et al. 2009). For the MAC assemblies of S. cf. sulcatum, RNA-seq data were mapped to the assembly using HISAT2 (v2.1.0, --dta) (Kim et al. 2015). All contigs without telomeres or any RNA read mapping were discarded. Short (< 400 bp) contigs, except for those with both telomeres, were also removed as poorly supported sequences. For the MIC assemblies, three replicates of H. grandinella and six of S. cf. sulcatum were assembled separately and reassembled using CAP3 with strict overlap parameters (-o 50 -p 99) (Huang and Madan 1999) to obtain more genomic data (Lyu et al. 2023). Potential MAC contamination in the MIC assemblies, which are unavoidable during amplification, were identified using BLAST (Camacho et al. 2009) (v2.10.0 + , cutoff: E-value < 1e–10, identity > 97%, qcov > 95%) and removed by a custom Perl script (Code S2).
Transcriptome assembly and gene prediction of the S. cf. sulcatum MAC genome
Transcripts were assembled using StringTie (v2.2.1) (Pertea et al. 2015). The S. cf. sulcatum genome assembly with all the identified introns removed was aligned against the Swiss-Prot database using BLAST (v2.10.0 + , blastx -query_gencode 6 -evalue 1e–5) (Camacho et al. 2009) to identify open reading frames (ORFs). The conventional stop codons TAA and TAG were modified to encode glutamine, as in other oligotrich and hypotrich ciliates (Swart et al. 2016). For the transcripts without ORFs identified, TransDecoder (v5.5.0, -G Ciliate, https://github.com/TransDecoder/TransDecoder/) was employed to extract the long ORFs and predict the likely coding regions of them. The ORFs identified by BLAST alignments were then used for building a training set for AUGUSTUS (Stanke et al. 2008), which was used to perform de novo gene prediction for the genes without assembled transcripts. Ultimately, 2078 S. cf. sulcatum genes were used to train AUGUSTUS. The transcripts assembled by StringTie were used to produce additional constraints (hints) for AUGUSTUS. For gene prediction, AUGUSTUS was run with the following parameters: “--alternatives-from-evidence = true --enemodel = complete --min_intron = 18”. Predicted protein products were annotated by alignment to domains in the Pfam-A database by InterProScan (Jones et al. 2014) and to the SwissProt database by DIAMOND (Buchfink et al. 2015) (-evalue 1e–5). Gene Ontology (GO) terms were enriched by using the BINGO (Maere et al. 2005) plugin (v3.0.3) in the Cytoscape platform (v3.6.0) (Shannon et al. 2003). The corrected (corr) p-values were derived from a hypergeometric test followed by Benjamini and Hochberg false discovery rate (FDR) correction. An FDR ≤ 0.05 was regarded as significant.
TBE/Tec detection and estimation of substitution rates
Transposable elements were detected as described in other ciliates (Chen and Landweber 2016; Feng et al. 2022). Briefly, representative Oxytricha TBE ORFs (Genbank accession AAB42034.1, AAB42016.1, and AAB42018.1) were used as queries to search TBEs in the S. cf. sulcatum and H. grandinella MIC genomes by TBLASTN (-db_gencode 6 -evalue 1e–5 -max_target_seqs 100,000). Tec ORFs were similarly detected by using E. crassus Tec1 and Tec2 ORFs as queries (-db_gencode 10 -evalue 1e–5 -max_target_seqs 100,000, Genbank accessions of Tec ORFs are AAA62601.1, AAA62602.1, AAA62603.1, AAA91339.1, AAA91340.1, AAA91341.1, AAA91342.1). Regions containing three ORFs in proximity (within 2 kb of each other) and in the correct orientation were annotated as complete TBEs, while those that did not contain all three ORFs were annotated as partial TBEs. ParaAT (v2.0, -c 6 -f axt -k) (Wang et al. 2010) and KaKs_Calculator (-m GY) (Wang et al. 2010) were employed to calculate the ratios of nonsynonymous to synonymous rates (dN/dS).
Biased nucleotide distribution
All two-telomere contigs were rearranged according to gene direction after removal of telomeric sequences. Each contig was sliced into 250 bins from the 5ʹ end to the 3ʹ end, and the frequencies of A, T, G, and C in each segment were calculated. The average frequency of the four nucleotides for each segment was counted in all chromosomes. 50 bp of the subtelomeric region at each end were extracted and a sliding window of 10 bp with a step of 2 bp was used to calculate the AT content. The same regions were used to analyze base compositions using WebLOGO3 (Crooks et al. 2004).
Genome rearrangement analysis
The MAC assembly was aligned using BLAST against the MIC assembly to analyze the rearrangement patterns between MIC and MAC genomes after trimming telomeric repeats. The whole pipeline was similar to, but more moderate than, the MIDAS (http://knot.math.usf.edu/midas/), which was originally developed to annotate MDS/IES. The masked BLAST database was created with the MIC assembly using windowmasker (Morgulis et al. 2006). Two steps of BLAST and a custom Perl script (Code S3) were applied to generate High-Scoring Pairs (HSPs): megablast was employed first (-word_size 28 -ungapped -evalue 1e–5); then blastn-short (-word_size 12 -ungapped -evalue 1e–5) was used immediately to search smaller MDSs for any gap between HSPs after megablast. Briefly, the main steps of Code S3 were: (1) HSPs with canonical “MDS-pointer-MDS” structures were identified as MDSs; (2) MDSs which shared the same pointer were temporarily connected to longer ones; (3) all adjacent MDSs were checked, and nested ones were discarded; 4) finally, the previously merged HSPs were restored to the original status, and HSPs were identified as MDSs on both MAC and MIC. The MIC-limited sequences between two MDSs were identified as IESs. The overlaps of two adjacent MDSs on MAC sequences were denoted as pointers. Scrambled chromosomes referred to those with MDSs in different orders or orientations, and the genes encoded by them were identified as scrambled genes. Chromosome breakage sites (CBSs) were identified as the regions between the subtelomeric MDSs from different MAC chromosomes that mapped to the adjacent regions of the MIC genome. The composition of bases of the most abundant pointers and CBSs was displayed using WebLOGO3 (Crooks et al. 2004). The rearrangement patterns of chromosomes encoding the same single-copy homologous gene “Calcineurin-like phosphoesterase” in the four species were displayed using LINKVIEW (https://github.com/YangJianshun/LINKVIEW).
Alternative processing
Two types of alternative fragmentation were identified based on the architecture of MDSs and IESs on germline contigs: (A) the same MDS region in micronuclear DNA was processed into multiple, distinct somatic chromosomes; (B) the MDS from one chromosome was nested by IES from another chromosome. MAC contigs involved in alternative fragmentation were aligned with the NCBI non-redundant protein sequence database after the removal of introns (DIAMOND-blastx -query-gencode 6 -e 1e–2) (Buchfink et al. 2015). The best hit of each query contig was retained.
Identification of homologous IESs and genes
The homologous genes of S. cf. sulcatum, H. grandinella, E. vannus and O. trifallax were clustered using OrthoVenn2 (Xu et al. 2019) with an E-value threshold of 1e–5. To identify the IESs with homologous excised sites, the homologous proteins were aligned using MAFFT (v7.487, --globalpair) (Katoh and Standley 2013). The locations of pointers that locate at amino acids with the same position in alignments were identified as homologous IES-excised sites. All the IES-excised sites involved in alternative fragmentation were also removed. Another category of homologous IESs were identified based on their sequence similarities without considering their locations. All the IESs identified in the germline genomes were clustered using CD-HIT (v4.6.1, cd-hit-est -c 0.9 -s 0.9) (Fu et al. 2012) after the removal of IESs with homologous excised sites. The list of clusters outputted in a text file was processed using a custom Perl script (Code S4) to analyze the homologous IESs.
Results
Genome assembly and features
Following removal of low-quality reads, nearly identical contigs, and contaminating mitochondrial and bacterial genomes from the initial sequencing of S. cf. sulcatum, we assembled a somatic MAC genome of 71.3 Mb (Table 1; Fig. 1). The overall GC content (51.6% of all contigs and 50.7% for two-telomere contigs, Fig. 1C) is higher in this species than all other spirotrichs included in the present analysis (Table 1; Supplementary Table S1) (Chen et al. 2019; Swart et al. 2013; Zheng et al. 2021). The final assembly of the S. cf. sulcatum MAC genome contains 20,086 contigs with telomeric repeats of C4A4 and G4T4 at both ends (avg. 1611 bp, Fig. 1B), indicating a highly fragmented macronuclear genome. Complete (two-telomere) chromosomes in S. cf. sulcatum range from 257 bp to 15,209 bp. An additional 15,744 contigs contain only one telomere (avg. 1478 bp, Fig. 1B) and contigs with at least one telomere comprise 73.7% of the assembly. About half of the telomeres sequenced on the ends of each chromosome in S. cf. sulcatum are 20 bp, with a range from 12 to 141 bp (Supplementary Fig. S1A). Among all two-telomere chromosomes, ~ 78% are shorter than 2000 bp, a higher percentage than is seen in other spirotrichs with “nanochromosomes”, such as Strombidium stylifer (~ 54%), Halteria grandinella (~ 61%), Oxytricha trifallax (~ 35%) and Euplotes vannus (~ 50%).

Macronucleus (MAC) genome assembly and features of chromosomes and introns of Strombidium cf. sulcatum. A The schema illustrates the canonical structure of nanochromosomes in the MAC of S. cf. sulcatum. B The size distribution of contigs with 0, 1 or 2 telomeres in S. cf. sulcatum. C Correlation between the GC-content (%) and mean base depth of each contig. X-axis shows the GC-content (%) and Y-axis shows the mean base depth. Each dot represents one contig. Scaffolds with either 0/1/2 telomeres share a similar GC-distribution. D The size distribution of 6240 identified introns. E The base composition of 10 nt at both ends of each intron. F Nanochromosomes with complete genes were assessed for gene content; only 3.7% of nanochromosomes contain two genes and 0.7% contain three or more genes. G The proportion of cis-nanochromosomes (two genes located on the same DNA strand) and trans-nanochromosomes (two genes located on opposite strands)
The final somatic genome assembly is predicted to contain 47,569 protein-coding genes, along with 71 tRNA genes covering the standard 20 amino acids. A model for nanochromosome structure in the MAC of S. cf. sulcatum is shown in Fig. 1A. Although a small proportion of the nanochromosomes encode two (827 nanochromosomes) or more genes (109 nanochromosomes), 95.6% of S. cf. sulcatum nanochromosomes encode only one gene each (Fig. 1F). Most of the two-gene nanochromosomes of S. cf. sulcatum encode genes on the same strand (described as “cis- nanochromosomes”, Fig. 1G), which is similar to Euplotes (62–89%), but different from Tetmemena (57%), Halteria (49%), Oxytricha (41%) and Stylonychia (36%) (Supplementary Table S2). The single-gene nanochromosomes contain very short non-coding regions while protein-coding regions account for an average of 82% of the entire chromosome. In total, 87.7% of RNAseq reads could align with the final assembly. Potential transcription start sites (TSS) and transcription end sites (TES) of each gene were identified based on the mapping boundary of RNAseq reads. The region between the telomere and the TSS averages 48 bp and TESs are on average 47 bp from the downstream telomere (Supplementary Fig. S2A). Moreover, the introns of S. cf. sulcatum are also small in size, with a principal size distribution between 28 and 42 bp (Fig. 1D). Canonical splice site dinucleotides (5ʹ-GT…AG-3ʹ) exist at the end of these introns, with a weaker but still obvious bias of 5ʹ-AAGT-3ʹ at the 3rd to 6th nucleotides from the 5ʹ end (Fig. 1E).
Partial germline MIC genomes of S. cf. sulcatum and H. grandinella were acquired using single-cell whole-genome amplification techniques (Table 2). We obtained 135 Mb and 225 Mb of the MIC genome in S. cf. sulcatum and H. grandinella, respectively. The MIC assemblies contain 53.1% and 42.2% of the nucleotides assembled into MAC contigs in S. cf. sulcatum and H. grandinella, respectively. We also annotated 214 complete transposable elements (TEs) with telomere-bearing elements (TBEs) of the Tc1/mariner family, as well as 10,983 partial TBEs and 30 partial Tec elements (transposon identified in Euplotes) in the MIC assembly of H. grandinella (Supplementary Table S3). 101 partial TBEs were also found in the MIC assembly of S. cf. sulcatum. The overall dN/dS ratios of TEs are lower than 1 and mostly in the range of 0.1–0.4 (Supplementary Fig. S1E), suggesting purifying selection acting on them, which is similar to a previous study (Chen and Landweber 2016).
Orthologous macronuclear genes in the four species of the class Spirotrichea
We analyzed the orthologous MAC genes of H. grandinella, S. cf. sulcatum, O. trifallax and E. vannus using OrthoVenn2 (Xu et al. 2019) to gain an insight of their genetic relationships. The four species share 1309 orthologous clusters which include 9084 genes (Fig. 2A, B). Generally, H. grandinella, S. cf. sulcatum and O. trifallax share many more orthologous genes with each other than with E. vannus. Halteria grandinella and O. trifallax specifically share the most orthologous groups (1845 clusters with 5661 genes), followed by that between S. cf. sulcatum and O. trifallax (1125 clusters with 3640 genes). Comparatively, S. cf. sulcatum shares fewer orthologous genes with H. grandinella (90 clusters with 450 genes) and E. vannus (153 clusters with 807 genes). The result of pairwise comparison of orthologous genes among the four species also shows that H. grandinella and O. trifallax share the most orthologous genes (Fig. 2C). In H. grandinella, more than one third (34.3%) of the genes are homologues to those in O. trifallax. In S. cf. sulcatum, 17.7% of the genes are homologues to those in O. trifallax, 12.3% to those in H. grandinella and 9.2% to those in E. vannus.

Homologous genes of Halteria grandinella, Strombidium cf. sulcatum, Oxytricha trifallax and Euplotes vannus. A Homologous clusters of the four species. The digits indicate the number of homologous clusters. B Number of homologous genes and clusters in each combination between the four species. Blocks on the left represent the quantity of genes, the darker the blocks the higher the number of genes. Bar graph refers to the proportion of genes from species indicated by the right blocks. C Pairwise comparison of genes among the four species. Each block indicates the number of homologous genes. The digits indicate the percentage of homologous genes in species with an asterisk (*) labeled. D Morphology of the four species involved in this study. Arrows indicate micronuclei
Chromosome breakage sites are retained in the somatic genome during PGR
An important event that occurs during MAC development is extensive chromosome fragmentation at chromosome breakage sites (CBSs). CBSs have been found to be duplicated and retained in the MAC genome in E. vannus and O. trifallax (Chen et al. 2019). To investigate the chromosome breakage mechanism in S. cf. sulcatum and H. grandinella, we analyzed the subtelomeric region of their complete MAC chromosomes and the corresponding MIC sequences to identify the CBS boundaries, using the same method as described in the study of E. vannus (Chen et al. 2019). Briefly, the CBS region is delimited by the end of the upstream MAC chromosomes (denoted as “m”) and the beginning of the downstream MAC chromosomes (denoted as “n”) in the germline sequences. The distance between two adjacent CBS boundaries is calculated using the value of “n – m” (Fig. 3A). A positive value indicates that the CBS is not retained in the MAC genome whereas a negative value means that the CBS is retained in the MAC genome.

Chromosome breakage site (CBS) models of Halteria grandinella, Strombidium cf. sulcatum, Oxytricha trifallax and Euplotes vannus. A The cartoon shows models for CBS retention (top) or elimination (bottom). “m” and “n” denote the ends of two adjacent MAC chromosomes, corresponding to the breakage points “m” and “n” in the MIC genome. The CBSs are retained in somatic chromosomes if n–m < 0, while they are deleted in the case of n–m > 0. B–E The putative CBS size distribution and base composition of CBSs in the most abundant size and flanked sequences in the four species. Dashed boxes in (B–E) denote CBSs (B–E) and nearby consensus elements (B–D)
In total, we identified 95 and 1033 predicted CBS regions in S. cf. sulcatum and H. grandinella, respectively. For most CBS regions in both species, the value of “n–m” is negative, showing that the CBS region is retained in both adjacent MAC chromosomes following genome rearrangement (Fig. 3B–E). Halteria grandinella shares the most common motif (5ʹ-AT-3ʹ) found in CBSs of O. trifallax, but also possesses larger CBSs between 5 and 30 bp, which are not prominent in O. trifallax or E. vannus. However, similar to the 2 bp ‘AT’ pattern, most of these larger CBSs begin with an “A” and end with a “T”. The 30 bp flanking standard CBSs were analyzed for conserved sequence motifs using WebLogo (Crooks et al. 2004). In H. grandinella, these flanking regions are AT-rich, with two complementary motifs located upstream and downstream of the CBSs. This pattern is similar to those in O. trifallax and E. vannus, though the conserved motifs are different (Chen et al. 2019).
In this study we were unable to obtain as many CBSs for S. cf. sulcatum as for the other three species. However, an initial check of the regions surrounding conserved 17 bp and 22 bp motifs, for which there were more than five examples, showed reverse complement motifs at the flanking 9th to 11th bases in either direction.
To verify that the CBSs were indeed retained in the MAC genome, we focused on sequences in the subtelomeric regions of the complete nanochromosomes. The 5ʹ–3ʹ orientation of the gene found on each chromosome was determined, then the sequence was subdivided into 250 equal bins. The average content of the four nucleotides (A/T/C/G) in each bin was calculated (Fig. 4). An AT-rich bias was found in the subtelomeric regions, especially for the first and last 5% of the full length of chromosomes. We then analyzed the AT-content within the 100 bp adjacent to each telomere, revealing obvious AT hotspots at approximately 10 bp (S. cf. sulcatum), 20 bp (E. vannus), and 30 bp (H. grandinella and O. trifallax). These AT hotspots are independent of the gene orientation, and are present at both the 5ʹ and 3ʹ ends. The subtelomeric regions were extracted and searched for conserved sequences in each species. A highly conserved motif, 5 ʹ-GAA-3ʹ, was detected at the 9th to 11th nucleotides in S. cf. sulcatum chromosomes (Fig. 4A). A similar pattern, 5ʹ-TTGAA-3ʹ, was found in nucleotides 18–22 in E. vannus (Fig. 4B). By contrast, H. grandinella and O. trifallax also have an AT-rich region, but lack a conserved sequence motif at either end. These results match the sequences seen in the CBS regions in the MIC genome. Based on these observations, we conclude that all four spirotrichs share a similar retained mode of CBS breakage, with greater sequence specificity required for H. grandinella and S. cf. sulcatum than for O. trifallax and E. vannus.

The nucleotide bias among somatic nanochromosomes and subtelomeric regions. A–D The top panels of each species show the nucleotide bias of nanochromosomes. All contigs capped with 2-telomeres were split into 250 bins after the removal of telomere sequences. The nucleotide composition of each bin was calculated and is shown as a line chart. AT-rich regions were found at both ends of the chromosomes. The terminal 100 bp of the subtelomeric regions were extracted to count the AT-content using a sliding window of 10 bp and step size of 1 bp, shown as heatmaps in the middle panels. Each row of the heatmap reflects one chromosome. The AT-content of each window is represented by the color range from warm (red) to cold (blue). Seqlogos in the bottom panels show the conserved bases of the subtelomeric region. A distinct boundary (blue arrows in A, B) in the heatmaps indicates the highly-conserved sites at the 8th to 10th nucleotides (A) and 18th to 22nd nucleotides (B) from both ends of the vast majority of nanochromosomes
We also considered whether the size and location of these AT-rich regions in S. cf. sulcatum may play additional roles in either transcription or translation of nanochromosome genes. Most of these regions are very short, but appear to be larger at the end containing the 5ʹ portion of the gene (avg. 17 bp for 5ʹ end vs. avg. 11 bp for 3ʹ end, Supplementary Fig. S2). The mean distance from the TSS (transcription start site) to the 5ʹ-telomere in S. cf. sulcatum is 48 bp, and 47 bp from TES (transcription end site) to 3ʹ-telomere. For single-gene chromosomes where the AT-rich regions were sequenced at both ends, more than 80% (14,391 of 17,741) of the TSSs and at least 85% of the start codons (“AUG”) on these chromosomes are located after the AT-rich region. This result was also observed in other three species (Supplementary Table S4). Since ciliates in general appear to lack a conserved TATA-box element (Brunk and Sadler 1990) and these low-complexity AT-rich regions are instead found in the chromosome where it would be expected, we speculate that these are retained in the MAC chromosomes to regulate transcription after extensive chromosome fragmentation.
General patterns of genome rearrangements between somatic and germline genomes
The rearrangements that process the germline MIC genome during conjugation to produce the somatic MAC genome are a significant feature of ciliates (Chen et al. 2014). The patterns of rearrangement for S. cf. sulcatum and H. grandinella were determined and compared with those of two other spirotrichs (E. vannus and O. trifallax), whose somatic and germline genomes are both available. Considering that assemblies of both Strombidium and Halteria are based on short reads, we used the assemblies of Euplotes and Oxytricha that are also based on short-reads sequencing technology to make the results more comparable.
MDSs in the somatic genome assembly were identified by BLAST against the germline genome assembly. The median size of the identified MDSs is 648 bp in S. cf. sulcatum, 179 bp in H. grandinella, 348 bp in E. vannus, and 169 bp in O. trifallax. More than 80% of the MDSs are shorter than 500 bp in H. grandinella (83.5%) and O. trifallax (87.5%). However, that proportion is only 38.7% in S. cf. sulcatum and 64.3% in E. vannus (Fig. 5A). To estimate the density of MDSs, we calculated the MDS count per kilobase of a MAC contig (MPKA). The median value of MPKA is 1.45 in S. cf. sulcatum, 1.96 in E. vannus, 4.34 in H. grandinella, and 4.19 in O. trifallax, which indicates that the somatic nanochromosomes of E. vannus and S. cf. sulcatum generally consist of fewer and longer MDSs than H. grandinella and O. trifallax (Fig. 5B). This means that the somatic genomes of S. cf. sulcatum and E. vannus are less fragmented.

Comparison of genome rearrangements among Halteria grandinella, Strombidium cf. sulcatum, Oxytricha trifallax and Euplotes vannus. A, C, D The size distribution of scrambled and unscrambled MDSs, IESs and pointers of the four species. Data in (A, C) are displayed in two scales. The seqlogos in (D) refer to sequence motifs of the most abundant pointers in each species. B The distribution of MDS density of each MAC contig in the four species. MPKA: MDS count per kilo bases of a MAC contig
After characterizing MDSs in these species, we performed similar calculations for the IESs. The average length of an IES is shorter than an MDS, and the diversity in size among species is not as pronounced as it is for MDSs (Fig. 5C). The median IES size we identified was 180 bp in S. cf. sulcatum, 172 bp in E. vannus, 104 bp in H. grandinella and 73 bp in O. trifallax, though it is possible that the small size of IESs may be biased by short-reads sequencing. In order to understand the evolution of IESs in spirotrichs, we compared homologous IESs across the four species. We first identified IESs inserted at homologous sites in homologous genes among all four species (“conserved IESs”). Halteria grandinella (0.9%, 247 of 27,705) and S. cf. sulcatum (1.3%, 52 of 3857) contain few conserved IESs. However, both O. trifallax (14.9%, 34,741 of 232,861) and E. vannus (15.6%, 2471 of 22,317) show ten times of this number. The majority of conserved IESs in all species are found in paralogous genes within the same species (99.5% in E. vannus, 84.6% in S. cf. sulcatum, 73.4% in H. grandinella and 70.9% in O. trifallax). Comparing orthologous gene clusters with conserved IES across species, nearly all (77/78) are shared by O. trifallax and another species: O. trifallax and H. grandinella (61; 78.2%), O. trifallax and S. cf. sulcatum (9; 11.5%), and O. trifallax and E. vannus (7; 9.0%).
We then searched for IESs that share high sequence similarity, but are located in different (nonhomologous) loci (“mobile IESs”). There are few mobile IESs in H. grandinella (1.1%, 374 of 35,048), E. vannus (1.8%, 397 of 22,019) and S. cf. sulcatum (2.7%, 220 of 8173), but they make up a large portion of the IES population in O. trifallax (31.5%, 60,330 of 191,263). In total, we identified 207 mobile IESs shared among the four species, with more than a half (117, 56.5%) of these shared by H. grandinella, S. cf. sulcatum and E. vannus (Supplementary Table S5).
Pointers are short sequences repeated in the MDS-IES junctions that flank an IES in the MIC, but are present in only one copy in the MAC following IES removal. Pointers were identified based on overlaps of two adjacent MDSs on somatic sequences. Most pointers in these four species are AT-rich sequences shorter than 10 bp (Fig. 5D). Most E. vannus pointers show a highly conserved 5ʹ-TA-3ʹ motif that has been observed in Paramecium species (Steele et al. 1994). Halteria grandinella, S. cf. sulcatum, and O. trifallax pointers are all more diverse, though roughly half of the pointers in each species contain at least one 5ʹ-ANT-3ʹ motif (41.9%, 47.8%, 50.6%, respectively).
Variable levels of gene scrambling in the germline genome
Many MAC chromosomes (termed here “scrambled chromosomes”) in spirotrichs require a number of unscrambling events during PGR, defined as either insertion or inversion of MDSs (Fig. 6B–D). Scrambled chromosomes in the MAC genome were identified as those with one or more MDSs which are out of order in direction or position when compared with the MIC genome (Fig. 6B, C). Chromosomes with an inserted MDS from another MIC contig (Fig. 6D) are considered “insertions” (shown as Fig. 6C) in the following analysis.

Scrambled genome rearrangement patterns in Halteria grandinella, Strombidium cf. sulcatum, Oxytricha trifallax and Euplotes vannus. A The most common pattern of unscrambled gene rearrangement. Colorful blocks indicate the MDSs, grey boxes represent the IESs. B–D Scrambled gene rearrangement models. The MDSs in (B) are in different orientations and the ones in (C) are in reverse order. The MIC-1 and MIC-2 in (D) represent different MIC chromosomes, or may be interrupted as a result of insufficient sequencing and is regarded as a case of (C) in the downstream analysis. E The number of scrambled chromosomes as shown in (B–D). “Concurrent” refers to chromosomes that follow either the (B) or C/D pattern. F The patterns of alternative processing events. A-type: the same MDSs in the micronuclear DNA are processed into multiple distinct somatic chromosomes. B-type: the MDS from one chromosome is nested by, or overlaps with, the IES from another chromosome. G The proportion of alternatively processed genes involving A-type and B-type. H An example case of alternative fragmentation in Euplotes vannus. The MDSs on one MIC contig are shared by five MAC contigs (MAC 2–5 contained both telomeres; MAC-1 had only one telomere. MDS2 is nested by MDS3). I The rearrangement patterns of chromosomes encoding homologs of the single-copy gene “Calcineurin-like phosphoesterase” in the four species. Other areas of the MIC are represented by short lines that are not to scale
All four species in this study show some degree of gene scrambling, although the levels varied widely. The highest percentage of scrambled chromosomes is in O. trifallax (29.2%, 5285 of 18,111), followed by S. cf. sulcatum (15.9%; 2901), H. grandinella (12.9%, 1693), and E. vannus (4.2%, 1387). The ratio of inversion to insertion events also varies among species. Scrambled nanochromosomes in S. cf. sulcatum resulting from an inversion of MDSs make up 69.4%, with 17.6% insertions, and 13.0% “concurrent” events where both occur simultaneously (Fig. 6E). Inversions are also highest in E. vannus (45.0%) and H. grandinella (37.7%), with insertions higher than concurrent events. During development, however, O. trifallax shows a higher rate of insertion (63.8%) than inverted (21.4%) or concurrent (14.7%).
Scrambled MDSs (13,588) accounted for 28.6% of all MDSs in S. cf. sulcatum. This is higher than the proportion in O. trifallax (48,689, 19.4%), which contains far more MDSs. Scrambled MDSs make up 10.6% (9569) of the MDSs in H. grandinella and only 4.2% (5201) in E. vannus. Scrambled MDSs are typically shorter than unscrambled ones. No significant difference was observed between the size of scrambled and unscrambled IESs in the four species. All four species contain more MDSs in scrambled than unscrambled nanochromosomes (S. cf. sulcatum: 2.1/kb scrambled, 1.3/kb unscrambled; H. grandinella: 4.9/kb scrambled, 4.2/kb unscrambled; E. vannus: 2.8/kb scrambled, 1.9/kb unscrambled; O. trifallax: 6.4/kb scrambled, 3.6/kb unscrambled).
Gene Ontology (GO) analysis of scrambled genes was performed with BiNGO (Maere et al. 2005). In S. cf. sulcatum, scrambled genes are enriched significantly for signal transduction (GO:0007165, P-value 6.9943 e–7), transmembrane transporter activity (GO:0022857, P-value 8.9672 e–3) and kinase activity (GO:0016301, P-value 1.4298 e–2). The scrambled genes in E. vannus are enriched significantly in copper ion binding (GO: 0005507, P-value 3.0387 e–2) and those in O. trifallax are enriched in lipopolysaccharide biosynthetic process (GO: 0009103, P-value 1.8318 e–2). No significant enrichments of scrambled genes were found in H. grandinella.
Diverse frequency of alternative processing in Spirotrichea
To explore more diverse DNA rearrangement patterns, we identified a more complex form of DNA processing, i.e., “alternative processing”, which occurs during PGR and is analogous to RNA alternative splicing. Alternative processing was reported in O. trifallax (Braun et al. 2018; Chen et al. 2014, 2015; Lindblad et al. 2019) and the phyllopharyngean ciliate Chilodonella uncinata (Gao et al. 2014, 2015; Katz and Kovner 2010). We identified two types of alternative processing (Fig. 6F). In the “A-type”, the same MDS regions in micronuclear DNA are processed into multiple, distinct somatic chromosomes. In the “B-type”, the MDS from one chromosome is nested within the IES from another chromosome. We observed a relatively high frequency of alternative processing in O. trifallax and E. vannus, as well as some cases in S. cf. sulcatum and H. grandinella. Generally, the A-type cases are much more abundant than the B-type.
Among genes whose MDS structure has been defined in O. trifallax, 7130 involve A-type processing and 236 involve B-type (Fig. 6G). In E. vannus, only 4011 genes involve A-type processing and 16 involve B-type. Very few cases of alternative processing were observed in H. grandinella and S. cf. sulcatum. In H. grandinella, 269 involve A-type and 15 involve B-type. Strombidium cf. sulcatum has only 33 A-type processing events and 54 B-type. These numbers could be underestimated because we used a very strict pipeline to identify the IESs and only non-scrambled IESs are considered here. Furthermore, short contigs may prevent identification of some cases.
About 70%-90% alternative processing events disrupt the coding regions and about 75%-95% of the alternatively processed nanochromosomes are transcribed (Supplementary Fig. S3). The number of transcribed genes could be underestimated, considering that only vegetative cells were used for RNA-seq while some genes could have developmental or stress-induced expression. One of the most diverse cases we observed is shown in Fig. 6H, where one MIC contig is processed into five MAC contigs in E. vannus. Four of the five contigs sequenced have two telomeres, while the other has only one. Sequence similarity among them is low except for the regions from the same MIC contig. Of the five MAC contigs, four have only one detected coding region, which is predicted to encode a G1/S-specific cyclin-E protein. The other contains the G1/S-specific cyclin-E gene along with a gene predicted to encode a SWI/SNF complex component snf12 homolog. Cyclin and snf12 homologs are typically involved in the cell cycle. We also found that the MAC contigs alternatively processed from the same MIC contigs generally belong to the same gene families (80.6% in E. vannus, 92.4% in O. trifallax, 73.9% in H. grandinella and 68.4% in S. cf. sulcatum), probably due to the repeated usage of the same gene regions.
Discussion
In this study, we assembled a high-quality MAC genome of the spirotrich ciliate Strombidium cf. sulcatum (which contains “gene-sized” nanochromosomes) and partial germline genomes of S. cf. sulcatum and Halteria grandinella. We characterized the PGR features of S. cf. sulcatum and H. grandinella by comparing their somatic genome to their germline genome. These features were compared with the two sequenced ciliates in the same class, Euplotes vannus and Oxytricha trifallax, to provide a holistic and evolutionary view of MIC and MAC structures in spirotrichs and to gain an insight of the diversity and architecture of PGR in ciliates. As a result, we demonstrated that: (1) somatic genomes comprised of “gene-sized” nanochromosomes are a common feature in a wide range of spirotrichs and those in oligotrichs are the most compact; (2) micronuclear chromosomes in spirotrichs are highly fragmented during macronuclear development at chromosome breakage sites which are duplicated and retained in the somatic genome; (3) the micronuclear-limited DNA may have originated as remnants of transposable elements or as degenerated duplicate sequences after gene duplication, some of which may have occurred prior to the last common ancestor of spirotrichs; (4) gene scrambling and alternative processing are widespread in spirotrichs and may play important roles in increasing genetic diversity.
Highly fragmented and compact macronuclear genomes are a common feature of spirotrichs
Based on the available somatic genome data, the extent of chromosomal fragmentation varies dramatically among ciliates, from limited, e.g., the well-studied genera in the class Oligohymenophorea, Tetrahymena (Sheng et al. 2020) and Paramecium (Aury et al. 2006), to extensively, e.g., species in the class Spirotrichea, Oxytricha (Swart et al. 2013), Euplotes (Chen et al. 2019) and Strombidium (present study). Gene-sized chromosomes have been reported in disparate classes other than Spirotrichea, e.g., Chilodonella uncinata in the class Phyllopharyngea and Metopus palaeformis and Nyctotherus ovalis in the class Armophorea (Riley and Katz 2001), indicating that gene-sized chromosomes originated multiple times within ciliates. Of the seven subclasses in Spirotrichea, somatic genome data have been reported in three subclasses, including Oxytricha, Tetmemena, Urostyla, Paraurostyla, Laurentiella, Sterkiella and Stylonychia in the subclass Hypotrichia (Aeschlimann et al. 2014; Chen et al. 2015; Feng et al. 2022; Swart et al. 2013), Euplotes in the subclass Euplotia (Chen et al. 2019, 2021; Feng et al. 2022; Jin et al. 2023; Mozzicafreddo et al. 2021; Vinogradov et al. 2012; Wang et al. 2016), and Strombidium in the subclass Oligotrichia (Li et al. 2021 and present study), as well as in Halteria, an extremely specialized “oligotrich‐like” hypotrich (Wang et al. 2019; Zheng et al. 2021) (Supplementary Table S1). Each of these somatic genomes is about 50 to 110 Mb in size and is composed of gene-sized chromosomes that are highly enriched with coding sequences.
In the present study, we assembled the 71.3 Mb somatic MAC genome of S. cf. sulcatum comprising more than 20,000 gene-sized nanochromosomes, of which nearly 16,000 contigs contain at least one telomere. Compared with other reported ciliates with extensively fragmented somatic genomes, S. cf. sulcatum has significantly shorter nanochromosomes, with an N50 of only 1721 bp (Supplementary Fig. S1B; Table 1). Conversely, the average gene size in S. cf. sulcatum (1121 bp) is larger than in most species, except in O. trifallax (1930 bp, Supplementary Fig. S1C). The untranscribed region before the transcription start site (TSS) averages 48 bp, with 47 bp found after the transcription end site (TES) on single-gene chromosomes (Supplementary Fig. S2A). This means that there are fewer “extraneous” nucleotides at the ends of nanochromosomes in this species. Moreover, the majority of genes (~ 86%) in S. cf. sulcatum are intron-free, and each gene contains only 0.16 introns on average, which is less than that of H. grandinella (0.34), Tetmemena (1.09), O. trifallax (1.74), E. woodruffi (2.23) and E. vannus (4.05) (Chen et al. 2019; Feng et al. 2022; Swart et al. 2013; Zheng et al. 2021). This makes the macronuclear genome of S. cf. sulcatum the most compact and efficient described to date in spirotrichs. Considering that both S. cf. sulcatum and H. grandinella are typically planktonic, these observations highlight the apparent importance of genome compaction for growth in planktonic ciliates, although the evolutionary pressures that have led to these extremes are not known.
The coding sequence-rich nature of S. cf. sulcatum helps to explain the high (over 50%) GC content of its macronuclear genome, which is higher than that of other reported ciliates (i.e., 20–40%, Supplementary Table S1). Except for the telomeric and subtelomeric regions, where the AT content increases sharply, the GC composition is relatively uniform. Previous research about codon usage in Strombidium sulcatum has also shown a bias toward more GC-rich codons than others that encode the same amino acid, which may be driven by its high GC composition (Knight et al. 2001; Wang et al. 2019). In addition, the GC contents of both H. grandinella and S. cf. sulcatum MIC assemblies are also higher than those of O. trifallax and E. vannus (Table 2). This suggests that both MAC and MIC genomes of H. grandinella and S. cf. sulcatum feature high GC composition, which may be also related to their typical planktonic life style.
The highly fragmented macronuclear genome is generated by developmental fragmentation of the micronuclear chromosomes at chromosome breakage sites
The levels of chromosome breakage during the development of somatic genomes vary dramatically among ciliates, and result in different levels of chromosomal fragmentation in the MAC genome. For spirotrichs, whose MAC genomes are composed of gene-sized chromosomes, chromosome breakage has to occur much more frequently than those with more limited fragmentation. The chromosome breakage mechanisms also differ among ciliates. In the well-studied model organism Tetrahymena, chromosomal breakage occurs at a conserved 15 bp sequence designated as the chromosome breakage sequence (CBS), which is necessary and sufficient to specify a breakage site (Yao et al. 1987, 1990). The CBSs and some flanking micronuclear DNA is eliminated during this process. Similarly, chromosome breakage in another model ciliate (Paramecium) occurs every several hundred kilobases. However, no conserved CBS has been identified and the breakage is considerably less precise, resulting in variable fragmentation sites and heterogeneity of the resulting MAC chromosomes (Baroin et al. 1987; Caron 1992). By comparison, previous findings focusing on E. vannus suggest that CBSs are duplicated and retained in the somatic genome (Baird and Klobutcher 1989; Chen et al. 2019; Klobutcher et al. 1998).
In addition to the consensus CBS motifs, there is a highly conserved motif flanking both sides of the CBS in an overall palindrome structure. Similar models are revealed in H. grandinella and O. trifallax in the present study, although the consensus CBS and the flanking conserved motifs are slightly different (Fig. 3). No obvious CBS and flanking conserved motif were identified for S. cf. sulcatum, although this may be due to the fact that we were unable to obtain as many CBSs as we did for the other three species. However, for the CBSs we did manage to identify, most are retained in the MAC genome. For all the studied spirotrichs in the present work, the CBS and its flanking conserved motif are also detected in the subtelomeric regions in their somatic nanochromosomes (Fig. 3). Although no obvious CBS was found in S. cf. sulcatum, a 5ʹ-GAA-3ʹ motif in the 9th–11th nucleotides at the subtelomeric regions of the MAC nanochromosomes is very obvious, and shares the same sequence with the two most abundant CBSs found in this species (Figs. 3E, 4). As the CBSs and their flanking regions are generally AT-rich (Cavalcanti et al. 2004), they might serve as potential TATA-boxes to initiate transcription (Zheng et al. 2018), with heterogeneity in these sequences allowing for the coregulation of specific genes.
The compact macronuclear genome results from extensive removal of micronuclear-limited DNA
In addition to chromosome fragmentation, IES excision is another important event during genome rearrangements from germline to somatic genomes. Previous studies indicate that ~ 30% of the germline genome of Tetrahymena is eliminated during genome rearrangements (Hamilton et al. 2016), whereas ~ 90% of the germline genomes in spirotrichs is eliminated (Chen et al. 2014; Prescott 1994). The number and size of MDSs/IESs also show great variability among ciliates. Approximately 12,000 IESs, ranging from 136 bp to 43.4 kb with a median length of 2.8 kb, were predicted in Tetrahymena (Hamilton et al. 2016). Comparatively, the number of IESs in the spirotrich O. trifallax (> 225,000) is much higher than that in Tetrahymena, but the length is much smaller, with a median length of about 70 bp (Chen et al. 2014). Similarly, E. vannus, S. cf. sulcatum and H. grandinella also feature smaller IESs, some of which are even less than 20 bp (Fig. 5), though it is possible that the small size of IESs may be biased by short-reads sequencing. Moreover, the IESs in spirotrichs are mostly located in the protein-coding regions that require precise elimination. This contrasts with IESs in Tetrahymena, most of which reside in non-coding regions and are imprecisely excised from the genome, although a small number of scrambled IESs were reported recently (Sheng et al. 2020),
The high number of IESs dispersed throughout the germline genome fragments it into a large number of MDSs. Comparatively, the nanochromosomes in H. grandinella and O. trifallax contain more but shorter MDSs (about four MDSs per kb) than are seen in E. vannus and S. cf. sulcatum (1.5–2.0 MDSs per kb). One example that highlights these differences is the recombination patterns of the single-copy homologous chromosome encoding “Calcineurin-like phosphoesterase” in the four species. The MAC nanochromosomes containing this gene in both S. cf. sulcatum and E. vannus consist of only one MDS, while the nanochromosomes in H. grandinella and O. trifallax are separated into six and four MDSs, respectively (Fig. 6I).
The high number of IESs in spirotrichs makes the origin and evolution of IESs complex. It was previously proposed that IESs might be degenerated remnants of transposable elements (TEs) (Klobutcher and Herrick 1995, 1997), or that they could have originated from degraded duplicate sequences following gene duplication events (Ehrenfeucht et al. 2007; Feng et al. 2022; Gao et al. 2015). The 8 bp consensus 5ʹ-TAYAGYNR-3ʹ on the boundary of IESs in Paramecium (Sellis et al. 2021) and domesticated transposase genes from the piggyBac family in Paramecium and Tetrahymena lineages (Baudry et al. 2009; Cheng et al. 2010) support the proposal of Klobutcher and Herrick (1995) that many IESs could have resulted from transposons via “IBAF” (invasion, bloom, abdicate, and fade). The identification of several families of mobile IESs that generated tens to thousands of new copies in the Paramecium germline genome supports the hypothesis that TEs account for the massive proliferation of IESs (Sellis et al. 2021). In O. trifallax, the discovery that some transposase genes are expressed during PGR, combined with the observation that the silencing of these genes leads to abnormal DNA rearrangement, also indicates a transposon-related origin for PGR in ciliates (Nowacki et al. 2009). In the present study, we detected TEs (Supplementary Table S3) and “mobile IESs” (Supplementary Table S5) in spirotrichs, especially in O. trifallax, which further supports the origin of IESs from transposons. We also identified mobile IESs shared among the four spirotrichs. Considering that IESs generally evolve rapidly, and that these shared mobile IESs have a high level of sequence similarity, they must have resulted from the recent invasion of mobile elements, which may be acquired via horizontal transfer as revealed in Paramecium (Sellis et al. 2021). In addition to mobile IESs, we detected some other IESs inserted at homologous sites in protein-coding sequences in spirotrichs, which was also reported by Feng et al. (2022). This indicates that these shared IESs may have arisen in the germline genome of the last common ancestor of spirotrichs. However, the number of shared IESs among all spirotrichs are very limited, while hypotrichs share more conserved IESs, e.g., Oxytricha and Halteria (present study), or Oxytricha and Tetmemena which both contain a high copy number of telomere-bearing element transposons and share conserved DNA rearrangement junctions (Feng et al. 2022). These findings further support the origin of IESs from transposons and indicate that the ancestor of hypotrichs gained some IESs after its divergence from the other spirotrichs. Furthermore, many IESs are found at homologous sites within paralogous genes of a given species, which indicates that these IESs were gained after speciation but before gene duplication.
It is noteworthy that the vast majority of IESs in spirotrichs are single copy and much smaller in size than those in oligohymenophoreans (Chen et al. 2019). One explanation for this may be the rapid evolution rate of IESs. Long IESs introduced as transposons, for example, may degenerate into much smaller, unrecognizable sequences over a short period of time. Another explanation is that different types of IESs may have different origins. The IESs in spirotrichs are mostly located in the protein-coding regions, whereas TEs are generally not inserted here due to the pressure of counterselection. According to the model proposed by Gao et al. (2015) and Chen et al. (2015), gene scrambling begins with gene duplication in the germline genome, followed by partial and reciprocal degradation of the resulting duplicate sequences over time. In this scenario, alternative processing chooses different pieces from the duplicated sequences. Parts of the gene copies that are not used will become IESs and eventually be eliminated from the somatic genome. A high frequency of alternative processing and gene scrambling events during PGR in spirotrichs and other groups has been detected in the present and previous studies (Ardell et al. 2003; Chen et al. 2014, 2019; Gao et al. 2014; Smith et al. 2020), which again support the above model and imply that the IESs in the protein-coding regions are more likely from the degradation of the duplicate sequences.
Gene scrambling and alternative processing are extensive in the core spirotrichs
Gene scrambling has been recognized in multiple ciliate lineages, such as Spirotrichea (Ardell et al. 2003; Chen et al. 2019; Smith et al. 2020), Heterotrichea (Maurer-Alcalá et al. 2018b), Karyorelictea (Maurer-Alcalá et al. 2018b) and Phyllopharyngea (Gao et al. 2015; Maurer-Alcalá et al. 2018a). The degree of scrambling varies greatly among species, and to date large-scale scrambling has only been observed in species that also show extensive fragmentation of their somatic genome. Among species where scrambled genes have been identified previously, ~ 31% of the putative genes identified in Chilodonella uncinata were scrambled (Maurer-Alcalá et al. 2018a), and scrambled genes account for 15.6% of all genes in O. trifallax, 13.6% in Tetmemena sp., and 7.3% in E. woodruffi (Feng et al. 2022). The proportion of scrambled genes has also been estimated in E. vannus (4.2%), S. cf. sulcatum (15.9%) and H. grandinella (12.9%) in the present study. These proportions are not as high as in O. trifallax, Tetmemena sp. and C. uncinata, which may be due to the fact that their germline genomes are still largely unsequenced. Consequently, the distal scrambled MDSs might not have been recovered or were assembled into different contigs and considered as “nonscrambled”.
Alternative processing is another complex form of chromosome rearrangement that has been proposed as an intermediate stage in the evolution of gene scrambling (Gao et al. 2015). It was previously revealed that alternative processing is extensive among gene families within C. uncinata (Gao et al. 2014). It has also been reported that alternative processing of MDSs usually occurs together with scrambling (Braun et al. 2018) and produces multiple genes and chromosomes in O. trifallax (Chen et al. 2014). In the present study, we revealed that nearly one third (28.8%) of the non-scrambled genes with MDS annotated in O. trifallax and 9.6% in E. vannus are produced by alternative processing. Only a small portion of genes were alternatively processed in S. cf. sulcatum and H. grandinella, which might be due to the low quality of their germline genomes. Most of the alternative processing events observed are ‘‘MDS shuffling’’, while some reuse the IES of one chromosome as the MDS in another. Moreover, most alternative processing events disrupt the coding regions of the genes, the majority of which are transcribed (Supplementary Fig. S3).
Both gene scrambling and alternative processing are novel processes that enable ciliates to efficiently explore protein space by constructing modular proteins, similar to exon shuffling in other eukaryotes (Patthy 1996). Scrambled nanochromosomes are produced by more and shorter MDSs than unscrambled nanochromosomes. The disparity of MDS density is most obvious in S. cf. sulcatum (2.1 per kb for scrambled vs. 1.3 per kb for unscrambled), which indicates that scrambled genes are fragmented more frequently, and IES gains may occur during this process. The higher MDS density in turn provides more opportunity for MDS shuffling to create more proteins. Considering that chromosomes which result from alternative processing tend to encode homologous proteins of the same family, these proteins may play roles at different life stages or in various biochemical processes. Therefore, alternative processing may also play important roles in regulating expression of genes on gene-sized chromosomes.
Data availability
The Illumina sequencing data and assemblies were deposited in GenBank (Bioproject PRJNA1035826). All custom scripts used in the current study have been deposited in GitHub repository (https://github.com/Liping-L/Spirotrichs-PGR).
References
Aeschlimann SH, Jönsson F, Postberg J, Stover NA, Petera RL, Lipps H-J, Nowacki M, Swart EC (2014) The draft assembly of the radically organized Stylonychia lemnae macronuclear genome. Genome Biol Evol 6:1707–1723
Ardell DH, Lozupone CA, Landweber LF (2003) Polymorphism, recombination and alternative unscrambling in the DNA polymerase alpha gene of the ciliate Stylonychia lemnae (Alveolata; class Spirotrichea). Genetics 165:1761–1777
Arnaiz O, Mathy N, Baudry C, Malinsky S, Aury J-M, Wilkes CD, Garnier O, Labadie K, Lauderdale BE, Le Mouël A, Marmignon A, Nowacki M, Poulain J, Prajer M, Wincker P, Meyer E, Duharcourt S, Duret L, Bétermier M, Sperling L (2012) The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet 8:e1002984
Aury J-M, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, Arnaiz O, Billaut A, Beisson J, Blanc I, Bouhouche K, Câmara F, Duharcourt S, Guigo R, Gogendeau D, Katinka M et al (2006) Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444:171–178
Baird SE, Klobutcher LA (1989) Characterization of chromosome fragmentation in two protozoans and identification of a candidate fragmentation sequence in Euplotes crassus. Genes Dev 3:585–597
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477
Baroin A, Prat A, Caron F (1987) Telomeric site position heterogeneity in macronuclear DNA of Paramecium primaurelia. Nucleic Acids Res 15:1717–1728
Baudry C, Malinsky S, Restituito M, Kapusta A, Rosa S, Meyer E, Bétermier M (2009) PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes Dev 23:2478–2483
Braun J, Nabergall L, Neme R, Landweber LF, Saito M, Jonoska N (2018) Russian doll genes and complex chromosome rearrangements in Oxytricha trifallax. G3-Genes Genom Genet 8:1669–1674
Brunk CF, Sadler LA (1990) Characterizaton of the promoter region of Tetrahymena genes. Nucleic Acids Res 18:323–329
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinform 10:421
Caron F (1992) A high degree of macronuclear chromosome polymorphism is generated by variable DNA rearrangements in Paramecium primaurelia during macronuclear differentiation. J Mol Biol 225:661–678
Cavalcanti ARO, Dunn DM, Weiss R, Herrick G, Landweber LF, Doak TG (2004) Sequence features of Oxytricha trifallax (class Spirotrichea) macronuclear telomeric and subtelomeric sequences. Protist 155:311–322
Chen X, Landweber LF (2016) Phylogenomic analysis reveals genome-wide purifying selection on TBE transposons in the ciliate Oxytricha. Mob DNA 7:2
Chen X, Bracht JR, Goldman AD, Dolzhenko E, Clay DM, Swart EC, Perlman DH, Doak TG, Stuart A, Amemiya CT, Sebra RP, Landweber LF (2014) The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158:1187–1198
Chen X, Jung S, Beh LY, Eddy SR, Landweber LF (2015) Combinatorial DNA rearrangement facilitates the origin of new genes in ciliates. Genome Biol Evol 7:2859–2870
Chen S, Zhou Y, Chen Y, Gu J (2018) fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:884–890
Chen X, Jiang Y, Gao F, Zheng W, Krock TJ, Stover NA, Lu C, Katz LA, Song W (2019) Genome analyses of the new model protist Euplotes vannus focusing on genome rearrangement and resistance to environmental stressors. Mol Ecol Resour 19:1292–1308
Chen W, Zuo C, Wang C, Zhang T, Lyu L, Qiao Y, Zhao F, Miao M (2021) The hidden genomic diversity of ciliated protists revealed by single-cell genome sequencing. BMC Biol 19:264
Cheng C-Y, Vogt A, Mochizuki K, Yao M-C (2010) A domesticated piggyBac transposase plays key roles in heterochromatin dynamics and DNA cleavage during programmed DNA deletion in Tetrahymena thermophila. Mol Biol Cell 21:1753–1762
Crooks GE, Hon G, Chandonia J-M, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190
Ehrenfeucht A, Prescott DM, Rozenberg G (2007) A model for the origin of internal eliminated segments (IESs) and gene rearrangement in stichotrichous ciliates. J Theor Biol 244:108–114
Fass JN, Joshi NA, Couvillion MT, Bowen J, Gorovsky MA, Hamilton EP, Orias E, Hong K, Coyne RS, Eisen JA, Chalker DL, Lin D, Collins K (2011) Genome-scale analysis of programmed DNA elimination sites in Tetrahymena thermophila. G3-Genes Genom Genet 1:515–522
Feng Y, Neme R, Beh LY, Chen X, Braun J, Lu MW, Landweber LF (2022) Comparative genomics reveals insight into the evolutionary origin of massively scrambled genomes. eLife 11:e82979
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28:3150–3152
Gao F, Song W, Katz LA (2014) Genome structure drives patterns of gene family evolution in ciliates, a case study using Chilodonella uncinata (Protista, Ciliophora, Phyllopharyngea). Evolution 68:2287–2295
Gao F, Roy SW, Katz LA (2015) Analyses of alternatively processed genes in ciliates provide insights into the origins of scrambled genomes and may provide a mechanism for speciation. mBio 6:e01998–14
Gao F, Warren A, Zhang Q, Gong J, Miao M, Sun P, Xu D, Huang J, Yi Z, Song W (2016) The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci Rep 6:24874
Gao Y, Solberg T, Wang C, Gao F (2023) Small RNA-mediated genome rearrangement pathways in ciliates. Trends Genet 39:94–97
Gong R, Jiang Y, Vallesi A, Gao Y, Gao F (2020) Conjugation in Euplotes raikovi (Protista, Ciliophora): new insights into nuclear events and macronuclear development from micronucleate and amicronucleate cells. Microorganisms 8:162
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075
Hamilton EP, Kapusta A, Huvos PE, Bidwell SL, Zafar N, Tang H, Hadjithomas M, Krishnakumar V, Badger JH, Caler EV, Russ C, Zeng Q, Fan L, Levin JZ, Shea T, Young SK, Hegarty R, Daza R, Gujja S, Wortman JR (2016) Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. eLife 5:e19090
Huang X, Madan A (1999) CAP3: a DNA sequence assembly program. Genome Res 9:868–877
Jahn CL, Klobutcher LA (2002) Genome remodeling in ciliated protozoa. Annu Rev Microbiol 56:489–520
Jiang Y, Zhang T, Vallesi A, Yang X, Gao F (2019) Time-course analysis of nuclear events during conjugation in the marine ciliate Euplotes vannus and comparison with other ciliates (Protozoa, Ciliophora). Cell Cycle 18:288–298
Jin D, Li C, Chen X, Byerly A, Stover NA, Zhang T, Shao C, Wang Y (2023) Comparative genome analysis of three euplotid protists provides insights into the evolution of nanochromosomes in unicellular eukaryotic organisms. Mar Life Sci Technol 5:300–315
Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Katz LA (2001) Evolution of nuclear dualism in ciliates: a reanalysis in light of recent molecular data. Int J Syst Evol Microbiol 51:1587–1592
Katz LA, Kovner AM (2010) Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata. J Exp Zool Part B 314:480–488
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360
Klobutcher LA, Herrick G (1995) Consensus inverted terminal repeat sequence of Paramecium IESs: resemblance to termini of Tc1-related and Euplotes Tec transposons. Nucleic Acids Res 23:2006–2013
Klobutcher LA, Herrick G (1997) Developmental genome reorganization in ciliated protozoa: the transposon link. Prog Nucleic Acid Res Mol Biol 56:1–62
Klobutcher LA, Gygax SE, Podoloff JD, Vermeesch JR, Price CM, Tebeau CM, Jahn CL (1998) Conserved DNA sequences adjacent to chromosome fragmentation and telomere addition sites in Euplotes crassus. Nucleic Acids Res 26:4230–4240
Kloc M, Zagrodzinska B (2001) Chromatin elimination—an oddity or a common mechanism in differentiation and development? Differentiation 68:84–91
Knight RD, Freeland SJ, Landweber LF (2001) A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol 2:research0010.1-0010.13
Kubota S, Ishibashi T, Kohno S (1997) A germline restricted, highly repetitive DNA sequence in Paramyxine atami: an interspecifically conserved, but somatically eliminated, element. Mol Gen Genet 256:252–256
Lee JJ, Faber WWJ, Anderson OR (1991) Life cycles of foraminifera. In: Lee JJ, Anderson OR (eds) Biology of foraminifera, 1st edn. Academic Press, London, pp 285–334
Li C, Chen X, Zheng W, Doak TG, Fan G, Song W, Yan Y (2021) Chromosome organization and gene expansion in the highly fragmented genome of the ciliate Strombidium stylifer. J Genet Genom 48:908–916
Lindblad KA, Pathmanathan JS, Moreira S, Bracht JR, Sebra RP, Hutton ER, Landweber LF (2019) Capture of complete ciliate chromosomes in single sequencing reads reveals widespread chromosome isoforms. BMC Genom 20:1037
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW et al (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18
Lyu L, Asghar U, Fu J, Gao Y, Zhang X, Al-Farraj SA, Chen Z, Gao F (2023) Comparative analysis of single-cell genome sequencing techniques toward the characterization of germline and somatic genomes in ciliated protists. Eur J Protistol 88:125969
Maere S, Heymans K, Kuiper M (2005) BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21:3448–3449
Maurer-Alcalá XX, Knight R, Katz LA (2018a) Exploration of the germline genome of the ciliate Chilodonella uncinata through single-cell omics (transcriptomics and genomics). mBio 9:e01836-17
Maurer-Alcalá XX, Yan Y, Pilling OA, Knight R, Katz LA (2018b) Twisted tales: insights into genome diversity of ciliates using single-cell ’omics. Genome Biol Evol 10:1927–1939
Morgulis A, Gertz EM, Schäffer AA, Agarwala R (2006) WindowMasker: window-based masker for sequenced genomes. Bioinformatics 22:134–141
Mozzicafreddo M, Pucciarelli S, Swart EC, Piersanti A, Emmerich C, Migliorelli G, Ballarini P, Miceli C (2021) The macronuclear genome of the Antarctic psychrophilic marine ciliate Euplotes focardii reveals new insights on molecular cold adaptation. Sci Rep 11:18782
Müller F, Tobler H (2000) Chromatin diminution in the parasitic nematodes Ascaris suum and Parascaris univalens. Int J Parasitol 30:391–399
Nowacki M, Higgins BP, Maquilan GM, Swart EC, Doak TG, Landweber LF (2009) A functional role for transposases in a large eukaryotic genome. Science 324:935–938
Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA (2013) Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737
Papudeshi B, Haggerty JM, Doane M, Morris MM, Walsh K, Beattie DT, Pande D, Zaeri P, Silva GGZ, Thompson F, Edwards RA, Dinsdale EA (2017) Optimizing and evaluating the reconstruction of metagenome-assembled microbial genomes. BMC Genom 18:915
Patthy L (1996) Exon shuffling and other ways of module exchange. Matrix Biol 15:301–310
Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33:290–295
Prescott DM (1994) The DNA of ciliated protozoa. Microbiol Rev 58:233–267
Prjibelski A, Antipov D, Meleshko D, Lapidus A, Korobeynikov A (2020) Using SPAdes de novo assembler. Curr Protoc Bioinform 70:e102
Riley JL, Katz LA (2001) Widespread distribution of extensive chromosomal fragmentation in ciliates. Mol Biol Evol 18:1372–1377
Rzeszutek I, Maurer-Alcalá XX, Nowacki M (2020) Programmed genome rearrangements in ciliates. Cell Mol Life Sci 77:4615–4629
Sellis D, Guérin F, Arnaiz O, Pett W, Lerat E, Boggetto N, Krenek S, Berendonk T, Couloux A, Aury J-M, Labadie K, Malinsky S, Bhullar S, Meyer E, Sperling L, Duret L, Duharcourt S (2021) Massive colonization of protein-coding exons by selfish genetic elements in Paramecium germline genomes. PLoS Biol 19:e3001309
Seyfert HM, Cleffmann G (1982) Mean macronuclear DNA contents are variable in the ciliate Tetrahymena. J Cell Sci 58:211–223
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504
Sheng Y, Duan L, Cheng T, Qiao Y, Stover NA, Gao S (2020) The completed macronuclear genome of a model ciliate Tetrahymena thermophila and its application in genome scrambling and copy number analyses. Sci China Life Sci 63:1534–1542
Smith JJ, Timoshevskaya N, Ye C, Holt C, Keinath MC, Parker HJ, Cook ME, Hess JE, Narum SR, Lamanna F, Kaessmann H, Timoshevskiy VA, Waterbury CKM, Saraceno C, Wiedemann LM, Robb SMC, Baker C, Eichler EE, Hockman D, Sauka-Spengler T et al (2018) The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat Genet 50:270–277
Smith SA, Maurer-Alcalá XX, Yan Y, Katz LA, Santoferrara LF, McManus GB (2020) Combined genome and transcriptome analyses of the ciliate Schmidingerella arcuata (Spirotrichea) reveal patterns of DNA elimination, scrambling, and inversion. Genome Biol Evol 12:1616–1622
Song W, Pan B, El-Serehy HA, Al-Farraj SA, Liu W, Li L (2020) Morphology and molecular phylogeny of two freshwater oligotrich ciliates (Protozoa, Ciliophora, Oligotrichia), Pelagostrombidium fallax (Zacharias, 1895) Krainer, 1991 and Limnostrombidium viride (Stein, 1867) Krainer, 1995, with brief notes on stomatogenesis. J Eukaryot Microbiol 67:232–244
Stanke M, Diekhans M, Baertsch R, Haussler D (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24:637–644
Steele CJ, Barkocy-Gallagher GA, Preer LB, Preer JR (1994) Developmentally excised sequences in micronuclear DNA of Paramecium. Proc Natl Acad Sci 91:2255–2259
Swart EC, Bracht JR, Magrini V, Minx P, Chen X, Zhou Y, Khurana JS, Goldman AD, Nowacki M, Schotanus K, Jung S, Fulton RS, Ly A, McGrath S, Haub K, Wiggins JL, Storton D, Matese JC, Parsons L, Chang WJ et al (2013) The Oxytricha trifallax macronuclear genome: a complex eukaryotic genome with 16,000 tiny chromosomes. PLoS Biol 11:e1001473
Swart EC, Serra V, Petroni G, Nowacki M (2016) Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166:691–702
Tian M, Cai X, Liu Y, Liucong M, Howard-Till R (2022) A practical reference for studying meiosis in the model ciliate Tetrahymena thermophila. Mar Life Sci Technol 4:595–608
Timoshevskiy VA, Herdy JR, Keinath MC, Smith JJ (2016) Cellular and molecular features of developmentally programmed genome rearrangement in a vertebrate (sea lamprey: Petromyzon marinus). PLoS Genet 12:e1006103
Vinogradov DV, Tsoĭ OV, Zaika AV, Lobanov AV, Turanov AA, Gladyshev VN, Gel’fand MS (2012) Draft macronuclear genome of a ciliate Euplotes crassus. Mol Biol (Mosk) 46:361–6
Wang D, Zhang Y, Zhang Z, Zhu J, Yu J (2010) KaKs_Calculator 2.0: a toolkit Incorporating gamma-series methods and sliding window strategies. Genom Proteom Bioinform 8:77–80
Wang R, Xiong J, Wang W, Miao W, Liang A (2016) High frequency of +1 programmed ribosomal frameshifting in Euplotes octocarinatus. Sci Rep 6:21139
Wang C, Yan Y, Chen X, Al-Farraj SA, El-Serehy HA, Gao F (2019) Further analyses on the evolutionary key-protist Halteria (Protista, Ciliophora) based on transcriptomic data. Zool Scr 48:813–825
Wang G, Wang S, Chai X, Zhang J, Yang W, Jiang C, Chen K, Miao W, Xiong J (2021) A strategy for complete telomere-to-telomere assembly of ciliate macronuclear genome using ultra-high coverage Nanopore data. Comput Struct Biotechnol J 19:1928–1932
Wang C, Solberg T, Maurer-Alcalá XX, Swart EC, Gao F, Nowacki M (2022) A small RNA-guided PRC2 complex eliminates DNA as an extreme form of transposon silencing. Cell Rep 40:111263
Xu Y, Zhao F (2018) Single-cell metagenomics: challenges and applications. Protein Cell 9:501–510
Xu L, Dong Z, Fang L, Luo Y, Wei Z, Guo H, Zhang G, Gu YQ, Coleman-Derr D, Xia Q, Wang Y (2019) OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res 47:W52–W58
Yao M-C, Zheng K, Yao C-H (1987) A conserved nucleotide sequence at the sites of developmentally regulated chromosomal breakage in Tetrahymena. Cell 48:779–788
Yao M-C, Yao C-H, Monks B (1990) The controlling sequence for site-specific chromosome breakage in Tetrahymena. Cell 63:763–772
Zhang X, Lu X, Chi Y, Jiang Y, Wang C, Al-Farraj SA, Vallesi A, Gao F (2022) Timing and characteristics of nuclear events during conjugation and genomic exclusion in Paramecium multimicronucleatum. Mar Life Sci Technol 4:317–328
Zhao L, Gao F, Gao S, Liang Y, Long H, Lv Z, Su Y, Ye N, Zhang L, Zhao C, Wang X, Song W, Zhang S, Dong B (2021) Biodiversity-based development and evolution: the emerging research systems in model and non-model organisms. Sci China Life Sci 64:1236–1280
Zheng W, Wang C, Yan Y, Gao F, Doak TG, Song W (2018) Insights into an extensively fragmented eukaryotic genome: de novo genome sequencing of the multinuclear ciliate Uroleptopsis citrina. Genome Biol Evol 10:883–894
Zheng W, Wang C, Lynch M, Gao S (2021) The compact macronuclear genome of the ciliate Halteria grandinella: a transcriptome-like genome with 23,000 nanochromosomes. mBio 12:e01964-20
Acknowledgements
We acknowledge the computing resources provided on IEMB-1, a high-performance computing cluster operated by the Institute of Evolution & Marine Biodiversity, OUC. Many thanks are given to Prof. Weibo Song (Ocean University of China), for his help during the preparation of the manuscript. This work was funded by the Laoshan Laboratory (No. LSKJ202203202), the National Natural Science Foundation of China (32270539, 31922013, 31961123002), the Natural Science Foundation of Shandong Province (ZR2020JQ13), Royal Society/NSFC International Exchanges Cost Share Project (IEC\NSFC\201024), National Institutes of Health (P40OD010964), and the Fundamental Research Funds for the Central Universities (202141004). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conception and design of the study: FG and LL. Acquisition of data: LL, XZ, YG, TZ and JF. Analysis and interpretation of data: LL. Drafting the article: FG and LL. Revising the manuscript: LL, XZ, YG, TZ, JF, NAS and FG. Supervision and funding acquisition: FG. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that there are no conflicts of interest.
Animal and human rights statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Edited by Jiamei Li.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lyu, L., Zhang, X., Gao, Y. et al. From germline genome to highly fragmented somatic genome: genome-wide DNA rearrangement during the sexual process in ciliated protists. Mar Life Sci Technol 6, 31–49 (2024). https://doi.org/10.1007/s42995-023-00213-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-023-00213-x