
Abstract
Quantitative structure–activity relationship studies conducted on forty-five (45) derivatives of 2-anilino 4-amino substituted quinazolines as malaria inhibitors to determine the structures responsible for their antimalarial properties and design novel derivatives with improved activities. The molecular descriptors generated were selected to develop the theoretical model using the genetic approximation component of the material studio. The developed model found to be a function of ATSC8c, GATS8i, SpMin1_Bhi, JGI10, and TDB6u descriptors, shows excellent statistical parameters (R2 = 0.7913, R2adj = 0.7553, Q2cv = 0.7112, LOF = 0.2125, and R2pred = 0.7650). The mean effect (MF) analysis revealed the descriptor SpMin1_Bhi, as the most influential by its largest percentage contribution (54%) to the developed model. The descriptor decodes the information on the first ionization potentials and was found to have positive MF. Hence, activity increases with increases the descriptor value. Structural modifications of the template (compound 13; pEC50 = 7.387) using electron-withdrawing groups increases the descriptor value (first ionization potentials) of the template, which by extension increases the antimalarial activity lead to the design of ten (10) novel theoretical derivatives with improve antimalarial activities. Compound 3, N4-(3-bromo-5-fluorobenzyl)-N2-(4-fluorophenyl)-6,7-dimethoxyquinazoline-2,4-diamine was found to have the highest antimalarial activities among all the designed derivatives (pEC50 = 8.0515).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Malarial has maintained its status as one of the most devastating diseases in evolving countries, which increases several socioeconomic challenges [3]. Estimated malaria casualty in the year 2017 is said to be in the region of 200 million infections worldwide leading to over 400 thousand death [10], with children in the age bracket of 5 years mostly affected [22]. The protozoan, Plasmodium consisting of five species of the renowned parasite (P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi) is the genesis of the contagious disease, with P. falciparum as the deadliest of the species [10].
The use of drugs such as chloroquine and artemisinin to treat malaria remains the major means of getting rid of the disease [21], despite several efforts put in at producing a malaria vaccine [15]. The uses of drugs for the treatment are however not without its challenges, inform of the resistance of P. falciparum against the available antimalarial drugs. The optimization of novel antimalarial drugs using new target molecules, having the capacity of overcoming this resistance menace has become a most and urgent quest to embark [5]. Several compounds such as 2-anilino 4-amino substituted quinazolines have indicated antimalarial potency in recent times. Gibson et al. claimed that the 2-anilino 4-amino substituted quinazolines target Plasmodium dihydrofolate reductase (DHFR) [8]. Although no pattern or pharmacophores were reported to be responsible for their antimalarial activity, the unlimited substitution at specific positions may be responsible for their observed potency. Hence, designing derivatives of 2-anilino 4-amino substituted quinazoline through substitution of a variety of groups at specific positions will collaborate this claim.
The synthesis of compounds with improved activities in the drug industry has always been a huge task as it cost time and resources to carry out what is practically a trial and error procedure. Hence, an alternative method of predicting the activity of the compounds before their synthesis becomes a necessity. And quantitative structure–activity relationship (QSAR) is one of the methods available for the prediction of the antimalarial activity of designed derivatives of 2-anilino 4-amino substituted quinazolines.
Several applications of QSAR studies have been reported for lots of biological compounds. Da Silva and his co-researchers conducted a QSAR analysis on some series of arylsulfonamide derivatives on 5-hydroxytryptamine subtype 6 (5-HT6) receptor to design novel anti-Alzheimer disease using PLS and CoMFA. The results of the study show that 23 compounds were designed to have better activity than the original compounds [6]. Thirty inhibitors of the acetylcholinesterase enzyme were modeled using the QSAR method, results in a robust and predictive model for the prediction of anti-Alzheimer disease [11].
This research targeted the development a descriptive model to predict the activity of antimalarial compounds through the use of experimentally determined compounds with activity values. The descriptors used for this analysis were obtained from Padel software and the contribution of each descriptor was determined which played a role in the design of novel antimalarial derivatives.
2 Materials and methods
2.1 Data source and preparation
The forty-five (45) derivatives of 2-Anilino 4-Amino Substituted Quinazolines deployed for this research (Table 1), were extracted from literature [8]. The 2-D structures were drawn with the aid of ChemDraw Ultra-version 12.0 [19] and thereafter opened in the Spartan software in 3-D for full geometry optimization. The Padel software was used to calculate 1875 molecular descriptors for each of the optimized compounds. The activities of the derivatives were converted to \({-log}_{10}^{{EC}_{50}}\) (pEC50) for better QSAR studies. Furthermore, the data set was slashed into 35 for model construction (training set) and 10 for the model validation (test sets).
2.2 Feature selection
2.2.1 Data pre-treatment and selection
The molecular descriptors were subjected to some series of treatments such as discarding all constant value descriptors as well as those that are highly correlated with each other. Furthermore, descriptors containing empty cells are also discarded [2]. This kind of descriptors are normally referred to as noisy descriptors and could make the selection of the informative descriptors more difficult. The whole of data pre-treatment was carried out with the aid of the "Data Pre-Treatment GUI 1.2" tool that employs the V-WSP algorithm [16].
2.2.2 Data division
The forty-five (45) derivatives of 2-Anilino 4-Amino Substituted Quinazolines data set were slashed into 35 training set (77.7% of the data set) for model construction and 10 test set (22.2% of the data set) for model validation. In slashing the data into a test and training set, the Kennard Stone algorithm technique of "Dataset Division GUI 1.2" software was deployed [4].
2.3 QSAR model development and validation
The predictive models were initiated from the training set where the activities serve as the response variable (pEC50), and the descriptor values as the explanatory variable by subjecting them to the material studio’s genetic approximation (GA) component. The genetic algorithm (GA) is a heuristic search algorithm that capitalized on the evolutional concept of natural selection and genetics [9]. The method has the advantage of addressing the challenges of constrained and unconstrained encountered during optimization. The GAs makes use of historical information through a definite search for excellent performance [17]. A genetic algorithm may find tremendous use in searching for a large pool of descriptors. The crossovers, smoothness values, are fixed at 800,000 and 1.00 respectively during the algorithm, as well as allowing other default settings. To construct the model, between five (5) to seven (7) descriptors were set as possible numbers in a model. The GA accessed the model fitness through evaluation of the Friedman lack of- fit (LOF) expressed by Eq. 1 below;
where SSE = sum of squares of errors, z = number of terms other than the constant in the model, d, represents the smoothing parameter, n, represents the model descriptor count, and M, representing the training set counts.
2.3.1 Model validation-internal
The leave-one-out (LOO) cross-validation was applied in model validation. As the name implies, the method involves leaving a molecule out of the data set before model development, which is then used to calculate the activity of the compound left out. The procedure is repeated again and again until all the data were left out once and were predicted with the constructed model. Equation 2, ensured the determination of the cross-validated correlation coefficient squared, R2cv (Q2).
where \({Y}_{obs}\) stands for the training set activity, \({Y}_{pred}\) stands for the training set predicted activity while \({{\hat{Y}}}\) stands for the training set observed mean activity of the.
2.3.2 Model validation-external
The predictive strength of the developed model is determined through the validation of the model constructed externally. The method involves splitting the data into training and test sets, where the training set is used to develop a model. The developed model is used to predict the activity of the test set, thereby estimating the value of the predictive R2 (R2pred) of the test set expressed by the formula presented in Eq. 3.
where \({\mathrm{Y}}_{\mathrm{Pred }(\mathrm{Test})}\) and \({\mathrm{Y}}_{\mathrm{Test}}\) respectively represent predicted and observed activity of the test compounds.\({{\hat{Y}}}_{{{\text{Training}}}}\) represents the training set mean activity value.
2.4 Y-Randomization
The application of the Y-Randomization technique is to test the robustness of the model developed. The techniques, the values of the activity (Y) are randomized while the descriptors remained unchanged [2] and used in developing a model. The value of \({\mathrm{R}}_{\mathrm{m}}^{2}\) parameter, expressed in Eq. 4 [1] measures the disparity in the squared mean correlation coefficient values of the randomized model, \({\mathrm{R}}_{\mathrm{rand}}^{2}\) from the values of the squared correlation coefficient of the non-random model (R2).
where \({\mathrm{R}}_{\mathrm{m}}^{2}\) represents the squared coefficient of regression of the randomized activity, \({R}^{2}\), the squared coefficient of regression in non-randomized activity, and \({R}_{r}^{2}\), the average value of \({R}^{2}\).
2.5 Model applicability domain
The ability of the developed QSAR model to make an excellent prediction of the activity of compounds is greatly a function of the applicability domain. The model predicts the activity of the compounds under its domain more correctly than those outside the domain [20]. Building the model applicability domain involves plotting the leverages of each compound against their respective standardized residuals. The diagonals of the hat matrix, \({H}_{i}={X}_{i}{\left({X}_{i}^{T}{X}_{i}\right)}^{-1}{X}_{i}^{T}\) produces the leverages for each of the compounds [12], with \({H}_{i}\), the training/test hat matrix, \({X}_{i}\) as the initial matrix of test/training set, and \({X}_{i}^{T}\) as the training/test set transpose matrix. The domain has warning leverage, \({h}^{*}=3(t+1)/z\), where z and t stand for the training set and model descriptors counts respectively. Beyond the warning leverage, a compound is regarded as an outlier and not reliably predicted by the model.
2.6 Mean effect (MF)
The relative contributions of each molecular descriptor to the development of a model are analyzed through the mean effect (MF) estimation. The size and sign of the mean effect respectively show the relevance and the direction of descriptors effect in the developed model. The mean effect can be evaluated using Eq. 5.
where \({\beta }_{j}\) stands for the coefficient of j, Dj stands for the training set value of the matrix descriptors and m, the descriptors sum in the model, and n is the training set count [12].
2.7 Molecular design
The careful analyzes of the molecular features play a significant role in designing compounds with improved antimalarial activities. Structural modifications of the molecule with the highest activity (template) were carried out by substitutions of groups with others using the information retrieved from the mean effect analysis, thereby designing several derivatives. The theoretical activities of the designed derivatives were estimated after optimizing the derivatives and their molecular descriptors calculated.
3 Results and discussions
3.1 Models developed
The results of the QSAR analysis of 2-anilino 4-amino substituted quinazolines derivatives were presented in Table 2.
3.2 Model selection
The statistical parameters of the generated models (Table 2) show the significant and dependable nature of the models. From these, model C was selected as the best model due to its highest external validation, the predictive R2 (R2pred = 0.765), despite not having the highest coefficient of determination (R2 = 0.7913), and internal validation coefficient (Q2 = 0.7112). The selected model is a five parametric equation with ATSC8c, GATS8i, SpMin1_Bhi, JGI10, and TDB6u as the contributive descriptors, whose definitions are provided in Table 3.
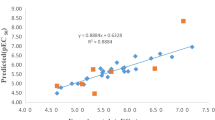
The plots of the predicted antimalarial activities of the 2-anilino 4-amino substituted quinazolines derivatives against their experimental activities (Fig. 1), shows the linearity of the data set around the training set legendary line. The correlation matrix of the selected model descriptors, Table 4, shows low values of the correlation coefficient between descriptors which may indicate a lack of collinearity among the descriptors. Further analysis of the selected model ensured that the determined variation inflation factors (VIF), which study the collinearity existing between the descriptors revealed that the VIF values of the model descriptors all fall below 2 (Table 4). These low VIF values which ensure the orthogonality among the descriptors were found to be within the allowed VIF range of 1 < VIF ≤ 5 [13], 18], hence indicates the model acceptability.

The predicted against the experimental pEC50 values for the training as well as the test sets
3.3 Model validation
Model C, selected to be the best model was internally as well as externally validated. The internal validation involves the determination of both the leave-one-out (LOO) and leave-5-out (L5O) cross-validations, as well as the Y-randomization. The LOO and L5O cross-validation produced correlation coefficients, R2 = 0.7144 and 0.6754 respectively. The high values of the results are an indication that the model does not occur by chance and can predict reasonably. The results of the Y-randomization after 10 different randomization trials, show the R2 and Q2 to have low values compared to those of the original model as reflected in Table 5, which shows the robustness of the model. In the external validation, the square of the regression coefficient (coefficient of determination) was calculated to be R2pred = 0.765, which is again greater than the minimum value for model acceptance.
3.4 Model applicability domain (AD)
The end-use of a developed QSAR model is for property prediction purposes and will predict effectively only compounds found within the model applicability domain. The applicability domain, Fig. 2, shows that all the data set were found to fall the domain with no outliers except compound 10 having the standardized residual greater than 3σ. The threshold (h*) of the model was calculated to be h* = 0.514, and no compound was found beyond the threshold, which points to the model strength of prediction.

The graph of standardized residuals of the data set against their leverage values
3.5 Descriptors mean effect
The percentage contribution of the model descriptors towards the antimalarial activity was determined and the results were displaced in a chart presented in Fig. 3. The first descriptor in the model is ATSC8c, a centered Broto-Moreau autocorrelation—lag 8/weighted by charges contributes 39% to the activity. The mean effect shows the descriptor to have a positive charge, indicating an increase in activity with an increase in the descriptor. The second descriptor, GATS8i, Geary autocorrelation—lag 8/weighted by first ionization potential has about 6% contribution to the activity, and since it is negatively charged, decreasing the descriptor value, increases the activity of the compound. Descriptor SpMin1_Bhi (smallest absolute eigenvalue of Burden modified matrix – n 1/weighted by relative first ionization potential) was found to contribute more (54%) to the antimalarial activity and is positively charged, hence, increasing the descriptor values increases the activity. The next descriptor determines charge transference between two atoms separated by 10 bonds [7], JGI10, with 1% contributions, increases the activity by increasing its values. The last descriptor is the positively charged TDB6u that belongs to the TDB descriptors. These descriptors sum up the products of bonds found between two atoms i and j [14]. Having a positive mean effect, the activity increases with increasing the value of the descriptor.

Descriptors Mean Effect 3-D Pie
3.6 Molecular design
The structural modification of compound 13, N4-benzyl-N2-(4-fluorophenyl)-6,7-dimethoxyquinazoline-2,4-diamine, Fig. 4 the most active compound (pEC50 = 7.387) as the template could be exploited in the design of several derivatives with improved antimalarial activities. The modification was done using the most contributive descriptor, SpMin1_Bhi by increasing the first ionization potentials of the template. The first ionization potentials of a substituted system are increased by electron attracting groups such as –CN, –CF3, –COCH3, –Cl, –F, –I, –COOH, –COCl2, –CN, etc. substituting these electron attracting groups at the meta positions of the substituted compounds produce derivatives of the template with improved malarial activities. Ten (10) theoretical derivatives of the template with improved theoretical activities were designed as shown in Table 6. The activities of five (5) of such compounds (3, 4, 5, 6, and 8) were found to be better than that of chloroquine standard with compound 3, N4-(3-bromo-5-fluorobenzyl)-N2-(4-fluorophenyl)-6,7-dimethoxyquinazoline-2,4-diamine as the most active of the theoretical compounds.

Design template, N4-benzyl-N2-(4-fluorophenyl)-6, 7-dimethoxyquinazoline-2,4-diamine (pEC50 = 7.3870)
3.7 Conclusion
This research aims to design enhanced antimalarial derivatives of 2-Anilino 4-Amino Substituted Quinazolines from a developed activity model. Molecular descriptors ATSC8c, GATS8i, SpMin1_Bhi, JGI10, and TDB6u were discovered to be significant to the antimalarial property of the compounds. The mean effect revealed SpMin1_Bhi, as the most influential descriptor and was essential in designing the ten (10) by-products through structural modifications of the template N4-benzyl-N2-(4-fluorophenyl)-6,7-dimethoxyquinazoline-2,4-diamine. The derivatives show better activities than that of the design template, with compounds 3, 4, 5, 6, and 8 showing even better activities than the chloroquine standard drug. When clinically validated, these compounds may pave way for more potent malarial inhibitors.
References
Ambure P, Roy K (2017) CADD Modeling of multi-target drugs against Alzheimer’s disease. Curr Drug Targets 18(5):522–533. https://doi.org/10.2174/1389450116666150907104855
Arthur DE, Uzairu A, Mamza P, Stephen AE, Shallangwa G (2016) Quantum modeling of the structure-activity and toxicity relationship studies of some potent compounds on SR leukemia cell line. Chem Data Collect 5–6:46–61. https://doi.org/10.1016/j.cdc.2016.10.004
Azmi HF, Lhaksmana KM, Kurniawan I (2020) QSAR study of fusidic acid derivative as anti-malaria agents by using artificial neural network-genetic algorithm. 2020 8th international conference on information and communication technology (ICoICT). doi:https://doi.org/10.1109/icoict49345.2020.9166158
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Garraway LA (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483(7391):603–607. https://doi.org/10.1038/nature11003
Chakraborty A (2016) Emerging drug resistance in Plasmodium falciparum: a review of well-characterized drug targets for novel antimalarial chemotherapy. Asian Pac J Trop Dis 6(7):581–588. https://doi.org/10.1016/s2222-1808(16)61090-3
Da Silva AP, de Angelo RM, de Paula H, Honório KM, da Silva ABF (2020) Drug design of new 5-HT6 antagonists: a QSAR study of arylsulfonamide derivatives. Struct Chem. https://doi.org/10.1007/s11224-020-01513-z
Freitas HF, Barros TF, Castilho MS (2013) 2D Chemometric studies of a series of Azole derivatives active against Fluconazole-ResistantCryptococcus gattii. J Braz Chem Soc. https://doi.org/10.5935/0103-5053.20130122
Gilson PR, Tan C, Jarman KE, Lowes KN, Curtis JM, Nguyen W, Sleebs BE (2017) Optimization of 2-Anilino 4-Amino substituted Quinazolines into potent antimalarial agents with oral in vivo activity. J Med Chem 60(3):1171–1188. https://doi.org/10.1021/acs.jmedchem.6b01673
Habibi-Yangjeh A, Pourbasheer E, Danandeh-Jenagharad M (2008) Application of principal component-genetic algorithm-artificial neural network for prediction acidity constant of various nitrogen-containing compounds in water. Monatshefte Für Chemie Chem Month 140(1):15–27. https://doi.org/10.1007/s00706-008-0049-7
Hadni H, Elhallaoui M (2020) 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. New J Chem. https://doi.org/10.1039/c9nj05767f
Hammoudi NEH, Benguerba Y, Sobhi W (2019) QSAR Modeling of thirty active compounds for the inhibition of the Acetylcholinesterase enzyme. Curr Res Bioinform 8(1):62–65. https://doi.org/10.3844/ajbsp.2019.62.65
Ibrahim ZY, Uzairu A, Shallangwa G, Abechi S (2020) In-silico design of Aryl and Aralkyl Amine-Based Triazolopyrimidine derivatives with enhanced activity against resistant plasmodium falciparum. Chem Africa. https://doi.org/10.1007/s42250-020-00199-4
Kim JH (2019) Multicollinearity and misleading statistical results. Korean J Anesthesiol 72(6):558–569. https://doi.org/10.4097/kja.19087
Klein CT, Kaiser D, Ecker G (2004) Topological distance based 3D descriptors for use in QSAR and diversity analysis. J Chem Inf Comput Sci 44(1):200–209. https://doi.org/10.1021/ci0256236
Ouattara A, Laurens MB (2015) Vaccines against malaria. Clin infect dis An Off Publ Infect Dis Soc Am 60(6):930–936. https://doi.org/10.1093/cid/ciu954
Panagos P, Meusburger K, Ballabio C, Borrelli P, Alewell C (2014) Soil erodibility in Europe: a high-resolution dataset based on LUCAS. Sci Total Environ 479–480:189–200. https://doi.org/10.1016/j.scitotenv.2014.02.010
Pourbasheer E, Aalizadeh R, Ardabili JS, Ganjali MR (2015) QSPR study on solubility of some fullerenes derivatives using the genetic algorithms — Multiple linear regression. J Mol Liq 204:162–169. https://doi.org/10.1016/j.molliq.2015.01.028
Senaviratna NAMR, Cooray TMJA (2019) Diagnosing multicollinearity of logistic regression model. Asian J Prob Stat 5(2):1–9
Srivastav VK, Tiwari M (2017) QSAR and docking studies of coumarin derivatives as potent HIV-1 integrase inhibitors. Arab J Chem 10:S1081–S1094. https://doi.org/10.1016/j.arabjc.2013.01.015
Tropsha A, Gramatica P, Gombar V (2003) The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 22(1):69–77. https://doi.org/10.1002/qsar.200390007
Wellems TE, Plowe CV (2001) Chloroquine-resistant malaria. J Infect Dis 184(6):770–776. https://doi.org/10.1086/322858
WHO (2017) World malaria report, Geneva Switzerland
Funding
The authors for this research did not receive any funding concerning this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Human and animal rights
No human or animal subjects were involved in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ibrahim, Z.Y., Uzairu, A., Shallangwa, G.A. et al. Computer-aided molecular design of 2-anilino 4-amino substituted quinazolines derivatives as malarial inhibitors. SN Appl. Sci. 3, 767 (2021). https://doi.org/10.1007/s42452-021-04748-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04748-5