
Abstract
Diabetes mellitus (DM) is the most predominant group of metabolic disorders wreaking havoc on the wellbeing of man, with type 2 diabetes mellitus (type 2 DM) accounting for most DM related cases. This study, hence, investigated the antidiabetic potential of Gongronema latifolium leaf fractionated compounds against proteins implicated in different molecular pathways related to the onset and progression of type 2 DM. A total of fifteen proteins that can act as type 2 DM therapeutic targets were identified from the literature and downloaded/modelled using respective repositories. After docking the compounds with the fifteen proteins, glycogen synthase kinase 3 beta (GSK 3β), glucagon-like peptide-1 receptor (GLP-1R) and human aldose reductase were chosen as the ideal targets due to their high binding affinities with the compounds. Subsequent in silico analysis like binding free energy, ADMET predictions using different servers, and machine-learning predictive models (QSAR) using kernel partial least square regression were employed to identify promising compounds against the three targets. The eleven identified compounds (Luteonin, Kampferol, Robinetin, Gallocatechin, Baicalin, Apigenin, Genistein, Rosmaric acid, Chicoric acid and Naringenin) formed stable complexes with the proteins, showed moderation for toxicity, drugability, GI absorptions and drug-drug interactions, though structure modifications may be needed for lead optimization. The predictive QSAR models with reliable correlation coefficient (R2) showed the potency of the compounds to act as inhibitors (pIC50) of aldose reductase and GSK 3β, and act as agonists (pEC50) of GLP-1R. Thus, this study experimental framework can be used to design compounds that can modulate proteins related to type 2 DM without inducing off-target effects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
After many years of medical progress in finding cure/therapeutics for multi-factorial chronic health diseases like diabetes mellitus (DM), the incidence of people living with DM remains as prevalent as ever, and it continues to make a major contribution to the reduction in life expectancy around the globe [1, 2]. A statistical report on diabetes compiled by WHO showed that the number of patients with diabetes has increased fourfold since the early 1980s to about 422 million people, and this figure is forecasted to hit 693 million in the next four decades [2,3,4]. DM is a group of pathological diseases presented with insulin deficiency due to β-cell destruction or gradual loss of β-cell insulin secretion as major clinical features. Type 1 diabetes mellitus (type 1 DM) and type 2 diabetes mellitus (type 2 DM) are the major/prevalent types of diabetes, with T2DM contributing 90–95% of diabetes-related cases [1]. Complications of diabetes are prevalent among people living with type 1 DM and type 2 DM which are responsible for alarming death rates; complications such as stroke, diabetic ulcer, diabetic nephropathy, retinopathy and neuropathy, cardiovascular diseases have higher prevalence [5, 6].
Presently, there is no effective cure for DM. however, DM can be managed and controlled through several therapies which may require lowering the blood level of sugar or delaying the onset or progression of its complications [7]. There are several FDA approved drugs that act as first-line pharmacotherapy for T2DM. The orally available Metformin is the most common antidiabetic drug, which functions by alleviating blood glucose levels and modulating insulin sensitivity of tissues [8]. However, Metformin like other antidiabetic drugs is often accompanied by moderate to severe side effects [9]. The use of drug monotherapy for controlling the glucose level in patients living with diabetes has proven unsuccessful; therefore therapies involving the combination of different antidiabetic drugs that act on protein targets in molecular pathways involved in diabetes progression have provided more satisfactory results [10,11,12]. The major problems posed by this combinational approach are unwanted side effects, drug-drug interactions, hepatotoxicity and another type of toxicities [13].
A less toxic and more effective therapy is the combination of the active components into a single molecule/compound that could selectively modulate different diabetes target proteins and pathways with proven efficacy and safety relative to single target drugs. The multi-target ligand drug design strategy prioritizes the selection of suitable targets implicated in a disease state, with the relative potency of the compound towards each receptor [14]. In recent years, the Computational pipeline is one of the screening methods proposed as an interesting route for multi target-based drug design, which allows high-throughput virtual screening (HTVS) of diverse compounds against several targets of interest through molecular docking studies, quantitative structure–activity relationship (QSAR) and other machine learning methods.
This study thus employs different computational tools to identify multi-target ligands from characterized compounds of Gongronema latifolium against selective targets for combating the ill-effect of type 2 DM. Their choice as therapeutic targets was based on the most studied type 2 DM related proteins [15].
2 Materials and methods
2.1 Retrieval of respective diabetes protein targets
A total of 15 (fifteen) proteins implicated in the onset, progression and pathogenesis of diabetes and its complications were collected from literature and downloaded from the protein database, other proteins without crystal structures were modelled using the SWISS homology modelling server. The human pancreatic alpha-amylase (PDB: 1B2Y), glycogen synthase kinase 3 beta (PDB ID:1UV5), human DPP-IV (PDB ID: 3BJM), PPAR- gamma (PDB ID:5U5L), glucagon-like peptide-1 (GLP-1) receptor (PDB ID: 6×1a), human glutamine-fructose-6-phosphate transaminase 1 (PDB ID: 2V4M), insulin receptor kinase (PDB ID: 1GAG), human aldose reductase (PDB ID; 2R24), alpha-glucosidase (PDB ID: 5ZCB), 11beta-hydroxysteroid dehydrogenase type 1 (PDB ID: 3CH6), human diacylglycerol O-acyltransferase 1 (PDB ID: 6VZ1), protein Tyrosine Phosphatase 1B (PDB ID: 2CM3). These crystal PDB structures were chosen because they had a significant resolution of 2.00 Å or more. The three-dimensional structure of other proteins not deposited in the protein databank repository which include sodium-glucose cotransporter-2 (SGLT-2), G protein-coupled receptors-120 (GPR120), G protein-coupled receptors-119 (GPR1190 were modelled by utilizing their fasta sequence.
2.2 Protein preparation and grid generation
The protein crystal structures were prepared using the Schrödinger protein preparation wizard to add missing hydrogen atoms, optimize hydrogen bonds, delete water molecules, and create disulfide bridges if necessary, fill missing side chain via Prime refinement, and minimize the structures using the OPLS3 force field. The grid files for defining the protein binding pockets were generated by picking the co-crystal ligand within the protein binding pockets. The active sites of modelled proteins were predicted by Sitemap before generating the grid file.
2.3 Ligand library and preparation
Different phytochemical fractions of Gongronema latifolium leaf have been isolated using a gas chromatographic flame Ionization detector [16]. Therefore, this study built a manually curated library by downloading the 2D structure of the compounds from the Pubchem chemical database. The ligand library contains thirty-nine (39) Alkaloids, thirty-eight (38) flavonoids, twenty-five (25) Terpenes, nine (9) total phenolics, eight (8) carotenoids, six (6) Hydroxycinnamic acids, seven (7) saponins and seven (7) Sterols, and was minimized using ligprep panel.
2.4 Molecular docking studies
Molecular docking studies were performed by docking the prepared compounds of Gongronema latifolium leaf with retrieved diabetes implicated proteins. Firstly, glide high throughput virtual screening (HTVS) was assigned as the scoring algorithm to screen the 134 compounds, the top 10% ranked compounds were selected for extra precision (XP) docking as a scoring algorithm to perform more expensive docking simulation with the proteins [17].
2.5 Employment of PRIME MM-GBSA for binding free energy calculation
The compound-protein complexes were evaluated for their stability via a post docking analysis tool called Prime MM-GBSA by taking a maestro pose viewer file. The major contributors for the stability complex are ligand binding energies and ligand strain energies [18].
2.6 ADME/T prediction
The selected hit compounds were screened for toxicity and drug-likeness using SwissADME and ADMETsar online server. The parameters for filtering the compounds include Lipinski rule of five (RO5), gastrointestinal (GI) absorption, inhibition of CYP450 isoenzymes, hepatotoxicity, eye irritation and corrosion, biodegradation etc.
2.7 Predictive QSAR modelling
The top criterion for building a predictive quantitative structure–activity relationship (QSAR) model is retrieving experimental compounds with known biological activities against the desired drug target. Therefore, the chemical and bioactivity (IC50) profile of a given set of experimental data sets of GSK-3β and Aldose reductase from ChEMBL database. Also, the agonists of GLP-1 alongside their EC50 were retrieved from ChEMBL database by blasting the PDB fasta sequence of GLP-1R. The predictive models were generated by AutoQSAR modelling which calculated the topological and physicochemical descriptors, in addition to the binary fingerprints.
3 Results and discussion
Polypharmacology is an exciting approach in drug development and discovery and has gained increased recognition over combinational therapies in the last two decades [19]. Complex diseases such as diabetes require a more modern treatment because they are implicated in several molecular pathways, therefore identifying efficacious small molecules capable of modulating a network of interacting proteins implicated in diabetes with little or no side effect is a legendary magic bullet than the combination of multiple drugs [20].
3.1 Molecular docking studies
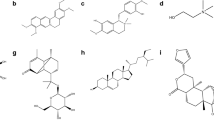
Molecular docking is an exciting technique in computer-aided drug design that employs a computer algorithm (e.g. Glide) to find the best ligand that fits into the binding pocket of a receptor at the atomic and sub-atomic levels, thereby simulating intermolecular interaction between them [21, 22]. Recent studies have shown the antidiabetic properties of Gongronema latifolium in rat model [23,24,25]. In order to investigate the binding capacity of the bioactive compound in Gongronema latifolium leaf on proteins related to diabetes in humans, the fractions of Gongronema latifolium leaf consisting of different phytochemicals were docked with fifteen protein targets associated with type 2 DM to discover the possible hit compounds. The results of the compounds docking scores against different proteins implicated in different diabetes molecular pathways are shown in Table S1. After analyzing the results, the compounds had more favourable binding affinities with a number of proteins. However, this study identified the top three interacting proteins which are GLP-1R, GSK-3β and aldose reductase. The compounds with good docking scores against the three top-ranked proteins were sorted out. They are Quercetin, Luteonin, Kampferol, Robinetin, Gallocatechin, Baicalin, Apigenin, Genistein, Rosmaric acid, Chicoric acid and Naringenin; the three-dimensional structure of the compounds is illustrated in Fig. 1. The selected small molecular weight compounds showed varied binding affinities with the proteins (Table 1). Compounds Quercetin, Chicoric acid and Gallocatechin with lowest binding energy against GLP-1 receptor recorded docking scores of − 12.860 kcal mol−1, − 12.339 kcal mol−1 and − 11.888 kcal mol−1. Quercetin, Chicoric acid, Luteonin and Baicalin had highest binding affinities with GSK-3β, exhibiting docking scores of − 13.138 kcal mol−1, − 11.839 kcal mol−1, − 11.236 kcal mol−1 and − 11.22 kcal mol−1, respectively. Also, the results from the docked compounds with aldose reductase showed that the docking scores ranged from − 10.220 to − 15.385 (kcal/mol) as seen in Table 1. Impressively, Rosmaric acid, Chicoric acid had an incredible binding affinity and lowest binding energy with a glide XP docking scores of 15.385 kcal mol−1 and − 14.522 kcal mol−1. The antidiabetic properties of the compounds with a good docking score (Table 1) selected as hits have been reported in several studies [26,27,28]. This demonstrates their therapeutics in alleviating diabetes by modulating different molecular pathways.

Three dimensional structure of top ranked compounds against the diabetes protein targets
3.2 Prime MM-GBSA
Calculation of ligand-bound protein stability complexes are known as binding free energy is one of the most reliable post-docking analysis techniques for confirming the docking score results. A negative value denotes favourable results. The results of the binding free energy calculated for the compounds in complex with GSK3β, GLP-1R and aldose reductase are enlisted in Tables S2–S4. All of the compounds formed stable stability with the protein targets. Naringenin formed the most stability with GSK3β (− 57.793 kcal mol−1), Kampferol, Quercetin and Luteonin had ΔG binding scores of − 62.616 kcal mol−1, − 62.400 kcal mol−1 and − 62.122 kcal mol−1 with aldose reductase. In addition, Chicoric acid had the most stability with GLP-1R (− 62.0383 kcal mol−1). The major contributors to the binding free energy are Coulomb energy, covalent energy, van der Waals energy, hydrogen bonding and lipophilic binding.
3.3 Interacting profiles
GLP-1, a glucose-dependent hormone, is known for its role in insulin secretion through the enhancement of beta-pancreatic cells to secrete insulin upon binding to Glucagon-like peptide-1 Receptor (GLP-1R) [29, 30]. To analyze the backbone, aromatic, side chain and hydrophobic interactions of the compounds with the receptor, Ligprep module of Schrödinger was employed. Pi-pi stacking and hydrogen bonding were the major intermolecular interactions between the compounds and GLP-1R (Table 1). The residue Phe381 made major intermolecular contacts with the compounds by forming Pi-pi stacking with the compounds phenyl rings. In addition, residues Thr207, Ser31, Lys197 Gln234 and Gln221 made most of the hydrogen interactions with the compounds (Fig. 2). The rich hydrogen bond donor and acceptor as a mode of interaction between the compounds and GLP-1R may contribute to the compounds high docking scores. Similar amino acid interactions are seen in potential GLP-R receptor agonists for simulating insulin secretion from Phyllanthus emblica phytocompounds [31].

Two dimensional interactions of a Quercetin, b Luteonin, c Kampferol, d Robinetin, e Gallocatechin, f Baicalin, g Apigenin, h Genistein, i Rosmaric acid, j Chicoric acid, k Naringenin with residues at the binding pocket of GLP-1
GSK-3β, a target for beta cell regenerative therapies, is one of the protein kinases implicated in the pathogenesis of several diseases including type 2 DM [32]. The GSK-3β binding site of small molecule inhibitors comprises hydrophilic amino acid residues Lys85, Asp200, and Glu51 known for ligand-ATP recognition [32]. While Quercetin and Luteonin formed H-bond interaction with Lys85, Genistein and Naringenin were observed to make intermolecular interaction with Asp200 via hydrogen bonds. The compounds Naringenin, Quercetin, Luteonin and Rosmaric acid have the most hydrogen donor and acceptor contact with the residues within the binding pockets of GSk-3β. Recognition of residues Val135, Gln185, Lys183, Ile62, Asn186 and Arg141 and Asp133 as a medium of hydrogen bonding with a range of GSk-3β inhibitors has been observed in different studies [33,34,35]. This study also showed similar interactions by the selected hits (Table 1, Fig. 3), in addition to polar interactions with residues such as Pro136 Thr138, Val70, Leu132 and Asn186 [36].

Two dimensional interactions of a Quercetin, b Luteonin, c Kampferol, d Robinetin, e Gallocatechin, f Baicalin, g Apigenin, h Genistein, i Rosmaric acid, j Chicoric acid, k Naringenin with residues at the binding pocket of GSK-3β
A number of structural diverse compounds either from natural origin or chemically synthesized molecules have demonstrated potency against aldose reductase in in vitro and in silico experiments [37, 38]. Plants bioactive compounds exhibiting substantial inhibitory attributes against aldose reductase are classified into flavonoids, tannins, phenolics, alkaloids, terpenoids chemical groups—interestingly the investigated Gongronema latifolium leaf compounds fall into these categories of chemical groups. In addition, these compounds showed very high docking scores with aldose reductase than other target proteins (Table S1). The interacting profiles of the compounds with aldose reductase showed that intermolecular interactions were formed mainly by residues Gln183, Trp219, Trp111, Trr48, Asn160, Cys298, Asp43, Ile260, Hip110, Asn150 and Asp43 (Table 1, Fig. 4). However, a distinct molecular interaction with Trp111 by all the compounds either via hydrogen interaction of pi-pi stacking was observed. The compounds interactions with the active site residues were similar to zenaresta, an FDA approved drug for aldose reductase inhibitor, which occupies nearly the hydrophobic region of the binding pocket and elicits conformational changes [39].

Two dimensional interactions of a Quercetin, b Luteonin, c Kampferol, d Robinetin, e Gallocatechin, f Baicalin, g Apigenin, h Genistein, i Rosmaric acid, j Chicoric acid, k Naringenin with residues at the binding pocket of Aldose reductase
3.4 Analysis of in silico ADMET prediction
All the eleven selected compounds showing the different degrees of binding affinities with the protein targets were screened for their pharmacokinetics, drug-likeness, and toxicity. This prediction is crucial in drug discovery and development because it eliminates drugs or potential drug candidates with unwanted side effects and off-target effects. To cross-check the properties of the compounds in the biological environment, ADMETsar and SwissADME servers built using different machine learning-based methods are used for the prediction. The physicochemical properties of the compounds showed that they are low molecular weight compounds (270.24–445.35 KDA), with total polar surface area (TPSA) ranging between 90.9 and 213.78. Lipinski rule of five (RO5) is a ssrule formulated by Christopher Lipinski and colleagues for determining the oral bioavailability of active drug substances [40]. All of the compounds except for Chicoric acid and Baicalin were in accordance with RO5, and this correlates with the compounds high GI absorption as listed in Table 2. Furthermore, the compounds pharmacokinetics properties were calculated. Four of the principal isoenzymes (CYP1A2, CYP2C19, CYP2C9 and CYP2D6) of cytochrome p450 are responsible for drug metabolism and determinant of drug interactions that can lead to drug toxicities were considered [41]. The server returns “Yes” if it’s an inhibitor of investigated CYP or returns “No” if otherwise. Gallocatechin, Baicalin, Rosmaric acid and Rosmaric acid showed the cleanest profiles against the isoenzymes. Other compounds are substrates for either CYP1A2 or CYP2D6.
The in silico prediction tool for toxicity offers a fast and reliable method of investigating compounds toxicity before further experimental validation through preclinical and clinical testing. Toxicity prediction showed that the compounds are non-carcinogens, biodegradable and do not induce eye corrosion. However, they can induce a significant level of hepatotoxicity, which may be dose-dependent as they showed different classes of acute oral toxicity (Table 2). In addition, a large number of the compounds had the potential to cause eye irritation and bind to aromatase. Overall, the compounds showed significant moderation for ADMET, though structural modifications may be required to optimize the molecules.
3.5 QSAR prediction
The predictive models for calculating the bioactivities of the compounds against the functional proteins are shown in Table 3. The models were constructed using kernel partial least squares regression with binary fingerprint (radial, molprint2d as descriptors). The statistical models showed a reliable correlation coefficient (R2) of 0.8467, 0.8600 and 0.7537 for GSK-3, aldose reductase and GLP-1R. [42]. The compounds had significantly predicted pIC50 (inhibitory activities) against GSK-3 and aldose reductase, recording pIC50 between the range of 6.762 and 5.661 (Table 4). The potency of the compounds to act as GLP-1R agonists is also demonstrated through their pEC50. Overall, the plant compounds selected as hits can act as inhibitors of GSK-3 and aldose reductase while showing moderation to activate GLP-1 receptors, thereby stimulating the secretion of insulin in the beta-pancreatic cells. The scatter plots containing both test and training set for the construction of predictive models are shown in Fig. 5. Details of AutoQSAR predicted activities compared with the observed activities for the investigated proteins are listed in Tables S6–S8.

Scatter plots of best QSAR model for GSK-3β, Aldose reductase and GLP-1
4 Conclusion
This study investigated the therapeutic potential of chemical constituents of Gongronema latifolium leaf against fifteen proteins related to type 2 DM. The docking scores of the compounds denote that they have high binding affinities with the proteins. However, the compounds showed more promising features with aldose reductase, GLP-1 and GSK 3β. The selected compounds with high docking scores interacted with amino acid residues crucial for the protein inhibitions or activation formed stable complexes upon binding with the proteins and showed moderation for ADMET parameters. In addition, the constructive prediction model showed considerable bioactivities of the molecules against aldose reductase, GLP-1 and GSK 3β. This study demonstrated the antidiabetic potential of fractionated compounds Gongronema latifolium leaf in slowing down or alleviating type 2 DM, therefore further experimental analysis are needed to confirm the therapeutic properties of these compounds.
Data availability
The data that supports the findings of this study are available in the supplementary material of this article.
References
American Diabetes Association (2018) Classification and diagnosis of diabetes: standards of medical care in Diabetes 2018. Diabetes Care 41:13–27
Artasensi A, Pedretti A, Vistoli G, Fumagalli L (2020) Type 2 diabetes mellitus: a review of multi-target drugs. Molecules 25(8):1987. https://doi.org/10.3390/molecules25081987
Cho NH, Shaw JE, Karuranga S, Huang Y, Fernandes JDR, Ohlrogge A, Malanda B (2018) IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138:271–281
WHO Diabetes Programme. WHO. 2019. https://www.who.int/diabetes/en/. Accessed 27 Jan 2019
Deshpande AD, Harris-Hayes M, Schootman M (2008) Epidemiology of diabetes and diabetes-related complications. Phys Ther 88(11):1254–1264. https://doi.org/10.2522/ptj.20080020
Tuttolomondo A, Maida C, Pinto A (2015) Diabetic foot syndrome as a possible cardiovascular marker in diabetic patients. J Diabetes Res. https://doi.org/10.1155/2015/268390.268390
Kasole R, Martins HD, Kimiywe J (2019) Traditional medicine and its role in the management of diabetes mellitus: “patients’ and herbalists’ perspectives.” Evid Based Complement Altern Med. https://doi.org/10.1155/2019/2835691
Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60(9):1577–1585. https://doi.org/10.1007/s00125-017-4342-z
McGovern A, Tippu Z, Hinton W, Munro N, Whyte M, De Lusignan S (2018) Comparison of medication adherence and persistence in type 2 diabetes: a systematic review and meta-analysis. Diabetes Obes Metab 20:1040–1043
Mudaliar S, Henry RR (1999) Combination therapy for type 2 diabetes. Endocr Pract 5(4):208–219. https://doi.org/10.4158/EP.5.4.208
Prabhakar PK, Kumar A, Doble M (2014) Combination therapy: a new strategy to manage diabetes and its complications. Phytomedicine 21(2):123–130. https://doi.org/10.1016/j.phymed.2013.08.020
Prabhakar PK, Prasad R, Ali S, Doble M (2012) Synergistic interaction of ferulic acid with commercial hypoglycemic drugs in streptozotocin induced diabetic rats. Phytomedicine 20(6):488–494. https://doi.org/10.1016/j.phymed.2012.12.004
Morphy R, Rankovic Z (2005) Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 48(21):6523–6543
Zimmermann GR, Lehár J, Keith CT (2007) Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today 12(1–2):34–42
Bhowmick A, Banu S (2017) Therapeutic targets of type 2 diabetes: an overview. MOJ Drug Des Develop Ther 1(3):60‒64. https://doi.org/10.15406/mojddt.2017.01.00011
Imo C, Uhegbu FO (2015) Phytochemical analysis of gongronema latifolium benth leaf using gas chromatographic flame ionization detector. Int J Chem Biomol Sci 1(2):60–68
Kikiowo B, Ogunleye J, Iwaloye O, Ijatuyi T (2020) Journal of biomolecular structure and dynamics iSSN: (Print) (Therapeutic potential of Chromolaena odorata phyto-constituents against human pancreatic α- amylase Therapeutic potential of Chromolaena odorata phyto-constituents against human pancreatic a-amylase. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1833758
Elekofehinti O, Iwaloye O, Famusiwa C, Akinseye O, Rocha J (2020) Identification of main protease of coronavirus SARS-CoV-2 (Mpro)Inhibitors from Melissa officinalis. Curr Drug Discov Technol. https://doi.org/10.2174/1570163817999200918103705
Mangiatordi GF, Intranuovo F, Delre P, Abatematteo FS, Abate C et al (2020) Cannabinoid receptor subtype 2 (CB2R) in a multitarget approach: perspective of an innovative strategy in cancer and neurodegeneration. J Med Chem 263(23):14448–14469. https://doi.org/10.1021/acs.jmedchem.0c01357
Oyinloye BE, Iwaloye O, Ajiboye BO (2021) Polypharmacology of Gongronema latifolium leaf secondary metabolites against protein kinases implicated in Parkinson’s disease and Alzheimer’s disease. Sci Afr 12(3):e00826. https://doi.org/10.1016/j.sciaf.2021.e00826
Alsamghan AS, Alwabli AS, Abadi M, Alsaleem SA, Anbari DM, Alomari AS, et al (2020) From sequence analysis of DPP-4 to molecular docking based searchingofits inhibitors. Bioinformation 16(6):444–451. https://doi.org/10.6026/97320630016444
Morris GM, Lim-Wilby M (2008) Molecular docking. Methods Mol Biol 443:365–382. https://doi.org/10.1007/978-1-59745-177-2_19
Ojo OA, Okesola MA, Ekakitie LI, Ajiboye BO, Oyinloye BE, Agboinghale PE, Onikanni AS (2020) Gongronema latifolium Benth. leaf extract attenuates diabetes-induced neuropathy via inhibition of cognitive, oxidative stress and inflammatory response. J Sci Food Agric 100(12):4504–4511.
Ojo OA, Osukoya OA, Ekakitie LI et al (2020) Gongronema latifolium leaf extract modulates hyperglycaemia, inhibits redox imbalance and inflammation in alloxan-induced diabetic nephropathy. J Diabetes Metab Disord 19:469–481. https://doi.org/10.1007/s40200-020-00533-0
Ajiboye BO, Oyinloye BE, Agboinghale PE, Onikanni SA, Asogwa E, Kappo A (2019) Antihyperglycaemia and related gene expressions of aqueous extract of Gongronema latifolium leaf in alloxan-induced diabetic rats. Pharm Biol 57(1):604–611. https://doi.org/10.1080/13880209.2019.1657907
Xincheng Y, Ling Z, Yuxin C, Jun T, Youwei W (2013) In vivo and in vitro antioxidant activity and α-glucosidase, α-amylase inhibitory effects of flavonoids from Cichorium glandulosum seeds. Food Chem 139:59–66. https://doi.org/10.1016/j.foodchem.2012.12.045
Vinayagam R, Xu B (2015) Antidiabetic properties of dietary flavonoids: a cellular mechanism review. Nutr Metab (Lond) 12:60. https://doi.org/10.1186/s12986-015-0057-7
Eid HM, Haddad PS (2017) The antidiabetic potential of quercetin: underlying mechanisms. Curr Med Chem 24(4):355–364. https://doi.org/10.2174/0929867323666160909153707
Wessel J, Chu AY, Willems SM et al (2015) Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun 6:16. https://doi.org/10.1038/ncomms6897
Aroda VR (2018) (2018) A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diab Obes Metab 20:22–33. https://doi.org/10.1111/dom.13162
Sharma P, Joshi T, Joshi T, Chandra S, Tamta S (2019) In silico screeningof potential antidiabetic phytochemicals from Phyllanthus emblica against therapeutic targets of type 2diabetes. J Ethnopharmacol https://doi.org/10.1016/j.jep.2019.112268.
Iwaloye O, Elekofehinti OO, Oluwarotimi EA et al (2020) Insight into glycogen synthase kinase-3β inhibitory activity of phyto-constituents from Melissa officinalis: in silico studies. In Silico Pharmacol 8:2. https://doi.org/10.1007/s40203-020-00054-x
Witherington J, Bordas V, Gaiba A, Naylor A, Rawlings AD, Slingsby BP, Smith DG, TakleAK WRW (2003) 6-heteroaryl-pyrazolo[3,4-b]pyridines: potent and selective inhibitors of glycogen synthase kinase-3 (GSK-3). Bioorg Med Chem Lett 13:3059–3062. https://doi.org/10.1016/S0960-894X(03)00646-2
Padavala A, Chitti S, Rajesh B, Vinukonda V, Jayanti R, Vali R (2010) In silico based ligand design and docking studies of GSK-3β inhibitors. Chem Bioinform J 10:1–12. https://doi.org/10.1273/cbij.10.1
Buescher JL, Phiel CJ (2010) A non catalytic domain of glycogen synthase kinase-3 (GSK-3) is essential for activity. J Biol Chem 285:7957–7963. https://doi.org/10.1074/jbc.M109.091603
Kramer T, Schmidt B, Fabio LM (2012) Small-molecule inhibitors of GSK-3: structural insights and their application to alzheimer’s disease models. Int J Alzheimer’s Dis. https://doi.org/10.1155/2012/381029
Reddy TN, Ravinder M, Bagul P, Ravikanti K, Bagul C, Nanubolu JB et al (2014) Synthesis and biological evaluation of new epalrestat analogues as aldose reductase inhibitors (ARIs). Eur J Med Chem 71:53–66. https://doi.org/10.1016/j.ejmech.2013.10.043
Kawanishi K, Ueda H (2003) Moriyasu M (2003) Aldose reductase inhibitors from the nature. Cur Med Chem 10(15):1353–1374. https://doi.org/10.2174/0929867033457304
Kinoshita T (2002) The structure of human recombinant aldose reductase complexed with the potent inhibitor zenarestat. Acta Crystallogr D Biol Crystallogr 58(4):622–626. https://doi.org/10.1107/S0907444902002378
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46(1):3–26. https://doi.org/10.1016/s0169-409x(00)00129-0
Ogu CC, Maxa JL (2000) Drug interactions due to cytochrome P450. Proc (Bayl Univ Med Cent) 13(4):421–423
Iwaloye O, Elekofehinti OO, Kikiowo B, Oluwarotimi E, Fadipe T (2020) Machine learning-based virtual screening strategy reveals some natural compounds as potential PAK4 inhibitors in triple negative breast cancer. Curr Proteomics. https://doi.org/10.2174/1570164618999201223092209
Author information
Authors and Affiliations
Contributions
BEO and BOA designed and supervised the study. BEO, BOA and OI carried out the study and wrote the first draft of the manuscript. OVO, JNE, AO, OOJ and AEU contributed to the study design, analysis and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
This manuscript reports the authors’ original work, which has not been previously published elsewhere or is currently being considered for publication elsewhere. The paper reflects the authors’ research and analysis truthfully and completely. The paper properly credits the meaningful contributions of co-authors and co-researchers.
Human and animal rights
No human or animal subjects were involved in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ajiboye, B.O., Iwaloye, O., Owolabi, O.V. et al. Screening of potential antidiabetic phytochemicals from Gongronema latifolium leaf against therapeutic targets of type 2 diabetes mellitus: multi-targets drug design. SN Appl. Sci. 4, 14 (2022). https://doi.org/10.1007/s42452-021-04880-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04880-2