
Abstract
There have been significant declines in population numbers of many bat species in the United Kingdom, including Natterer’s bats Myotis nattereri, over the last century, largely due to anthropogenic changes. The philopatry, which temperate-zone bats often exhibit to their natal landscapes, in combination with anthropogenic threats, can lead to fragmentation, isolation and sub-division of populations. This may result in bottlenecks and declines in genetic diversity. Multi-scaled research is required to disentangle how the variation in the physical traits of bat species (e.g. affecting flight), as well as their social and behavioural traits (e.g. community size, migration, breeding systems), may affect the genetic health of populations and provide a potential buffer against fragmentation. We used microsatellite markers to characterise the genetic diversity and population structure present in Natterer’s bat colonies to determine whether summer roosting bat colonies were spatially differentiated or part of a meta-population. Analyses of population structure and measures of genetic relatedness suggest spatially differentiated populations of bats exhibit long term site fidelity to summer roosting sites, whilst high genetic diversity at sites indicates gene exchange occurs via swarming sites. Natterer’s bats in northern England may travel greater distances to swarming sites than has been previously documented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The maintenance of genetic diversity in populations is important in conservation biology as it allows adaptation to environmental change (Lande 1988). Gene flow helps to maintain genetic diversity and is enabled by connectivity between populations while also allowing the recolonisation of available habitats where species have previously become extirpated (Fischer and Lindenmayer 2007). Emerging diseases pose a significant threat to many species, either as a result of environmental change altering the epizootiology of endemic pathogens (Munson et al. 2008), or through the natural or anthropogenic arrival of new pathogens (Fitzpatrick et al. 2018; Marino et al. 2017). In this context, genetic diversity and gene flow are the principal agents of evolutionary rescue for species facing new diseases (Gonzalez et al. 2013; Langwig et al. 2017) although high gene flow may also enable the rapid spread of disease through increased mixing of individuals, e.g. (Wilder et al. 2015).
Some temperate bats appear more vulnerable to genetic and demographic effects (Kerth and Van Schaik 2012) than many terrestrial species, despite flight permitting rapid and long-distance movement. This is largely due to strong natal philopatry and limited mixing of individuals between colonies, sub-populations or populations (Kerth et al. 2000; Dixon 2011). This philopatry appears commonplace in many bats and, in combination with many anthropogenic threats, it can lead to fragmentation, isolation and sub-division of populations due to bottlenecks, leading to a decline in genetic diversity (Frankham et al. 2002). Research is required to disentangle how the contrasting physical traits (flight) and behavioural traits (philopatry and aversion to anthropogenic features in landscapes) may affect the genetic health of populations and provide a potential buffer against fragmentation. This should be integrated across the variety of geographic scales used by bats and the contrasting sites from which they may be sampled. It should also accommodate the seasonally sedentary character of some social systems in temperate bats, alongside spatially dynamic breeding/migratory behaviours.
Molecular genetic techniques provide an invaluable resource for obtaining information about bat species, such as population boundaries, evidence of seasonal migration and the identification of geographical barriers to geneflow (Burland and Wilmer 2001). Recently, they have been used to describe many aspects of behaviour (Atterby et al. 2010; Burland et al. 1999; Entwistle et al. 2000; Moussy et al. 2015; Pritchard et al. 2000; Rivers et al. 2005) enabling researchers to assess the distribution and spatial dynamics of different species, which should allow an improvement in broad-scale conservation.
The Natterer’s bat (Myotis nattereri) is a temperate Vespertilionid (Salicini et al. 2011), currently listed as least concern on the International Union for the Conservation of Nature (IUCN) Red List (2019). It is relatively scarce in the UK and as it is associated with woodland, possesses a relatively quiet echolocation with late emergence after dark, it is difficult to monitor (BCT 2017). Although the Natterer’s bat is currently maintaining a stable population size, there have been significant declines in population numbers of many UK bat species including Natterer’s bats over the last century, largely due to anthropogenic changes. As with all UK bats, Natterer’s bats and their roosts are protected under the UK Wildlife and Countryside Act, 1981 and are European Protected Species under Annex IV of the European Habitats Directive (Directive 1992).
There is limited information about Natterer’s bats movement around the landscape beyond their summer colonies. They are thought to undertake relatively short migratory movements at regional scales (e.g. < 200 km; Hutterer 2005), which may have been a driver for the presence of independent lineages in mitochondrial and nuclear DNA throughout Europe, indicating that M. nattereri is a complex of four cryptic species (Salicini et al. 2013). Also, in common with most other temperate Myotids, females are considered faithful to summer colonies, although males are thought to be locally dispersive (Kerth et al. 2002a; Rivers et al. 2005) but see August (2014) for evidence of potentially philopatric adult males. Myotid bats are a relatively long-lived mammal (McCracken and Wilkinson 2000) and are thought to form kin-based colonies (Kerth and Van Schaik 2012; August et al. 2014). Both sexes of Natterer’s bats undertake annual autumn swarming events during which large numbers of bats from colonies across a wide geographical area aggregate prior to hibernation. If significant mating does occur during swarming, this is likely to have an important function in maintaining gene flow between otherwise discrete spatially segregated summer colonies (Burns et al. 2014).
Natterer’s bat, which are thought to show limited female dispersal from their summer colonies, should demonstrate a differentiated population structure at a fine spatial scale, e.g. shared mitochondrial haplotypes within a summer community. In addition, previous work in the UK suggests that the regional-scale catchments around swarming sites may be relatively small, e.g. with radii of 65 km; (Rivers et al. 2006), and this should produce a well-structured national population at scales greater than this. This assumption is based on the premise that gene flow between swarming site catchments is limited and summer colonies associate mainly with a single swarming site. However, if gene flow occurred between catchments, either because of extensive male dispersal, or because of non-exclusive association with multiple swarming sites, populations might appear mixed at supra-regional scales, i.e. across multiple catchments.
We hypothesise that there are three hierarchical geographic and social scales at which Natterer’s bats associate, summer colonies (100 s of bats), catchment areas around traditional swarming sites (1000–10,000 s of bats), and a supra-regional scale, i.e. between catchments, (100,000 s of bats). The current understanding of the biology and behaviour of Natterer’s bats suggests that individual summer colonies should host stable social groups (August et al. 2014) of relatively closely related bats and each may even be composed of a limited number of extended kin groups (Scott et al. 2018). Colonies associated within an exclusive swarming catchment should be less closely related but differentiated from colonies in distant catchments. Catchments will also be differentiated at the supra-regional scale with relatively little gene flow between them. We sampled five colonies across northern England, three in Yorkshire, one in Northumberland and one in Lancashire. Previous evidence suggests that the three colonies in Yorkshire are all associated with a single swarming site (Rivers et al. 2006) and that more distant sites should represent additional exclusive catchment areas. The aim of our study was to explore the relatedness of individuals within and across each of these scales. We test the hypotheses using a suite of microsatellite markers developed for Natterer’s bat (Puechmaille et al. 2012; Rivers et al. 2005; Scott et al. 2013) to characterise the genetic diversity and structure present in colonies of Natterer’s bat in northern England. We assessed gene flow to consider the scale at which conservation management needs to be considered. In addition, we sequenced individuals at two mitochondrial genes (Cytochrome B and NADH Dehydrogenase 1) to compare UK Natterer’s bats to haplotypes identified in European populations (Salicini et al. 2013).
Materials and methods
Sample collection
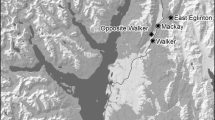
All bat surveying and sampling was performed with the approval of the ethical review bodies at The Animal and Plant Health Agency (APHA) and Newcastle University and under licence from the UK Home Office (Animals in Scientific Procedures Act 1986) and Natural England (Habitats Regulations, 2010). Natterer’s bats were captured from free flight or from known roosts at five sites (Fig. 1) using harp traps (Austbat), handmade pole mounted harp traps, mist nets and hand nets. Captured bats were described noting: sex, age (adult/juvenile) (Dietz et al. 2009), reproductive condition (pregnant or lactating; by palpation of the abdomen), forearm length (0.1 mm) weight (to 0.1 g) and any existing mark. Unmarked bats were marked with a unique ring (2.9 mm Alloy; BCT, England). A wing tissue sample was taken using a 3 mm biopsy punch and stored at − 20 °C in screw-topped rubber-sealed tubes containing 1 ml of 90% ethanol (Analytical Reagent Grade) until DNA extraction (Burland et al. 1999). Fifty-two Natterer’s individuals were sampled from Wallington, Northumberland, 86 from Yorkshire; Wintringham (34), Harpham (25) and Low Catton (27), and 28 from Lancashire (Wycoller) (Supplementary Table 4) using identical methods (Fig. 1). All bats were released otherwise unharmed within a few minutes of the procedure at their place of capture.

Summer sampling sites (filled black circles) and known swarming sites (red ellipses) of Natterer’s bats in Northern England shown on a hill-shaded topographical map for context: YD is an extensive complex of karstic caves in the Yorkshire Dales; WP are the ‘windy pit’ slip fault caves in the North Yorkshire Moors; Wallington is located in Northumberland, Wintringham, Harpham and Low Catton are located in Yorkshire and Wycoller is located in Lancashire
DNA extraction
Genomic DNA was extracted using the Dynamite kit for difficult cells (Microzone), (Robson et al. 2009).
Amplification and genotyping of nuclear DNA
Twenty dinucleotide microsatellite loci have previously been characterised in temperate bats (Supplementary Table 1). These loci were tested individually to determine their optimum annealing temperatures. After optimization, five loci were removed due to either difficulties in amplification or difficulties in scoring, resulting in 15 loci remaining for amplification and informative analysis. To improve efficiency and reduce costs, primer sets were combined and amplified together in multiplex PCR panels selected using Multiplex Manager (Holleley and Geerts 2009). The selected multiplex sets of loci were amplified together using the QIAGEN Multiplex PCR kit following the manufacturer’s instructions and using various optimized primer concentrations (Supplementary Table 1). Polymerase chain reactions (PCR) were then carried out in 5 μl reactions with 2 μl DNA and 3 μl of the primer mix. PCR amplifications were performed in an Applied Biosystems 2400 thermal cycler using one cycle of 95 °C for 15 min to activate the QIAGEN HOTSTAR Taq DNA polymerase, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 48 °C (Set 1) or 55.9 °C (Set 2 and 3) for 90 s, extension at 72 °C for 90 s, and a final extension step at 72 °C for 30 min (Supplementary Table 1). Amplified DNA (2 μl) and a mix of 10 μl of HiDi (Applied Biosystems, Foster City, California)/ROX 500 size standard (Applied Biosystems) at a concentration of 12:1. Amplicons were separated and analysed on an ABI 3130XL Sequencer. Genotypes were analysed using Genemapper 23 software (Prism 1999). Only individuals that amplified allele sizes within the expected size range based on the sequences allele and published data (± 50 bp) at more than eight loci were included in the analysis. This resulted in 152 individuals with a sufficient number of typed loci to be used for further analyses (mean number of loci scored per individual, 13; range 8-15).
Amplification and Sanger sequencing of mtDNA
To compare UK Natterer’s bats to haplotypes identified in European populations, twelve individuals were randomly selected and sequenced at two mitochondrial genes: Cytochrome b (CytB) and NADH Dehydrogenase 1 (ND1) using the primer sets, Molcit-F (Ibáñez et al. 2006) Molcit-R (Salicini et al. 2011) and ND1F2–ND1R (Kawai et al. 2002). After PCR amplification using MyTaq (Bioline, London, UK) using manufacturer’s instructions, the PCR product was purified using ExoSAP (Thermo Fisher, Massachusetts. USA) and Sanger sequenced using Big Dye terminators (BigDye Terminators Sequencing Kit V3.1. Thermo Fisher, Massachusetts. USA). Sequencing products were purified using ethanol precipitation following Kareem et al. (2018) and visualised on an ABI3130XL Gene Analyser.
Statistical analysis of microsatellite data
Data were prepared for analysis using GenAlex 6.503 (Peakall and Smouse 2006). Estimated null allele frequencies were calculated using Cervus 3.0 (Kalinowski et al. 2007). GenePop 4.0 (Raymond and Rousset, 1995) was used to test loci for deviation from Hardy–Weinberg equilibrium and linkage disequilibrium. P values from the linkage disequilibrium tests were corrected for multiple tests (Verhoeven et al. 2005). Heterozygosity, number of alleles per locus, FIS per sample site and pairwise FST estimates between sites (significance tested using 10,000 permutations) were estimated using Microsatellite Analyser 4.05 (MSA) (Dieringer and Schlötterer 2003). Differences in genetic diversity between sites were calculated using a one-way ANOVA.
Relatedness was assessed using the R package Related (Pew et al. 2015). To determine which relatedness estimator was most appropriate, 100 individuals of known relatedness were simulated using the existing genotype data and the relatedness estimates for four different estimators was compared. The Pearson’s correlation coefficient between observed and expected relatedness for each estimator was then calculated to determine the best fit, and the relatedness estimator with the highest correlation was selected to use in further analysis. To determine if individuals at sites were more related than randomly distributed individuals, the average relatedness for each of the sites was estimated and compared to a simulated data set. The observed relatedness values were compared to the expected distribution of relatedness values for each site from the simulated data using the function ‘grouprel’. For simulated data, the group membership counts were preserved for each site, but individuals were shuffled randomly across 1000 iterations, providing a distribution of expected relatedness assuming random distribution of individuals across sites. P values were calculated as the percentage of iterations where the expected relatedness was greater than or equal to the observed relatedness for a given site. Results were considered significant at the 5% level.
The null hypothesis of no population structure among sites was tested by conducting global tests of genetic differentiation. Hierarchical pairwise comparisons were then conducted between all pairs of sites at a local level and also at a regional level, by combining the three North Yorkshire sites (Harpham, Low Catton and Wintringham) and comparing those to the site at Lancashire (Wycoller) and the site in Northumberland (Wallington). Differentiation was assessed using a G test implemented in ‘Hierfstat’ (Goudet 2006). The G test is thought to be more powerful than FST permutation tests for microsatellites (Goudet et al. 1996). Statistical significance was determined through 10,000 random permutations of the data set.
Two different clustering methods, STRUCTURE 2.3.1 (Falush et al. 2003; Pritchard et al. 2000) and TESS (Chen et al. 2007), were used to determine whether sites supported differentiated populations of bats. Whilst STRUCTURE has been shown to be better at individual assignments to clusters, TESS is more effective at determining the number of clusters or populations, especially for spatial models (Chen et al. 2007). Therefore, using both methods, we aimed to have a more robust understanding of the true population structure.
STRUCTURE 2.3.1 (Falush et al. 2003; Pritchard et al. 2000) was used first. The program uses Markov chain Monte Carlo sampling to find the most likely model describing the population structure. Estimates of the number of independent genetic clusters were obtained using the admixture algorithm, correlated allele frequencies and a separate alpha for each population with and without sampling location used as a prior. A burn-in of 1 × 104 followed by 1 × 106 permutations were input to test the number of populations (K). K = 1 to K = 10 were tested with ten iterations each to calculate q, the membership coefficient. q represents proportional individual memberships to different inferred population clusters, indicating if an individual showed admixture of two or more population clusters. Results were summarised using STRUCTURE HARVESTER v 0.9.93 (Earl 2012). To identify the likely number of clusters, the ΔK method was applied. Finally, CLUMPP v.1.1.2 (Jakobsson and Rosenberg 2007) and DISTRUCT v1.1 (Rosenberg 2004) were used to average individual and population membership q values over the ten runs and to visualise the Bayesian assignment. To identify admixed individuals, we applied a threshold q-value of 0.8 to assign an individual as belonging to a specific group. A q-value of less than 0.8 therefore indicated an admixed individual. To quantify the differentiation of the defined clusters suggested by the optimum value of K, following Arteaga et al. (2018), an AMOVA was performed on the genotype data for all un-admixed individuals in GenAlex 6.503 (Peakall and Smouse 2006). The AMOVA partitions the total sum of the squares into components representing variation among and within clusters. The significance of the FST values was then evaluated by comparing the observed value with the distribution of the values obtained from 999 random permutations among clusters.
The second clustering method was implemented in TESS (Chen et al. 2007) with the correlated allele frequencies model and the spatial option. Ten independent runs of 1 × 106 MCMC iterations were performed with thinning set to 100. K values were set from 1 to 10. The most likely K value was decided based on the median K inferred in the models with the highest log posterior density. Finally, TESS was run again with the same parameters as the spatial model but with the inferred K value fixed according to the most likely value, to accurately estimate the membership of each of the individuals.
The effective population size (Ne) of each geographic population identified from STRUCTURE and TESS was calculated using NeEstimator software version 2.0.1 (Do et al. 2014), based on the linkage disequilibrium (LD) model. This estimate was calculated from the deviation of expected compared to observed degree of LD in the populations.
The relative migration network was calculated using the function divMigrate of the diveRsity package (Sundqvist et al. 2016). The network was estimated using GST statistics. GST values could be interpreted as a measure of the level of allele fixation in populations and also reflect the population’s demographic properties, e.g. the number of migrants per generation under assumed mutation rate (Alcala et al. 2014). To test whether migration between populations was asymmetrical (significantly higher in one direction than the other), 95% confidence intervals were calculated from 50,000 bootstrap iterations.
Isolation by distance, the process of increasing genetic differentiation correlated with increasing geographic distance (Wright 1943), for all sites was tested via Mantel’s test (Mantel, 1967) from the comparison of all pairwise FST/(1 − FST) values with pairwise geographic distances using the R package ‘ecodist’ (Mira et al. 2017) with 10,000 permutations. The test was performed for the whole dataset as well as within the North Yorkshire region consisting of three populations (Harpham, Low Catton and Wintringham).
Statistical analysis of mtDNA data
Geneious V10 (Kearse et al. 2012) was used to manually check sequences, to generate alignment using ClustalW and detect haplotypes. Sequences were compared to homologous sequences obtained from other Natterer’s bat populations (Salicini et al. 2011; Puechmaille et al. 2012). The haplotypes were imported in MEGA X (Kumar et al. 2018) as a text file and then exported as Phylip alignment. Haplotype sequence networks were generated using the programme PopART (Leigh and Bryant 2015) using the Phylip alignments and the trait function to assign haplotypes to populations. The mitochondrial DNA sequences obtained were submitted to GenBank (Sequence accession numbers: MH933715–MH933719).
Results
Microsatellite analysis
Tests for LD revealed significant linkage between three pairs of loci at Harpham and one pair of loci at Wallington. However, as these loci were not linked in at least three out of the five populations, they were included for the rest of the analyses. Departure from Hardy–Weinberg equilibrium (HWE) (after Bonferroni correction) was low (P = 0.052) over all loci. Departures were significant for locus Mnatt-10 in three out of five populations. This may be caused by the presence of null alleles. However, Mnatt-10 was retained in the analysis as the locus has been found to have no null alleles in other studies involving Natterer’s bats (Halczok et al. 2017; Scott et al. 2013; 2018; Stumpf et al. 2017). In addition, analyses with and without this locus lead to the same conclusion. Other loci that deviated from HWE were Mnatt-11, D15 and Paur2-Mluc but these loci only deviated in one out of five of the populations, therefore were kept in the analysis. Genetic diversity, as measured by expected heterozygosity and number of alleles, was high at all sites (mean (±SD) expected heterozygosity = 0.78±0.01, Table 1) and there was no difference in the level of genetic diversity among sites (One-way ANOVA F = 0.12, P = 0.975). There was no evidence of inbreeding within sites (FIS) as estimates did not differ significantly from zero.
When sites were considered independently, all pairwise FST values between sites were low but statistically significant from zero, ranging 0.02–0.07 (Table 2). The highest genetic differentiation was found between Harpham and Low Catton (pairwise FST = 0.07) and Harpham and Wallington (pairwise FST = 0.06), whilst the most genetically similar sites were Wintringham and Wycoller (pairwise FST = 0.02) (Table 2). There was no evidence of genetic differentiation between the regions of Northumberland, Yorkshire and Lancashire (FST = 0.01, P = 0.08).
The output values from the Queller and Goodnight estimator, most closely correlated to the expected values of relatedness (e.g. 0.5 parent–offspring or full siblings, 0.25 half siblings, 0 unrelated) (correlation coefficient 0.88) and therefore this estimator was used in further analysis.
There were positive average pairwise relatedness estimates across individuals from all sites suggesting a small chance of individuals having shared alleles through common descent (Table 3). Pairwise relatedness was highly variable between all individuals sampled (− 0.52 to 0.74), although individuals within each site were more closely related to each other than would be expected by chance (P < 0.001 at all sites) which shows some support for female philopatry. Bats from Harpham had the highest averaged relatedness followed by Wintringham, Low Catton and Wycoller. Those from Wallington showed the lowest pairwise relatedness (0.03), although this low level of relatedness in Myotis sp. is not unusual (Kerth et al. 2002b). The sex ratio was fairly similar between sites, F:M—Harpham 25:0, Low Catton 25:1, Wallington 29:9, Wintringham 33:2 and Wycoller 24:4 although slightly more males were caught at Wallington than the other sites so this could partly explain the lower level of relatedness estimated at Wallington. These low levels of relatedness at all sites however are still very low for exclusive female philopatry and therefore support the hypothesis of some female dispersal.
There was a slight positive correlation between genetic distance (FST) and geographic distance between all the sites (r = 0.14, P < 0.01, Fig. 2) and a stronger correlation within the North Yorkshire region (r = 0.35, P < 0.01, Fig. 3) indicating some support for isolation by distance.

Plot of geographic distance (km) v FST Statistics for all samples, the density of data points is indicated by colours

Geographic distance (km) compared to genetic distance (FST) of Natterer’s bats sampled in North Yorkshire only (Harpham, Low Catton and Wintringham)
Hierarchical F-statistics revealed that overall genetic differentiation was low and not significant at the regional level (FRegion/Total = 0.0005; P = 0.082), while there was significant genetic differentiation at the sampling location level (FLocality/Region = 0.0196, P = 0.009) suggesting that locality rather than region explained the distribution of genetic variability.
The optimal configuration determined from the STRUCTURE analysis characterised individuals belonging to four groups based on the ΔK values (Table 4) when sample site was not used as a prior.
K-group assignment resulted in a structure where individuals from Wycoller, Harpham, Low Catton and Wallington formed four differentiated groups with a small number of admixed individuals, whilst individuals at Wintringham seemed to be mostly admixed (Fig. 4a). When the sample sites were used as a priori, the optimal value for K was 2 (Fig. 4b) and it grouped samples from Wallington and Low Catton together in one cluster and samples from Harpham, Wintringham and Wycoller in the other cluster. Further analysis with STRUCTURE on these initial two clusters identified 5 individual clusters representing the 5 sample sites (Fig. 4c). TESS analysis with the geographic locations resulted in the optimal value of K = 5 (Fig. 4d) which corresponds with the site geographic locations.

Bayesian posterior probability of membership of Natterer’s bats to populations obtained by STRUCTURE without (a) and with population info as prior (b, c) and TESS spatial model (d)
Considering the optimal grouping from STRUCTURE when sample site was not used as a prior (K = 4) (Supplementary Table 5, Supplementary Fig. 1), clusters were all significantly different from each other, with pairwise FST values ranging 0.04–0.06 (Table 4). The highest genetic differentiation was found between cluster 1 (with the majority of individuals from Harpham) and cluster 4 (with individuals from Low Catton and Wallington (pairwise FST = 0.06), whilst the most genetically similar clusters were 1 (majority Harpham) and 2 (majority Wycoller) (pairwise FST = 0.04) (Table 4).
Effective population size (Ne) ranged from 42.2 at Wallington with a 95% confidence interval (CI) of 29.2–69.9 to 101.2 (CI 52.9–570.5) at Low Catton (Fig. 5). The average Ne was 71.8 (CI 66.2–77.4). The wide CI at Harpham suggests very little inference can be drawn from the Harpham estimate.

Mean effective population size Ne and 95% credible intervals of Natterer’s bats at five sample sites in Northern England. Note the alternative scale for the Harpham site
On average, relative migration rates were high (average: 0.611; range 0.24–1) in the context of the estimation of demographic parameters (Table 5). Analysis of the migration dynamics revealed gene flow between most populations, with high bidirectional gene flow between Wallington and Low Catton, and also between Wintringham and Wycoller. Wintringham was the only population that had relatively high (above 0.7) migration rates to or from all other populations. The only migration to or from Harpham above 0.7 was from Wintringham (Fig. 6).

Bidirectional estimate of relative migration rate (GST) between all sample sites. Figure shows relative migration rates above 0.7 to aid visualisation
Sequencing of mtDNA
Comparison of both the CytB and the ND1 sequences with those in the NCBI nr sequence database confirmed that all 14 individuals sampled were the species M. nattereri. CytB sequences (535–600 bp) were obtained from 12 individuals (Wallington n = 3, Wintringham n = 2, Low Catton n = 1, Harpham n = 2, Wycoller n = 4) and three haplotypes were distinguished (Fig. 7). Six individuals (four from Wycoller, one from Wallington and one from Low Catton) shared a haplotype (Sequence accession number MH933717), identical to six M. nattereri samples from France, Montenegro, Serbia and the UK on GenBank, i.e. sequence accession record JN591505.1 (Salicini et al. 2011). A second haplotype was found in one individual from Wallington (sequence accession number MH933716), which was very similar to the previous haplotype, with a T to C substitution at nucleotide 54. The third haplotype was shared by all three individuals from Harpham and the two from Wintringham (Sequence accession number MH933715). The third haplotype was also very similar to the middle/northern European haplotype, but with a G to A substitution at nucleotide 237.

Network of haplotypes from CytB sequences from 12 sampled individuals, coloured by the geographic origin of the samples. The size of the pie depicts the total number of individuals sampled with that haplotype and the colours of the pie depict the populations those samples came from
ND1 sequences (600–995 bp) were obtained from fourteen individuals (Harpham n = 7, Wallington n = 1, Wintringham n = 1, Wycoller n = 1 and Low Catton n = 4). Two ND1 haplotypes were found (Fig. 8). Again, the most prevalent haplotype (eleven out of twelve samples, sequence accession number MH933719) was identical to the haplotype found for M. nattereri by Salicini et al. (Salicini et al. 2013) in France, Montenegro, Serbia and the UK (e.g. GenBank JN591538.1). A second ND1 haplotype was found in three of the seven Harpham samples, with a C to T substitution at nucleotide 204 (Sequence accession number: MH933718). Combining the CytB and ND1 sequences indicate that the sampled individuals at Harpham are genetically different to the other populations.

Network of haplotypes from ND1 sequences from 14 sampled individuals, coloured by the geographic origin of the samples. The size of the pie depicts the total number of individuals sampled with that haplotype and the colours of the pie depict the populations those samples came from
Discussion
Our work suggests that the previous hierarchical generalisation of bat geography and social structure may be an oversimplification of a more complex network of sites and associations; at least for the Natterer’s bat in northern England. We found that gene flow between distant sites may be more common than previously thought, which is important for conservation as this may mitigate the potential effects of anthropogenic fragmentation of the population through changes in land use. However, we also identified a summer community (Harpham), geographically proximal to others, as well as to a known autumnal swarming site (WP), which is differentiated from the expected genetic structure without any obvious geographical explanation. Unexpectedly, we find that at the finest geographical scale, extended groups of highly related bats did not dominate Natterer’s bat colonies as overall relatedness at site level was low and varied between sites.
We had hypothesised that the Natterer’s bat in northern England would have followed a general pattern established by previous work on the same or similar European woodland bats (Linton and Macdonald 2019). Whereby, they would form socially discrete, long-lasting geographically stable colonies in their natal summer landscapes, with little significant female dispersal, e.g. Linton and Macdonald (2019). That concept agrees with the extensive work by Zeus et al. (2018), describing Natterer’s bats as well as Bechstein’s bats (Myotis bechsteinii). These species are considered to be phylogenetically closely related and ecologically similar (Ruedi and Mayer 2001). Both appear to form closely related kin-based summer colonies with limited female dispersal, e.g. Kerth and Van Schaik (2012). Similarly, examples of this pattern are demonstrated by congeners from North America, e.g. M. sodalis (Kurta and Murray 2002), M. septentrionalis (Patriquin et al. 2013) and M. lucifugus (Dixon 2011). Though for this latter species, philopatry may be slightly facultative (Norquay et al. 2013). Our hypothesis was that closely knit colonies, based on kin relationships, would share resources, such as access to swarming sites and hibernacula, which would require pups to learn the location and function of distant sites from their mothers (Stumpf et al. 2017). The establishment and maintenance of such learned traditions in close colonies of bats is not firmly established, but reports suggest it is likely, e.g. Stumpf et al. (2017). As the current limited evidence suggests individuals do not visit more than one autumnal swarming site (or complex of geographically proximal sites), we assumed that individual colonies of Natterer’s bat were more or less exclusively associated with individual swarming sites, even though those sites might serve a geographical catchment comprising many colonies. Extending this further, we assumed an individual summer community would be associated with its closest (or at least most accessible) swarming site, which would support the common reports of isolation by distance in studies of population genetics for the Natterer’s bat or its close relatives (Rivers et al. 2006; Halczok et al. 2017) or the similar patterns more distantly related North American congeners demonstrate towards the cold edges of their range, e.g. (Norquay et al. 2013; McLeod et al. 2015; Davy et al. 2015). Therefore, we expected three of our sites (Low Catton, Wintringham and Harpham), considered by previous authors to be associated through WP as their known proximal autumnal swarming site, to show a relatively high gene flow between them compared to the two other sites, which were either closer to an alternative known swarming complex (Wycoller and cave systems in the Yorkshire Dales; Fig. 1) or were at much greater distance (Wallington). However, this was not the case, as gene flow was higher between Wallington and Low Catton (distant sites) compared to Low Catton, Wintringham and Harpham. This suggests some Natterer’s bats may either not use the closest swarming site or they may attend multiple geographically distant swarming sites over their life-times. In this respect, our findings have more in common with those reported for some north American Myotids which may travel considerable distances to their swarming sites or hibernacula, e.g. > 500 km for M. lucifugus (Norquay et al. 2013) M.sodalis (Kurta and Murray 2002). Also, it could be possible that some swarming sites and hibernacula may be separate (e.g. Norquay et al. 2013), or that swarming may include visits to sites beyond the closest (e.g. Johnson et al. 2015). As natural swarming sites are often spatially and temporally cryptic, it seems unlikely that bats discover these sites individually (i.e. opportunistically) after travelling 100’s of kilometres from their natal summer landscapes. Swarming sites may be spatially cryptic to individual bats because they often present as very small entrances to underground spaces surrounded by vegetation, and may be temporally cryptic because their use for mating is often ephemeral (e.g. a few hours only on suitable nights in autumn). Learning the location and timing for the use of swarming sites or hibernacula, therefore seems to imply the transmission of information (i.e. a tradition or behaviour), especially if some bats go on to migrate to multiple swarming or hibernation sites in their lifetimes.
Summer roost population structure
For the first time, we have found genetically differentiated populations of Natterer’s bats in the UK corresponding to the summer roost sites. The differentiation identified by the STRUCTURE analysis supports this finding of at least four evolutionary units, with some mixing of individuals between sites. Within summer colonies, we also found unexpectedly low rates of close kin relationships. This is contrary to the theory of female philopatry at summer colonies, which would likely result in close relationships, such as half siblings and mother–offspring. This suggests a higher rate of female dispersal than previously described (Castella et al. 2001; Kerth et al. 2000, 2002a) which may be more similar to the observations made for M. lucifugus at it natal roosts where a higher number of females than males moved between maternity sites (Norquay et al. 2013). Scott et al. (2018) also found low levels of relatedness in Natterer’s bat summer community boxes (sampled once per year) but suggested that this may be due to bats using a network of roosts and being dispersed throughout the network rather than female dispersal. We agree with the hypothesis that bats use a network of roosts, but at Wallington we sampled from a wide variety of roosts as well as from free flight and therefore accounted for a large proportion of the population (38 individuals at Wallington from an estimated effective population size of 42). Despite this, we still found low levels of relatedness (0.03 at Wallington), which supports the possibility of some female dispersal from their summer landscape. We also note that our study was conducted towards the cold edge of the range for this species (i.e. < 150 km from the most northerly summer colonies in Scotland) as well as in a more variable climate than some previous studies on similar species. In principal, the population dynamics of some individual summer colonies may be marginal, i.e. sink sub-population (Hanski and Gilpin 1991), especially towards range edges. At the same time, the structure of the gene flow across the population may also be temporally dynamic. In this context, sink sites associated with increased mortality and decreased productivity may show increases in an index of relatedness and reductions in genetic diversity where immigration is low (e.g. Harpham) or the converse of this where immigration may be high (e.g. Wallington). Using fine-scale population genetic approaches, to identify source and sink sub-populations within a wider meta-population system, may permit the environmental characteristics of each (e.g. differential foraging and roost provision) to be identified more quickly than the lengthy process of estimating traditional demographic parameters (e.g. mortality, fecundity) in slow living cryptic species, such as bats. In this way, prescriptions for interventions to improve the conservation status of bat species, might be directly linked to the defining characteristics of successful (source) sub-populations, rather than inferred from studies on sub-populations in marginal (sink) locations.
Regional population structure
We found limited evidence to support the hypothesis that there is regional structure of Natterer’s bat populations sharing a swarming site. Rivers et al. (2005), found a negative correlation between genetic assignment of Natterer’s bats to WP and geographical distance to capture sites in summer colonies. This coincides with the limited evidence for isolation by distance here (at relatively low levels) at both the regional and northern England scale, to suggest that the likelihood of a summer community using a swarming site declines with the increasing distance between them, and consequently that a variable proportion of more distant colonies may use one or more additional swarming sites. However, here we also find evidence that geographically proximal sites (the three Yorkshire sites; Wintringham, Low Catton and Harpham), with a known association to the WP complex of swarming sites (Rivers et al. 2006) are genetically differentiated, whilst substantially more distant sites are gentically similar (Wallington in Northumberland and Low Catton in Yorkshire). Therefore, some Natterer’s bats in Northern England may travel further than previously thought (Halczok et al. 2017; Rivers et al. 2006) to alternative swarming sites rather than visiting the closest (Dekeukeleire et al. 2016; Johnson et al. 2015). This is also supported with relatively high migration rates between relatively distant sites, e.g. Wallington and Low Catton (Fig. 6). In addition to this, our findings of admixed individuals at summer sites, support the hypothesis that bats from the same summer community may visit different swarming or hibernation sites (Dekeukeleire et al. 2016; Johnson et al. 2015; Norquay et al. 2013; Kurta and Murray 2002). Swarming site catchments may therefore be much larger than previously described, with this study suggesting that individual Natterer’s bats in northern England travel up to 115 km to mate, approaching the maximum migration distances described for this species in Europe (Hutterer 2005), though much less than the distances travelled by similar congeners in North America (Kurta and Murray 2002; Norquay et al. 2013).
Although this study, and that of Rivers et al. (2005) sampled across parts of the same landscapes, distinct differences in our interpretation is evident. As up to 15 years elapsed between the field work, it is not clear if the differences in interpretation between that study and this are the consequence of changes in the bats across this period, or the more precise description facilitated by the increased number of loci available here (fifteen compared to seven). Although individual bats might still associate exclusively with a single swarming site, it is now clear that a number of differentiated swarming sites may be associated with a single community (Dekeukeleire et al. 2016; Norquay et al. 2013). The contribution of each swarming site to the genetic character of the summer community may not be temporally stable. For example, it is not yet clear how juvenile bats find either their swarming or hibernation sites when these can be so cryptic and distant to their natal landscapes. Current speculation suggests that they either follow their mothers to these sites (Stumpf et al. 2017), or other experienced adults from their natal roosts (Rivers et al. 2006) producing a transmissible tradition. Small sustained changes in the quality of these distant sites (either as mating sites or survival where they are also used as hibernacula) would promote changes in the relative proportions of individuals and their offspring returning to the natal landscape year on year. Thus, Wycoller and Wallington, though geographically distant from the WP swarming complex, appear to share a strong traditional connection to this gene-exchange system, despite Wycoller being much closer to another known swarming complex (Yorkshire Dales) and Wallington being over 115 km from WP. Similarly, the site at Low Catton, despite its relative proximity to WP, appears to support a substantial tradition in the use of an alternative unknown site (Divergent FST relationships with most other sites).
National/international population structure
Mitochondrial DNA (mtDNA), being maternally inherited is used to estimate genetic variation within maternal lineages. The mtDNA population diversity is known to reflect maternally-directed natal-site fidelity, whereas genome-wide bi-parentally inherited nuclear DNA markers assist in quantifying levels of gene flow among subpopulations for both sexes (Hoffman et al. 2009). The mtDNA analysis, revealed that haplotypes were identical, or very similar, to those of Natterer’s bats from middle and northern Europe (Salicini et al. 2013), including their two UK samples (Cumbria and Essex), and also the middle and northern European samples from their later studies. The new haplotype for CytB found at Harpham and Wintringham, with one SNP different from the most common haplotype, may indicate an exchange of females between Harpham and Wintringham. This is supported by the high relative migration values (GST) seen in the microsatellite analysis (Table 5). Also, three of the seven Harpham individuals have a new, unique, haplotype for ND1, differing by one SNP from the majority. Combining the CytB and ND1 data indicate that many of the individuals at Harpham are genetically different than the other populations and this may indicate that there are fewer (or no) females moving from Harpham to the other sites with the exception of Wintringham, which again is supported by GST values, but this needs larger sample sizes to confirm this. Although this study found more mitochondrial haplotypes in England than found by Salicini et al. (2011), this is not surprising since Salicini et al. included only two individuals from England in their study. More individuals of both sexes from the North of England populations need to be sequenced to draw firm conclusions on the number of haplotypes present in the UK.
Conclusion
The increased distance bats may travel to swarming areas compared to previous studies, is positive in terms of maintaining genetic diversity within species. In addition, the population structure observed here shows sites as genetically differentiated. This could be due to long-term site fidelity to summer roosting sites by both males and females with limited summer dispersal, and some fidelity to swarming sites (Rivers et al. 2006). Our results highlight the importance of protecting both swarming and summer roosting sites.
References
Alcala N, Goudet J, Vuilleumier S (2014) On the transition of genetic differentiation from isolation to panmixia: what we can learn from GST and D. Theor Popul Biol 93:75–84
Arteaga MC, Medellín RA, Luna-Ortíz PA, Heady PA, Frick WF (2018) Genetic diversity distribution among seasonal colonies of a nectar-feeding bat (Leptonycteris yerbabuenae) in the Baja California Peninsula. Mamm Biol 92:78–85
Atterby H, Aegerter JN, Smith GC, Conyers CM, Allnutt TR, Ruedi M, Macnicoll AD (2010) Population genetic structure of the Daubenton’s bat (Myotis daubentonii) in western Europe and the associated occurrence of rabies. Eur J Wildl Res 56:67–81
August TA, Nunn MA, Fensome AG, Linton DM, Mathews F (2014) Sympatric woodland myotis bats form tight-knit social groups with exclusive roost home ranges. PLoS One 9:e112225
BCT (2017) The State of UK’s bats 2017
Burland TM, Wilmer JW (2001) Seeing in the dark: molecular approaches to the study of bat populations. Biol Rev 76:389–409
Burland TM, Barratt EM, Beaumont MA, Racey PA (1999) Population genetic structure and gene flow in a gleaning bat, Plecotus auritus. Proc R Soc Lond Ser B Biol Sci 266:975–980
Burns LE, Frasier TR, Broders HG (2014) Genetic connectivity among swarming sites in the wide ranging and recently declining little brown bat (Myotis lucifugus). Ecol Evol 4:4130–4149
Castella V, Ruedi M, Excoffier L (2001) Contrasted patterns of mitochondrial and nuclear structure among nursery colonies of the bat Myotis myotis. J Evol Biol 14:708–720
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756
Davy CM, Martinez-Nunez F, Willis CKR, Good SV (2015) Spatial genetic structure among bat hibernacula along the leading edge of a rapidly spreading pathogen. Conserv Genet 16:1013–1024
Dekeukeleire D, Janssen R, Haarsma A-J, Bosch T, van Schaik J (2016) Swarming behaviour, catchment area and seasonal movement patterns of the Bechstein’s bats: implications for conservation. Acta Chiropterol 18:349–358
Dieringer D, Schlötterer C (2003) Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Notes 3:167–169
Dietz C, Von Helvers O, Nill D, Lina PHC, Hutson AM (2009) Bats of Britain, Europe and Northwest Africa. A & C Black London, London
DIRECTIVE, E. U. H. (1992) EU Habitats Directive (92/43/EEC). Consolidated text. Office for Official Publication of The European Union. CONSLEG: 1992LOO43-01.05-2004
Dixon MD (2011) Population genetic structure and natal philopatry in the widespread North American bat Myotis lucifugus. J Mamm 92:1343–1351
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Entwistle AC, Racey PA, Speakman JR (2000) Social and population structure of a gleaning bat, Plecotus auritus. J Zool 252:11–17
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fischer J, Lindenmayer DB (2007) Landscape modification and habitat fragmentation: a synthesis. Glob Ecol Biogeogr 16:265–280
Fitzpatrick LD, Pasmans F, Martel A, Cunningham AA (2018) Epidemiological tracing of Batrachochytrium salamandrivorans identifies widespread infection and associated mortalities in private amphibian collections. Sci Rep 8:13845
Frankham R, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Gonzalez A, Ronce O, Ferriere R, Hochberg ME (2013) Evolutionary rescue: an emerging focus at the intersection between ecology and evolution, vol 368. Philosophical Transactions of the Royal Society: Biological Sciences, p 20120404
Goudet J (2006) Estimation and tests of hierarchical F-statistics. R package
Goudet J, Raymond M, de Meeüs T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Halczok TK, Fischer K, Gierke R, Zeus V, Meier F, Treß C, Balkema-Buschmann A, Puechmaille SJ, Kerth G (2017) Evidence for genetic variation in Natterer’s bats (Myotis nattereri) across three regions in Germany but no evidence for co-variation with their associated astroviruses. BMC Evol Biol 17:5
Hanski I, Gilpin M (1991) Metapopulation dynamics: brief history and conceptual domain. Biol J Lin Soc 42:3–16
Hoffman JI, Dasmahapatra KK, Amos W, Phillips CD, Gelatt TS, Bickham JW (2009) Contrasting patterns of genetic diversity at three different genetic markers in a marine mammal metapopulation. Mol Ecol 18:2961–2978
Holleley CE, Geerts PG (2009) Multiplex Manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46:511–517
Hutterer R (2005) Bat migrations in Europe: a review of banding data and literature. Federal Agency for Nature Conservation, Bonn
Ibáñez C, García-Mudarra JL, Ruedi M, Stadelmann B, Juste J (2006) The Iberian contribution to cryptic diversity in European bats. Acta Chiropterol 8:277–297
IUCN (2019) The IUCN Red List of Threatened Species [Online]. IUCN. http://www.iucnredlist.org/search/link/5933e59e-14e27428. Accessed 4 June 2017
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Johnson LNL, McLeod BA, Burns LE, Arseneault K, Frasier TR, Broders HG (2015) Population genetic structure within and among seasonal site types in the little brown bat (Myotis lucifugus) and the northern long-eared bat (M. septentrionalis). PLoS One 10:e0126309
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Kareem AA, Port G, Wolff K (2018) Population structure of the glasshouse whitefly Trialeurodes vaporariorum (Hemiptera: Aleyrodidae) shows multiple introductions to the UK. Agric For Entomol 21(2):139–148
Kawai K, Nikaido M, Harada M, Matsumura S, Lin L-K, Wu Y, Hasegawa M, Okada N (2002) Intra-and interfamily relationships of Vespertilionidae inferred by various molecular markers including SINE insertion data. J Mol Evol 55:284–301
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Kerth G, van Schaik J (2012) Causes and consequences of living in closed societies: lessons from a long-term socio-genetic study on Bechstein’s bats. Mol Ecol 21:633–646
Kerth G, Mayer F, König B (2000) Mitochondrial DNA (mtDNA) reveals that female Bechstein’s bats live in closed societies. Mol Ecol 9:793–800
Kerth G, Mayer F, Petit E (2002a) Extreme sex-biased dispersal in the communally breeding, nonmigratory Bechstein’s bat (Myotis bechsteinii). Mol Ecol 11:1491–1498
Kerth G, Safi K, König B (2002b) Mean colony relatedness is a poor predictor of colony structure and female philopatry in the communally breeding Bechstein’s bat (Myotis bechsteinii). Behav Ecol Sociobiol 52:203–210
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Kurta A, Murray SW (2002) Philopatry and migration of banded Indiana bats (Myotis sodalis) and effects of radio transmitters. J Mamm 83:585–589
Lande R (1988) Genetics and demography in biological conservation. Science (Washington) 241:1455–1460
Langwig KE, Hoyt JR, Parise KL, Frick WF, Foster JT, Kilpatrick AM (2017) Resistance in persisting bat populations after white-nose syndrome invasion. Philos Trans R Soc B 372:20160044
Leigh JW, Bryant D (2015) popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Linton DM, Macdonald DW (2019) Roost composition and sexual segregation in a lowland population of Daubenton’s bats (Myotis daubentonii). Acta Chiropterol 21:129–137
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Marino J, Sillero-Zubiri C, Deressa A, Bedin E, Bitewa A, Lema F, Rskay G, Banyard A, Fooks AR (2017) Rabies and distemper outbreaks in smallest ethiopian wolf population. Emerg Infect Dis 23:2102
McCracken GF, Wilkinson GS (2000) Bat mating systems. Reprod Biol Bats 12:3–5
McLeod BA, Burns LE, Frasier TR, Broders HG (2015) Effect of oceanic straits on gene flow in the recently endangered little brown bat (Myotis lucifugus) in maritime Canada: implications for the spread of white-nose syndrome. Can J Zool 93:427–437
Mira Ó, Sánchez-Prieto CB, Dawson DA, Burke T, Tinaut A, Martínez JG (2017) Parnassius apollo nevadensis: identification of recent population structure and source–sink dynamics. Conserv Genet 18:1–15
Moussy C, Atterby H, Griffiths AGF, Allnutt TR, Mathews F, Smith GC, Aegerter JN, Bearhop S, Hosken DJ (2015) Population genetic structure of serotine bats (Eptesicus serotinus) across Europe and implications for the potential spread of bat rabies (European bat lyssavirus EBLV-1). Heredity 115:83–92
Munson L, Terio KA, Kock R, Mlengeya T, Roelke ME, Dubovi E, Summers B, Sinclair ARE, Packer C (2008) Climate extremes promote fatal co-infections during canine distemper epidemics in African lions. PLoS One 3:e2545
Norquay KJO, Martinez-Nuñez F, Dubois JE, Monson KM, Willis CKR (2013) Long-distance movements of little brown bats (Myotis lucifugus). J Mamm 94:506–515
Patriquin KJ, Palstra F, Leonard ML, Broders HG (2013) Female northern myotis (Myotis septentrionalis) that roost together are related. Behav Ecol 24:949–954
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pew J, Muir PH, Wang J, Frasier TR (2015) related: an R package for analysing pairwise relatedness from codominant molecular markers. Mol Ecol Resour 15:557–561
Prism ABI (1999) GeneMapper® Software v3. 7. Applied Biosystems
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ, Allegrini B, Boston ESM, Dubourg-Savage M-J, Evin A, Knochel A, le Bris Y, Lecoq V, Lemaire M, Rist D (2012) Genetic analyses reveal further cryptic lineages within the Myotis nattereri species complex. Mamm Biol 77:224–228
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Rivers NM, Butlin RK, Altringham JD (2005) Genetic population structure of Natterer’s bats explained by mating at swarming sites and philopatry. Mol Ecol 14:4299–4312
Rivers NM, Butlin RK, Altringham JD (2006) Autumn swarming behaviour of Natterer’s bats in the UK: population size, catchment area and dispersal. Biol Cons 127:215–226
Robson MA, Thomason JC, Wolff K (2009) Eight polymorphic microsatellite loci markers for the barnacle Balanus amphitrite (syn. Amphibalanus amphitrite) Darwin 1854. Mol Ecol Resour 9:368–369
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Ruedi M, Mayer F (2001) Molecular systematics of bats of the genus Myotis (Vespertilionidae) suggests deterministic ecomorphological convergences. Mol Phylogenet Evol 21:436–448
Salicini I, Ibáñez C, Juste J (2011) Multilocus phylogeny and species delimitation within the Natterer’s bat species complex in the Western Palearctic. Mol Phylogenet Evol 61:888–898
Salicini I, Ibáñez C, Juste J (2013) Deep differentiation between and within Mediterranean glacial refugia in a flying mammal, the Myotis nattereri bat complex. J Biogeogr 40:1182–1193
Scott DD, Fitzpatrick S, Bailie DA, Boston ESM, Lundy MG, Buckley DJ, Teeling EC, Montgomery WI, Prodöhl PA (2013) Isolation and characterization of eight polymorphic microsatellite loci for Natterer’s bat, Myotis nattereri (Vespertilionidae, Chiroptera). Conserv Genet Resour 5:643–645
Scott DD, Boston ESM, Lundy MG, Buckley DJ, Gager Y, Chaplain CJ, Teeling EC, Montgomery WI, Prodöhl PA (2018) Relatedness, parentage, and philopatry within a Natterer’s bat (Myotis nattereri) maternity colony. Popul Ecol 60:1–10
Stumpf M, Meier F, Grosche L, Halczok TK, van Schaik J, Kerth G (2017) How do young bats find suitable swarming and hibernation sites? Assessing the plausibility of the maternal guidance hypothesis using genetic maternity assignment for two European bat species. Acta Chiropterol 19:319–327
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475
Verhoeven KJF, Simonsen KL, McIntyre LM (2005) Implementing false discovery rate control: increasing your power. Oikos 108:643–647
Wilder AP, Kunz TH, Sorenson MD (2015) Population genetic structure of a common host predicts the spread of white-nose syndrome, an emerging infectious disease in bats. Mol Ecol 24:5495–5506
Wright S (1943) Isolation by distance. Genetics 28:114–138
Zeus V, Reusch C, Kerth G (2018) Long-term roosting data reveal a unimodular social network in large fission-fusion society of the colony-living Natterer’s bat (Myotis nattereri). Behav Ecol Sociobiol 72:99
Acknowledgements
We thank the UK Natural Environment Research Council (NERC) (Grant number: NBAF874), and the UK’s Department for Food Environment and Rural Affairs (DEFRA) for funding and support (SE0430). The laboratory work was performed at the NERC Biomolecular Analysis Facility at the University of Sheffield and Newcastle University. We thank Gavin Horsburgh, Helen Hipperson, Natalie dos Remedios and Kathryn Maher at Sheffield for guidance and support with the laboratory work and data analyses; and Samuel Logan for general laboratory assistance at Newcastle University. The National Trust at Wallington kindly provided access to summer bat roosts. We also gratefully acknowledge the funding provided to CC by the Brazilian Government Programme Science Without Borders. We also acknowledge the helpful comments of the reviewers. Data will be archived with Dryad on publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Handling editor: J. Paul Grobler.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mordue, S., Aegerter, J., Mill, A. et al. Population structure, gene flow and relatedness of Natterer’s bats in Northern England. Mamm Biol 101, 233–247 (2021). https://doi.org/10.1007/s42991-021-00102-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42991-021-00102-9