
Abstract
Doping is promising strategy for the alteration of nanomaterials to enhance their optical, electrical, and catalytic activities. The development of electrocatalysts for oxygen reduction reactions (ORR) with excellent activity, low cost and durability is essential for the large-scale utilization of energy storage devices such as batteries. In this study, MnO2 and Ni-doped MnO2 nanowires were prepared through a simple co-perception technique. The influence of nickel concentration on electrochemical performance was studied using linear sweep voltammetry and cyclic voltammetry. The morphological, thermal, structural, and optical properties of MnO2 and Ni-doped MnO2 nanowires were examined by SEM, ICP-OES, FT-IR, XRD, UV–Vis, BET and TGA/DTA. Morphological analyses showed that pure MnO2 and Ni-doped MnO2 had flower-like and nanowire structures, respectively. The XRD study confirmed the phase transformation from ε to α and β phases of MnO2 due to the dopant. It was also noted from the XRD studies that the crystallite sizes of pure MnO2 and Ni-doped MnO2 were in the range of 2.25–6.6 nm. The band gaps of MnO2 and 0.125 M Ni-doped MnO2 nanoparticles were estimated to be 2.78 and 1.74 eV, correspondingly, which can be seen from UV–Vis. FTIR spectroscopy was used to determine the presence of functional groups and M–O bonds (M = Mn, Ni). The TGA/TDA examination showed that Ni-doping in MnO2 led to an improvement in its thermal properties. The cyclic voltammetry results exhibited that Ni-doped MnO2 nanowires have remarkable catalytic performance for ORR in 0.1 M KOH alkaline conditions. This work contributes to the facile preparation of highly active and durable catalysts with improved catalytic performance mainly due to the predominance of nickel.
Article Highlights
-
MnO2 and Ni-doped MnO2 nanowires were synthesized via a facile co-perception approach.
-
Nickel doping in MnO2 induces the formation of wire-like nanostructures.
-
Nickel doping enhances the electrochemical activity and thermal stability of MnO2 nanoflowers.
-
The addition of nickel into MnO2 promoted the catalytic activity for oxygen reduction reaction.
-
A higher catalytic activity was achieved in 0.125 M Ni-MnO2 nanowires.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Globally, the focus on low-carbon emissions and the concept of sustainability has notably proposed the development of sustainable energy sources [1, 2]. Electrochemical energy storage tools such as metal-air batteries (MABs) and fuel cells are considered to play a significant impact in the transformation to a sustainable prospect [3,4,5]. Moreover, one of the main problems that restrict the application of lithium-ion batteries for electric vehicles is its low specific energy density [6, 7]. Recently, special attrition is given to the performance enhancement of MABs due to their extraordinary ideal energy density and low cost, mainly in lithium-air and zinc-air batteries (ZABs) [8,9,10]. Hence, ZABs have a low cost, comparatively higher energy density, and excellent safety [11, 12]. However, the fabrication with effective air electrodes is found to be the critical problem associated with ZABs [13, 14]. The challenges of air electrodes can be solved by preparing active, efficient, stable and durable oxygen catalysts for the ORR [15]. The ORR is a vital cathodic reaction in fuel cells and MABs [16, 17]. Precious metals such as Platinum (Pt) and Pt-based alloys are the most effective ORR electrode substances [18, 19]. However, the application of Pt and its derivatives as ORR catalysts is limited because of their poor stability, limited access, and high price [20, 21]. Hence, to address these concerns, strategies have been designed to prepare highly-active, durable, and more stable non-precious metal-based ORR catalysts [22, 23]. Transition metals, metal oxides, and carbon-based materials can be used as alternative electrocatalysts [24, 25]. Among transition metal oxides, MnO2 can be used as an air electrocatalyst for the ORR due to its excellent catalytic performance, abundance, low price, and environmental safety [26,27,28]. Moreover, MnO2 is particularly promising material because of its mild preparation, stable thermal behavior, and excellent overall performance [29, 30]. However, the performance of MnO2 is restricted by its inherently poor electronic conductivity, low structural stability, and ion conductivity [31, 32]. Hence, the emerging approach to increase the electronic conductivity and electrochemical performance of MnO2 nanoparticles (NPs) is to combine with high conductive substances such as graphene, carbon nanotubes, and carbon fibers [33, 34]. An alternative approach is defect engineering, in which foreign elements should be doped to improve the inherent electronic conductivity and enhance the concentration of redox-active cites [35, 36]. The incorporation of different foreign element into MnO2 NPs, particularly defect engineering, has been considered as an effective technique to enhance its electronic conductivity [37]. For enhanced ORR catalyst activity, different metals have been doped into MnO2 such as Iron (Fe), Nickel (Ni) [38], Chromium (Cr), Cobalt (Co) [39], Copper (Cu) [40], Molybdenum (Mo) [41], Aluminum (Al) [42], and Vanadium (V) [43]. Those results exhibited that doping improves the overall performance of MnO2 NPs [44]. One on hand different approaches have been investigated for the synthesis of MnO2 NPs such as emulsion, sol–gel, co-precipitation, thermal decomposition [45], simple reduction, and hydrothermal approach [46, 47]. However, those approaches are expensive, needs sophisticated instruments, and relatively longer preparation time [48, 49]. Among these, the co-precipitation approach is advantageous due to easy, fast, cost-effective, and easy to control during preparation [50, 51]. Many studies have been investigated on optical, electrical, and magnetic characteristics of Ni doped MnO2 nanoparticles [52]. However, only a few studies have been reported about the structural, morphological, thermal, and electrochemical characteristics of Ni-doped MnO2 NPs by defect engineering at ambient conditions. Herein, we examined the influence of nickel doping on structural, morphological, thermal, and electrochemical characteristics of MnO2 NPs prepared by a facile co-precipitation approach [53, 54].
2 Materials and methods
2.1 Materials
All chemicals and reagents were used without any refinement and of analytical grade. Ethanol, Vulcan XC 72 carbon powder, and isopropanol were obtained from India (Alpha Chemika). Potassium permanganate (KMnO4, 99.5%), nickel (II) nitrate hexahydrate (Ni(NO3)2·6H2O, 98%) and manganese sulfate monohydrate (MnSO4·H2O, 99%) were obtained from Fluka (Germany). The solutions were prepared by using distilled water.
2.2 Synthesis of Ni-doped MnO2 NPs
Nickel doped-MnO2 nanowires were developed using a facile co-precipitation technique. In a typical synthesis, 0.3 M of MnSO4·H2O and the preferred mole of (0.025, 0.05, 0.75, 0.1, and 0.125 M) Ni(NO3)2·6H2O were dissolved in 100 mL distilled water. Then, 0.2 M potassium permanganate solution dissolved in 100 mL of distilled water was added drop wise to the combination of Ni(NO3)2·6H2O and MnSO4·H2O. The mixture was stirred at 300 rpm at 60 °C for 5 h. The pH of the mixture was adjusted to 12 by adding 0.2 M NaOH solution. Then, the product was filtered and washed with distilled water and ethanol. Moreover, the as-prepared samples were dried at 80 °C for 8 h to evaporate water and organic materials. Finally, the products were annealed in a muffle furnace at 500 °C for 4 h. A similar technique was used to prepare pure MnO2. The detailed schematic illustration of Ni-doped MnO2 nanowires prepared by the co-precipitation route is showed in Fig. 1.

The synthesis route of Ni-doped MnO2 nanowires prepared via co-precipitation technique
2.3 Characterization
The functional groups were studied using Fourier transform infrared spectrometer (FT-IR, FT-IR 6660 (JASCO MODEL)) in the wavenumber range of 4000–400 cm−1, in a standard KBr pellet method. The crystal structures of the synthesized nanoparticles were examined using X-ray diffraction (MAXima_X XRD-7000, SHIMADZU) using radiation of Cu-Kα. XRD analysis at room temperature in theta–2 theta range between 10° and 80°. It was continuously scanned at a step size of 0.02° with a counting time of 0.30 s per step with a scan speed of 4.0 (deg/min) operating at 30 mA and 40 kV with a divergence slit of: 1.0 (°), scatter slit: 1.0 (°), receiving slit: 0.30 (mm). Scanning electron microscopy (SEM, INSPECT F50) was used to examine the morphological properties of as-prepared NPs. A thermal property study was performed using the TGA/DTA study. Ultraviolet–visible spectrophotometer (UV–Vis, Lambda 35 (PerkinElmer)) in the wavelength range of 200–800 nm was conducted to analyze the optical characteristics of prepared NPs. Brunauer–Emmett–Teller (BET, Quanta chrome Instruments version 11.0) was used to determine the specific surface areas of as-prepared nanoparticles. After dissolving the synthesized materials in HCl and HNO3 aqueous solution, the composition of Ni and Mn in the Ni-MnO2 product was analyzed via inductively coupled plasma optical emission spectrometry (ICP-OES, Perkin-Elmer 800). Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) were done for electrochemical analysis using CHI760E electrochemical workstation.
2.4 Preparation of working electrode
To examine the electrochemical characteristics of the developed MnO2 and Ni-doped MnO2 NPs, the working electrode was fabricated as follows: MnO2 and Ni-doped MnO2 nanoparticle samples were combined with carbon powder at a mass ratio of 30:70 to ensure adequate electronic conductivity. Then, 5 mg of MnO2 and Ni-doped MnO2 catalysts (dispersed in 1 mL mixed solution of water/isopropanol, 2.5:1 v/v) was sonicated for 30 min to form a uniform ink. Then 5 µL of the catalyst solution was pipetted onto a glassy carbon electrode (GCE) of 3 mm diameter and dried at room temperature.
2.5 Electrochemical characterization
Electrochemical analyses were accomplished using a computer-controlled potentiostat (CHI760E electrochemical workstation) equipped with a three-electrode conventional cell. The electrocatalytic characteristics were analyzed by LSV and CV. CV was estimated in a 0.1 M KOH electrolyte using platinum coil and Ag/AgCl as a counter and reference electrode, respectively. All potentials stated in this study were referenced to the Ag/AgCl electrode. For CV analyses, the working electrode was examined between + 0.1 to − 0.7 V at a sweep rate of 5, 10, 20, 50, and 100 mV s−1 at room temperature via saturating the electrolyte through O2 for 30 min. LSV analysis was done at a scan rate of 5 mV s−1 in + 0.1 to − 0.7 V (vs. Ag/AgCl) of potential range.
3 Results and discussion
3.1 Structure analysis
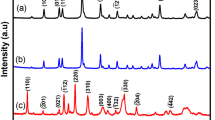
The crystalline size, structure, and phase purity, of the MnO2, and Ni-MnO2 samples were examined using XRD. The XRD patterns of the MnO2 and Ni-MnO2 nanoparticles are shown in Fig. 2. Figure 2a shows the diffraction peaks of MnO2 at 2θ angles of 37.03°, 42.65°, 56.44°, and 66.11° are attributed to the diffraction of (1 0 0), (1 0 1), (1 0 2), and (1 1 0) crystal planes of akhtenskite ε-MnO2 (JCPDS 00–030-0820). In contrast, the Ni-doped MnO2 sample in Fig. 2b, c shows sharp diffraction peaks at 28.55°, 37.03°, 42.75°, 56.44°, and 72.58°, which can be indexed to the (110), (101), (111), (211), and (112) crystal planes of β-MnO2 (JCPDS card 00-024-0735) and the peaks at 2θ values of 37.13°, 62.46°, and 76.44°, were corresponding to the (111), (220), and (311) planes of NiO (JCPDS card no: 00-047-1049), respectively. The XRD pattern with a sharp peak indicates the excellent crystallinity of the prepared sample. The sharp diffraction peaks at 2θ of 12.72°, 17.93°, 28.65°, 37.03°, 42.44°, 49.70°, 56.65°, 60.11°, 65.84°, 69.10°, and 72.98°, match to the (110), (200), (310), (211), (301), (411), (600), (521), (002), (541), and (312) are the crystal planes of α-MnO2 (JCPDS card NO. 00-044-0141) [55]. Whereas the diffraction peaks shown in Fig. 2e–f at 2θ angles of 37.03°, 42.65°, 56.44°, and 66.11° correspond to the (1 0 0), (1 0 1), (1 0 2), and (1 1 0) crystal planes of akhtenskite ε-MnO2 (JCPDS 00–030–0820) and the peaks at 2θ values of 37.13°,, 62.46°, and 76.44° are correspond to the (111), (220), and (311) crystal planes of NiO (JCPDS card no: 00–047-1049). It was also noted that nickel contents has an effect on crystal structure of MnO2. The slight change in the peak intensity of nickel-doped MnO2 samples are due to the size change of the nanoparticles. Thus, nickel doping promotes the formation of α, and β-MnO2 nanoparticles [56].

XRD patterns of (a) MnO2, (b) Ni-MnO2 (0.025 M), (c) Ni-MnO2 (0.05 M), (d) Ni-MnO2 (0.075 M), (e) Ni-MnO2 (0.1 M), (f) Ni-MnO2 (0.125 M) NPs
3.2 Crystallite size analysis
3.2.1 Scherrer technique
The average crystallite sizes of pure MnO2 and Ni-MnO2 NPs at different doping level were determined by using the Scherrer Eq. (1) [57].
where λ, K, D, θ, and β are wavelength of the x-ray sources, Scherer constant, crystallites size, peak position and full width at the half maximum intensity (FWHM), respectively [2]. The estimated crystallite size of MnO2, Ni-MnO2 (0.025 M), Ni-MnO2 (0.05 M), Ni-MnO2 (0.075 M), Ni-MnO2 (0.1 M), and Ni-MnO2 (0.125 M) NPs were 2.25, 4.03, 5.3, 6.6, 5.52, and 6.1 nm, correspondingly (Table 1). The dislocation density and strain of the MnO2 and Ni-MnO2 NPs are other vital determinants that can be calculated from the XRD analysis. The dislocation density (δ) values were estimated using Eq. (2).
where D and δ are crystallites size and dislocation density, respectively. Hence, the addition of dopant increases the dislocation density of the NPs.
3.2.2 Williamson–Hall method
A Williamson–Hall plot was drawn to determine the lattice strain and effective crystallite size using Eq. (3) [7]:
where K, β, D, λ and ε are shape factor, full width hall maximum, crystallite size, wavelength, and microstrain, respectively. Therefore, the graph was plotted within “4sinθ” on x-axes and “βcosθ” on the y-axis, and a straight line was obtained. The intercept of a straight line is the crystallite size whereas slope provides the value of the microstrain as presented in Fig. 3. The crystallite size of MnO2, Ni-MnO2 (0.025 M), Ni-MnO2 (0.05 M), Ni-MnO2 (0.075 M), Ni-MnO2 (0.1 M), and Ni-MnO2 (0.125 M) NPs were 1.43, 1.73, 3.24, 3.77, 1.39, and 4.92 nm, correspondingly (Table 1). It was noticed that both methods gave very close crystallite size.

Williamson Hall plots of (a) pure MnO2, (b) 0.025 M Ni-MnO2, (c) 0.05 M Ni-MnO2, (d) 0.075 M Ni-MnO2, (e) 0.1 M Ni-MnO2, and (f) 0.125 M Ni-MnO2 NPs
3.3 Morphology analysis
The morphologies of MnO2 and Ni-doped MnO2 were studied using SEM. Figure 4 displays the SEM image of MnO2 NPs, which have a flower-like structure and a diameter of 4.7 nm. This flower-like structure is composed of different self-assembled irregular nanosheets. On the other hand, 0.025 M Ni-doped MnO2 has agglomerated small nanoneedles of porous morphology with an average particle size of 7.13 nm (Fig. 5). This suggested that, the addition of nickel affected the surface morphology of MnO2, as shown in Fig. 5. Moreover, 0.125 M Ni-doped MnO2 consisted of nanowires with an average particle size of 7.07 nm, which implies the key role of Ni+2 is regulating the nanostructures of MnO2 (Fig. 6). The average diameter of as-prepared nanowires were analysed via particle size distribution histogram computed from SEM image via Image J software as shown in Fig. 7a–c. Generally, when Ni2+ were added the original flower-like structures started to dissolve and slowly change into the shape of the sphere and finally nanowires. It can be noted from the SEM images that there is an obvious distinction in morphologies between pure and Ni-doped MnO2 nanomaterials indicating that the doping level of Ni cations has a vital effect on the morphologies of MnO2 NPs. Thus, Ni-doped MnO2 NPs show high porosity and better particle dispersion than that of MnO2 NPs, which is a promising property to enhance the catalytic performance of the as-prepared NPs.

SEM images of a pure MnO2 NPs at 400 nm, b pure MnO2 NPs at 5 µm c pure MnO2 NPs at 10 µm

SEM images of a 0.0125 M Ni-doped MnO2 NPs at 500 nm, b 0.0125 M Ni-doped MnO2 NPs at 3 µm, and c 0.0125 M Ni-doped MnO2 NPs at 10 µm

SEM images of a Ni-doped MnO2 (0.125 M) NPs at 500 nm, b Ni-doped MnO2 (0.125 M) NPs at 1 µm, and c Ni-doped MnO2 (0.125 M) NPs at 3 µm

Histogram scheme showing a particle size distribution of a MnO2, b 0.025 M Ni-doped MnO2 and c 0.125 M Ni-doped MnO2 NPs
3.4 ICP-OES analysis
Elemental compositions studies of the as-prepared samples were examined by ICP-OES to confirm the amount of Mn and Ni loaded in Ni-doped MnO2 nanoparticles. ICP-OES analysis were conducted via dissolving the as-prepared nanoparticle in a concentrated acidic solution (HCl:HNO3, 3:1 v/v by volume). Table 2 displays the content of Ni and Mn in the Ni-doped MnO2 nanoparticles. Moreover, the Ni/Mn doped molar ratio in Ni-doped MnO2 nanoparticles were 0.025, 0.066, 0.086, 0.137 and 0.103 for Ni-MnO2 (0.025 M), Ni-MnO2 (0.05 M), Ni-MnO2 (0.075 M), Ni-MnO2 (0.1 M), and Ni-MnO2 (0.125 M) nanoparticles, respectively. Hence, the ICP-OES analysis results showed that 0.1 M Ni-MnO2 and 0.125 M Ni-MnO2 nanoparticles have the higher Ni/Mn molar ratio than the others, which affects the thermal, structural, and electrochemical properties of the nanoparticles [58].
3.5 Surface area analysis
BET analysis techniques have been utilized to determine the textural properties such as specific surface areas, pore volume and pore size of MnO2 and Ni-MnO2 nanoparticles. Table 3 presents the BET surface area, pore radius and pore volume of MnO2, Ni-MnO2 (0.1 M), and Ni-MnO2 (0.125 M) NPs. The BET analysis display a specific surface area of 95.619, 103.475, and 124.386 m2 g−1 for MnO2, Ni-MnO2 (0.1 M) and Ni-MnO2 (0.125 M) NPs, respectively. The corresponding pore radius was 34.056, 34.92, and 34.257 Å for MnO2, 0.1 M Ni-MnO2, and 0.125 M Ni-MnO2 NPs, respectively. Hence, 0.125 M Ni-MnO2 nanoparticles exhibited the largest BET surface area, which is ascribed to porous structure. The porous structure enables the transfer and diffusion of electrolyte ions through the charging and/or discharging process. Thus, the doping of nickel in MnO2 nanoparticles leads to an increment of the BET surface area [59].
3.6 FT-IR analysis
FT-IR spectroscopy was accomplished within 400 to 4000 cm−1 ranges of the electromagnetic spectrum. Figure 8 illustrates the typical FT-IR spectra of MnO2 and Ni doped MnO2 NPs at various doping level. As shown in Fig. 8, the band approximately at 3433 cm−1 shows O–H stretching in water molecules, and the small band nearly at 1633 cm−1 may be because of bending vibration of O–H groups in the adsorbed water molecules. Moreover, the bands approximately at 1386 and 1092 cm−1 match to the coordination of Mn by the O–H. Finally, the characteristic peak at 578 cm−1, and 518 cm−1 can be associated with the stretching vibrations of M–O or M–O–M (M = Ni, Mn). Hence, the appearance of M–O or M–O–M bond and O–H was confirmed from the FTIR band of MnO2 and Ni-MnO2 NPs. The FT-IR spectra observed in this study are consistent with previously reported results [59].

FTIR spectra of MnO2, Ni-MnO2 (0.025 M), Ni-MnO2 (0.05 M), Ni-MnO2 (0.075 M), Ni-MnO2 (0.1 M), and Ni-MnO2 (0.125 M) NPs
3.7 UV–visible analysis
To study optical characteristics of the as-prepared samples, UV–Vis analysis were performed between 200 and 800 nm. Figure 9a illustrates UV–visible spectrum of MnO2 and Ni-doped MnO2 NPs. It was observed that MnO2 showed absorption peaks at 318 nm [40]. Furthermore, the characteristics absorption bands at 378 nm corresponded to Ni-doped MnO2 nanoparticles sample. The optical band gap of MnO2 and Ni-doped MnO2 sample were computed using Tauc relationship (Eq. 1) [60].
where α, hν, and A are the absorption coefficient, photon energy and constant. Figure 9b shows the energy band gap values of MnO2 and Ni-MnO2 NPs computed by extrapolating the linear part of these plots of (αhν)2 axis to (hν) axis. The results showed that 2.78 and 1.74 eV were band gap energy of MnO2 and Ni-doped MnO2 NPs, respectively. UV–Vis investigation showed that Ni-doped MnO2 NPs with enhanced electronic conductivity.

UV–Vis spectra of a pure MnO2 and Ni-MnO2 NPs, and b Tauc scheme of pure MnO2 and Ni-MnO2 NPs
3.8 Thermal analysis
TGA and DTA analysis were investigated to study the thermal and phase evolution properties of pure MnO2 and Ni-MnO2 NPs. The as-prepared NPs were heated in the range of 25–950 °C with 10 °C min−1 heating rate in air atmosphere. Figure 10 presents the TGA/DTA curve of pure MnO2 and Ni-MnO2 NPs. In the TGA curve of pure MnO2, the first (0.47 mg) weight loss occurs between 25 and 278 °C, and the corresponding endothermic peak observed at 70 °C may be because of the loss of adsorbed water. The next weight loss of 0.43 mg in the range of 278–564 °C can be due to the phase change of MnO2 to Mn2O3. Thus, the weight loss of 0.19 mg in between 564 and 838 °C can be associated with the phase change of Mn2O3 to Mn3O4. Moreover, in the DTA curve of pure MnO2 the two sharp exothermic and endothermic peaks that arise at 588 °C and 815 °C, respectively that might be due to a variation in crystallinity or possibly due to a phase transition in the NPs. A final weight of 6.91 mg was obtained from the initial weight of 8 mg which corresponds to 13.6% weight loss when heated to 950 °C (Fig. 10a). Similarly, the 0.125 M Ni-MnO2 TGA curve, shows the first weight loss of 0.55 mg approximately at 385 °C, and the corresponding endothermic peak at 117 °C may be due to loss of physically adsorbed water (Fig. 10b). The next weight loss of 0.17 mg in the range of 385–569 °C could be due to the phase change of MnO2 to Mn2O3. Finally, the weight loss of 0.27 mg in between 569 and 882 °C can be associated with the phase change of Mn2O3 to Mn3O4. Moreover, in the DTA curve of pure MnO2, the two sharp exothermic and endothermic peaks that arise at 586 °C and 874 °C, respectively, which might be due to a variation in crystallinity or possibly due to a phase transition in the NPs. Hence, we observe that the study was begun with 8 mg of catalyst, and after complete heating up to 950 °C, the catalyst remained was 7.01 mg. Thus, the percentage weight loss for our analysis up to 950 °C, is 12.36%. Generally, in our analysis weight loss of 13.63% and 12.36% is associated to MnO2 and Ni-MnO2 NPs, correspondingly. Hence, our analysis showed that Ni-MnO2 NPs with improved thermally stability. The TGA and DTA result showed similarities with the previously reported results. However, slight change in the peak positions were observed at different stages, which may be due to the use of different reagents in the synthesis process [61].

TGA/DTA illustration of a MnO2 NPs, and b Ni-doped MnO2 (0.125 M) NPs
3.9 Electrochemical studies
3.9.1 CV study
The catalytic activity of un-doped MnO2 and Ni-MnO2 electrocatalysts were analyzed using CV in O2-saturated KOH (0.1 M) solution (Fig. 11). Figure 11a which exhibits the (CV) curves of pure MnO2, 0.1 M Ni-MnO2, and 0.125 M Ni-MnO2 in a 0.1 M KOH solution. For pure MnO2 electrode, the reduction peak was present at − 0.361 V (vs. Ag/AgCl). The ORR peak potential of 0.1 M Ni-MnO2, and 0.125 M Ni-MnO2 is placed at − 0.348 V, and − 0.348 V (vs. Ag/AgCl), correspondingly. The result showed that the 0.1 M Ni-MnO2 and 0.125 M Ni-MnO2 reduction potential were shifted by 13 mV to the positive direction compared to pure MnO2. Moreover, at the potential of − 0.348 V (vs. Ag/AgCl), the current of the ORR at the electrode with a catalyst 0.125 M Ni-MnO2 is 38.56 µA, which is higher than that of the electrode with pure MnO2 (32.17 µA). The ORR peak current of Ni-MnO2 catalyst is bigger than that of pure MnO2 catalyst [24]. The findings are Our work in agreement with previously reported results [44, 62]. It can be said that the as-prepared MnO2 and Ni-doped MnO2 electrocatalysts are to be an ideal air electrode material for metal-air batteries and fuel cells. Figure 11b, c illustrates the CV illustration of un-doped MnO2 and Ni-MnO2 nanowires at numerous scan rates. The effect of scan rate in between 5 and 100 mV/s was studied at pure MnO2 and 0.125 M Ni-MnO2 modified electrode. Figure 11b, c shows the largest variation between the four ORR curves where a negative peak potential shifts with increase of scan rate are observed, which is detected when the oxidation reaction is irreversible. Hence, the ORR activities of MnO2 and Ni-MnO2 catalysts are also dependent on the scan rates. The result showed that excellent ORR catalytic activity can be obtained at lower scan rate [63, 64]. Figure 12 shows the ORR activity of pure MnO2, and 0.125 M Ni-MnO2 modified electrode before saturation and after oxygen saturation. The result showed that un-saturated pure MnO2 with better catalytic performance but poor current intensity. Moreover, un-saturated 0.125 M Ni-MnO2 modified electrode showed poor catalytic performance and current intensity. It is notably seen that nickel doped (0.1 M Ni-MnO2 and 0.125 M Ni-MnO2) electrode displayed excellent reduction peak potential, which confirms a higher electrocatalytic activity to ORR due to the synergistic effect of nickel and MnO2.

CV curve of; a pure MnO2 and Ni doped MnO2 nanoparticles at 50mv/s scan rate, b 0.125 M Ni doped MnO2 at different scan rates, and c MnO2 at different scan rates

CV curve of saturated and unsaturated MnO2 and 0.125 M Ni-doped MnO2 NPs at 50 mv/s scan rate
3.9.2 LSV analysis
ORR activity of the un-doped MnO2 and Ni-MnO2 (0.125 M) NPs were evaluated using linear scanning voltammetry (LSV). Figure 13 shows the LSV profile of MnO2 and Ni doped MnO2 (0.125 M) nanoparticles in O2-saturated KOH (0.1 M) alkaline media at a scan rate of 5 mV s−1 from + 0.1 to − 0.7 V (vs. Ag/AgCl). As it can be seen in Fig. 13, the reduction peak potential of MnO2 and 0.125 M Ni doped MnO2 nanoparticles appear at − 0.389 V and − 0.372 V (vs. Ag/AgCl), respectively. Thus, 0.125 M Ni-MnO2 NPs shows relatively better ORR catalytic performance compared to that of un-doped MnO2 with more positive current and peak potential. Hence, more content of Mn3+ in MnO2 and large surface area leads to better electro-catalytic performance of Ni doped MnO2 [65].

LSV sketch of un-doped MnO2 and Ni-MnO2 (0.125 M) NPs at 5 mVs−1 scan rates
3.9.3 CV scans rate analysis
CV analyses were conducted by changing the scan rate between 5 and 100 mV/s (Fig. 14a). Scan rate analysis can reveal whether the electrocatalytic activity of Ni-doped MnO2 NPs on ORR is controlled by diffusion, adsorption and even these two processes. The plot of cathode peak current (ipc) with the scan rate (v)1/2 and the square root of the scan rate (v) are shown in Fig. 14b, c. It is observed that a linear relationship between the cathode peak current versus v1/2 and v values (Fig. 14b, c). The linear relationship of peak current and v1/2 shows a diffusion control process. Moreover, the peak current is also linearly related to v, which means that there is adsorbed electroactive species. These result show that the ORR on the Ni-MnO2/GCE is a combination of kinetic and diffusion control reactions, involving the adsorption of O2 on the surface of the electrode [66]. The involved numbers of electrons (n) in the catalytic reaction of irreversible CV behaviour have been calculated via Laviron equation. The linear relationship among logarithmic sweep rate and peak potential is shown in Fig. 14d. The logarithm of the reduction potential and the scan rate is in the range of 5–100 mV/s as shown in Eq. (5).
In addition, Laviron equation of irreversible CV characteristics is utilized to estimate the transferred number of electrons (n) in the electrocatalytic reaction as shown in Eq. (6) [67]:
where k0, E0′, v, n, α, F, R and T are standard heterogeneous rate constant of the reaction, formal redox potential, scan rate (V s−1), transferred number of electrons, coefficient of transfer, Faraday constant, gas constant and temperature (K), respectively. The slope of Ep versus log v is used to calculate the value of αn. The calculated value of αn was 1.498 taking the slope of the plot (0.03948), as well as considering the value of R, T, K and F are 8.314 J/K mol, 298 K, and 96,480 C/mol, respectively. According to Bard and Faulkner, α can be computed using Eq. (7) [68];
where Ep/2 is the potential when the current is at half the peak value. Therefore, the computed value of α is 0.62. Hence, the number of electrons transferred (n) during the reduction process of Ni-MnO2/GCE surface oxygen is computed to be 3.47. The finding shows that Ni-doped MnO2 NPs are an active electrocatalyst with high catalytic performance towards ORR. Table 4 displays the comparison the electrochemical characteristics of recently studied MnO2 based electrocatalysts. The surface area of the as-prepared Ni-MnO2 nanoparticles in this study is 124.4 m2 g−1 which is in close agreement those reported for Ni-α-MnO2 (52.3 m2 g−1) and Cu-α-MnO2 Nanowire (83.8 m2 g−1). Similarly, the number of transferred electron is slightly greater than that of Cu-α-MnO2 nanowires (n = 3.2–3.3) and Ni-MnO2 nanoneedles (n = 2.33), which show that the enhancement of the electrochemical performance of Ni-MnO2 nanoparticles was observed in this study.

a The CV sketches of Ni-MnO2 nanoparticles at different scan rates for ORR (modified GC electrode). b Sketch of the cathodic peak current with respect to v1/2. c Sketch of the cathodic peak current with respect to v. d Plot of Epc with respect to log v
4 Conclusions
Pure MnO2 and Ni-MnO2 electrocatalysts were prepared using a co-precipitation approach. The synthesized MnO2 and Ni-doped MnO2 electrocatalysts were analyzed using SEM, ICP-OES, FT-IR, XRD, UV–Vis, BET and TGA/DTA. Moreover, the electrochemical behaviors of the prepared catalysts were studied using LSV and CV. The characterization results demonstrated that highly active, cost-effective, and thermally stable electrode materials were achieved through simple co-preciptation approach. The XRD investigation showed that the doping of nickel promotes the formation of α and β-MnO2 NPs. The tetragonal and hexagonal crystal arrangement of α, β, and ε-MnO2 was verified via XRD investigation. The XRD examinations confirmed that the crystallite sizes of MnO2 and Ni-doped MnO2 were in the range of 2.25–6.6 nm. The morphological analyses showed that un-doped MnO2 and 0.125 M Ni-MnO2 have flower-like and nanowire structures with the estimated bandgap energy of 2.78 and 1.74 eV, respectively. It was also inferred that the thermal stability of the synthesized material was improved due to the incorporation of Ni on the MnO2 framework. The Ni-doped MnO2 NPs showed higher catalytic performance, as confirmed by LSV and CV analysis. Overall, Ni-doped MnO2 NPs exhibited excellent catalytic activities, which might be because of enhanced electronic conductivity in the doping process. Hence, the addition of Ni dopants had influenced the physical characteristics of MnO2 NPs and affected the catalytic performance of the pure MnO2 NPs.
References
Kumar A, Rout L, Achary LSK et al (2021) Solvent free synthesis of chalcones over graphene oxide-supported MnO2 catalysts synthesized via combustion route. Mater Chem Phys 259:124019. https://doi.org/10.1016/j.matchemphys.2020.124019
Zhang N, Guo G, He B et al (2020) Synthesis and research of MnO2–NiO composite as lithium-ion battery anode using spent Zn–Mn batteries as manganese source. J Alloys Compd 838:155578. https://doi.org/10.1016/j.jallcom.2020.155578
Bento FR, Corradini PG, Mascaro LH (2019) The critical effect of electrodes components mixing on efficiency of anode material for lithium-ion batteries. Mater Today Commun 21:100668. https://doi.org/10.1016/j.mtcomm.2019.100668
Wang J, Yang J, Sun Y et al (2019) Theoretical insights in Ni doped intrinsic magnetic CrCl3 monolayer. Mater Today Commun 21:100724. https://doi.org/10.1016/j.mtcomm.2019.100724
Yang Y, Meng H, Zhang Y et al (2021) Influence of deposition time on the electrocatalytic activities of core-shell Co(OH)2/NiS/NF electrode. Mater Today Commun 26:101890. https://doi.org/10.1016/j.mtcomm.2020.101890
Shen H, Kong X, Zhang P et al (2021) In-situ hydrothermal synthesis of δ-MnO2/soybean pod carbon and its high performance application on supercapacitor. J Alloys Compd 853:157357. https://doi.org/10.1016/j.jallcom.2020.157357
Salari H, Kohantorabi M (2020) Facile template-free synthesis of new α-MnO2 nanorod/silver iodide p–n junction nanocomposites with high photocatalytic performance. New J Chem 44:7401–7411. https://doi.org/10.1039/D0NJ01033B
Hu Q, Jiang X, He M et al (2020) Core-shell nanostructured MnO2@Co9S8 arrays for high-performance supercapacitors. Electrochim Acta 338:135896. https://doi.org/10.1016/j.electacta.2020.135896
Worku AK, Ayele DW, Habtu NG (2021) Recent advances and future perspectives in engineering of bifunctional electrocatalysts for rechargeable zinc–air batteries. Mater Today Adv 9:100116. https://doi.org/10.1016/j.mtadv.2020.100116
Mahmoud H (2019) Corrosion performance of carbon steel in N-doped mesoporous carbon spheres (NMCS)-containing alkaline medium in presence of chloride. Mater Today Commun 21:100677. https://doi.org/10.1016/j.mtcomm.2019.100677
de Cremonezzi JM, O, Tiba DY, Domingues SH, (2020) Fast synthesis of δ-MnO2 for a high-performance supercapacitor electrode. SN Appl Sci. https://doi.org/10.1007/s42452-020-03488-2
Kumar A, Maitra U (2020) Facile bile salt-induced synthesis of porous MnO2 nanoflowers: applications in dye removal and oxidation. SN Appl Sci. https://doi.org/10.1007/s42452-020-03731-w
Chen M, Cheng Q, Qian Y et al (2020) Alkali cation incorporated MnO2 cathode and carbon cloth anode for flexible aqueous supercapacitor with high wide-voltage and power density. Electrochim Acta 342:136046. https://doi.org/10.1016/j.electacta.2020.136046
Mohamed Racik K, Prabakaran KP, Madhavan J, Antony Raj MV (2019) Surfactant free hydrothermal synthesis, structural and dielectric studies of one-dimensional β-MnO2 nanorods. Mater Today Proc 8:162–168. https://doi.org/10.1016/j.matpr.2019.02.095
Wu X, Yang F, Dong H et al (2019) Controllable synthesis of MnO2 with different structures for supercapacitor electrodes. J Electroanal Chem 848:113332. https://doi.org/10.1016/j.jelechem.2019.113332
Kazemi SH, Kiani MA, Ghaemmaghami M, Kazemi H (2016) Nano-architectured MnO2 electrodeposited on the cu-decorated nickel foam substrate as supercapacitor electrode with excellent areal capacitance. Electrochim Acta 197:107–116. https://doi.org/10.1016/j.electacta.2016.03.063
Kumar Y, Chopra S, Gupta A et al (2020) Low temperature synthesis of MnO2 nanostructures for supercapacitor application. Mater Sci Energy Technol 3:566–574. https://doi.org/10.1016/j.mset.2020.06.002
Zhang M, Chen Y, Yang D, Li J (2020) High performance MnO2 supercapacitor material prepared by modified electrodeposition method with different electrodeposition voltages. J Energy Storage 29:101363. https://doi.org/10.1016/j.est.2020.101363
Mathur A, Halder A (2019) One-step synthesis of bifunctional iron-doped manganese oxide nanorods for rechargeable zinc–air batteries. Catal Sci Technol 9:1245–1254
Dubey M, Challagulla NV, Wadhwa S, Kumar R (2021) Ultrasound assisted synthesis of magnetic Fe3O4/ɑ-MnO2 nanocomposite for photodegradation of organic dye. Colloids Surf A Physicochem Eng Asp 609:125720. https://doi.org/10.1016/j.colsurfa.2020.125720
Li Y, Wang S, Salvador JR et al (2019) Reaction mechanisms for long-life rechargeable Zn/MnO2 batteries. Chem Mater 31:2036–2047. https://doi.org/10.1021/acs.chemmater.8b05093
Ye Z, Li T, Ma G et al (2017) Metal-Ion (Fe, V Co, and Ni)-doped MnO2 ultrathin nanosheets supported on carbon fiber paper for the oxygen evolution reaction. Adv Funct Mater 27:1704083. https://doi.org/10.1002/adfm.201704083
Cao Z, Xiao Q, Lei G, Li Z (2019) Excellent cyclic performance of electrolytic MnO2 in Li/MnO2 rechargeable batteries. SN Appl Sci. https://doi.org/10.1007/s42452-019-1585-y
Woon CW, Ong HR, Chong KF et al (2015) MnO2/CNT as ORR electrocatalyst in air-cathode microbial fuel cells. Procedia Chem 16:640–647. https://doi.org/10.1016/j.proche.2015.12.003
Zahan M, Podder J (2020) Structural, optical and electrical properties of Cu:MnO2 nanostructured thin films for glucose sensitivity measurements. SN Appl Sci. https://doi.org/10.1007/s42452-020-2191-8
Mothkuri S, Chakrabarti S, Gupta H et al (2020) Synthesis of MnO2 nano-flakes for high performance supercapacitor application. Mater Today Proc 26:142–147. https://doi.org/10.1016/j.matpr.2019.03.236
Yadav GG, Wei X, Huang J et al (2018) Accessing the second electron capacity of MnO2 by exploring complexation and intercalation reactions in energy dense alkaline batteries. Int J Hydrog Energy 43:8480–8487. https://doi.org/10.1016/j.ijhydene.2018.03.061
Worku AK, Ayele DW, Habtu NG et al (2021) Recent progress in MnO2-based oxygen electrocatalysts for rechargeable zinc-air batteries. Mater Today Sustain 13:100072. https://doi.org/10.1016/j.mtsust.2021.100072
Wu D, Xie X, Zhang Y et al (2020) MnO2/Carbon composites for supercapacitor: synthesis and electrochemical performance. Front Mater 7:2. https://doi.org/10.3389/fmats.2020.00002
Aghazadeh M, Maragheh MG, Ganjali MR et al (2016) Electrochemical preparation of MnO2 nanobelts through pulse base-electrogeneration and evaluation of their electrochemical performance. Appl Surf Sci 364:141–147. https://doi.org/10.1016/j.apsusc.2015.12.146
Boyom-Tatchemo FW, Devred F, Ndiffo-Yemeli G et al (2020) Plasma-induced redox reactions synthesis of nanosized α-, γ- and δ-MnO2 catalysts for dye degradation. Appl Catal B Environ 260:118159. https://doi.org/10.1016/j.apcatb.2019.118159
Wang J, Li H, Xu N, Qiao J (2018) Optimization of rechargeable zinc-air battery with Co3O4/MnO2/CNT bifunctional catalyst: effects of catalyst loading, binder content, and spraying area. Ionics (Kiel) 24:3877–3884. https://doi.org/10.1007/s11581-018-2562-y
Kumar R, Soam A (2020) Synthesis and characterization of sucrose derived carbon/MnO2 nanocomposite. Mater Today Proc. https://doi.org/10.1016/j.matpr.2020.02.957
Bin JS, Jeong JM, Son SG et al (2021) Synthesis of two-dimensional holey MnO2/graphene oxide nanosheets with high catalytic performance for the glycolysis of poly(ethylene terephthalate). Mater Today Commun 26:101857. https://doi.org/10.1016/j.mtcomm.2020.101857
Kong S, Gong Y, Liu P et al (2019) Synthesis of lithium rich layered oxides with controllable structures through a MnO2 template strategy as advanced cathode materials for lithium ion batteries. Ceram Int 45:13011–13018. https://doi.org/10.1016/j.ceramint.2019.03.231
Tara N, Siddiqui SI, Bach QV, Chaudhry SA (2020) Reduce graphene oxide-manganese oxide-black cumin based hybrid composite (rGO-MnO2/BC): a novel material for water remediation. Mater Today Commun 25:101560. https://doi.org/10.1016/j.mtcomm.2020.101560
Andris R, Ridley P, Byles BW et al (2021) Synthesis strategies toward improved ordering of [MnO6] octahedra in tunnel structured 2 × 3 and 2 × 4 MnO2. Scr Mater 195:113713. https://doi.org/10.1016/j.scriptamat.2020.113713
Hao J, Liu Y, Shen H et al (2016) Effect of nickel-ion doping in MnO2 nanoneedles as electrocatalyst for the oxygen reduction reaction Effect of nickel-ion doping in MnO2 nanoneedles as electrocatalyst for the oxygen reduction reaction. J Mater Sci Mater Electron. https://doi.org/10.1007/s10854-016-4606-2
Jadhav SM, Kalubarme RS, Suzuki N et al (2021) Cobalt-doped manganese dioxide hierarchical nanostructures for enhancing pseudocapacitive properties. ACS Omega 6:5717–5729. https://doi.org/10.1021/acsomega.0c06150
Su X, Yu L, Cheng G et al (2014) Controllable hydrothermal synthesis of Cu-doped δ-MnO2 films with different morphologies for energy storage and conversion using supercapacitors. Appl Energy 134:439–445. https://doi.org/10.1016/j.apenergy.2014.08.050
Uematsu T, Miyamoto Y, Ogasawara Y et al (2016) Molybdenum-doped α-MnO2 as an efficient reusable heterogeneous catalyst for aerobic sulfide oxygenation. Catal Sci Technol 6:222–233. https://doi.org/10.1039/c5cy01552a
Bodoardo S, Penazzi N, Spinelli P, Arrabito M (2001) Influence of aluminum doping on the electrochemical behaviour of manganese dioxide. J Power Sources 94:194–200. https://doi.org/10.1016/S0378-7753(00)00586-3
Hu Z, Xiao X, Huang L et al (2015) 2D vanadium doped manganese dioxides nanosheets for pseudocapacitive energy storage. Nanoscale 7:16094–16099. https://doi.org/10.1039/c5nr04682c
Lübke M, Sumboja A, McCafferty L et al (2018) Transition-metal-doped α-MnO2 nanorods as bifunctional catalysts for efficient oxygen reduction and evolution reactions. ChemistrySelect 3:2613–2622. https://doi.org/10.1002/slct.201702514
Huang A, Chen J, Zhou W et al (2020) Electrodeposition of MnO2 nanoflakes onto carbon nanotube film towards high-performance flexible quasi-solid-state Zn–MnO2 batteries. J Electroanal Chem 873:114392. https://doi.org/10.1016/j.jelechem.2020.114392
Zhang M, Yang D, Li J (2020) Supercapacitor performances of MnO2 and MnO2/ reduced graphene oxide prepared with various electrodeposition time. Vacuum 178:109455. https://doi.org/10.1016/j.vacuum.2020.109455
Jamil S, Rauf S, Bushra K et al (2018) Synthesis of saucer shaped manganese oxide nanoparticles by co- precipitation method and the application as fuel additive. J Clust Sci. https://doi.org/10.1007/s10876-018-1428-9
Min X, Guo M, Liu L et al (2021) Synthesis of MnO2 derived from spent lithium-ion batteries via advanced oxidation and its application in VOCs oxidation. J Hazard Mater 406:124743. https://doi.org/10.1016/j.jhazmat.2020.124743
Peng T, Dai X, Zhang Y et al (2020) Facile synthesis of SiO2@MnO2 nanocomposites and their applications on platforms for sensitively sensing antibiotics and glutathione. Sens Actuat B Chem 304:127314. https://doi.org/10.1016/j.snb.2019.127314
Fu X, Wang X, Chen Y et al (2019) Rapid oxidation-etching synthesis of low-crystalline δ-MnO2 tubular nanostructures under ambient with high capacitance. J Colloid Interface Sci 557:168–173. https://doi.org/10.1016/j.jcis.2019.09.024
Wang J, Yang H, Sun Q et al (2021) Synthesis of δ-MnO2/C assisted with carbon sheets by directly carbonizing from corn stalk for high-performance supercapacitor. Mater Lett 285:129116. https://doi.org/10.1016/j.matlet.2020.129116
Jiang J, Sun Y, Chen Y et al (2020) Design and fabrication of metal-organic frameworks nanosheet arrays constructed by interconnected nanohoneycomb-like nickel-cobalt oxide for high energy density asymmetric supercapacitors. Electrochim Acta 342:136077. https://doi.org/10.1016/j.electacta.2020.136077
Arena F, Di Chio R, Palella A et al (2021) Tailoring manganese oxide catalysts for the total oxidation of pollutants in gas and liquid phase. Appl Catal A Gen 610:117917. https://doi.org/10.1016/j.apcata.2020.117917
Huang A, Zhou W, Wang A et al (2021) Molten salt synthesis of α-MnO2/Mn2O3 nanocomposite as a high-performance cathode material for aqueous zinc-ion batteries. J Energy Chem 54:475–481. https://doi.org/10.1016/j.jechem.2020.06.041
Shi C, Zang GL, Zhang Z et al (2014) Synthesis of layered mno2 nanosheets for enhanced oxygen reduction reaction catalytic activity. Electrochim Acta 132:239–243. https://doi.org/10.1016/j.electacta.2014.03.150
Swain N, Mitra A, Saravanakumar B et al (2020) Construction of three-dimensional MnO2/Ni network as an efficient electrode material for high performance supercapacitors. Electrochim Acta 342:136041. https://doi.org/10.1016/j.electacta.2020.136041
Worku AK, Ayele DW, Habtu NG et al (2021) Structural and thermal properties of pure and chromium doped zinc oxide nanoparticles. SN Appl Sci. https://doi.org/10.1007/s42452-021-04682-6
Yuan Y, Zhan C, He K et al (2016) The influence of large cations on the electrochemical properties of tunnel-structured metal oxides. Nat Commun. https://doi.org/10.1038/ncomms13374
Tarimo DJ, Oyedotun KO, Mirghni AA et al (2020) Enhanced electrochemical performance of supercapattery derived from sulphur-reduced graphene oxide/cobalt oxide composite and activated carbon from peanut shells. Int J Hydrog Energy 45:33059–33075. https://doi.org/10.1016/j.ijhydene.2020.09.142
Rajabathar JR, Arunachalam P, Issa ZA, Ahmed MT (2020) Synthesis and characterization of novel metal chalcogenide modified Ni-Co-MnO2 nanofibers rolled with graphene based visible light active catalyst for nitro phenol degradation. Optik (Stuttg) 224:165538. https://doi.org/10.1016/j.ijleo.2020.165538
Zheng JY, Zhao WK, Wang X et al (2020) Electric-enhanced hydrothermal synthesis of manganese dioxide for the synergistic catalytic of indoor low-concentration formaldehyde at room temperature. Chem Eng J 401:125790. https://doi.org/10.1016/j.cej.2020.125790
Huang B, Zhang X, Cai J et al (2019) A novel MnO2/rGO composite prepared by electrodeposition as a non-noble metal electrocatalyst for ORR. J Appl Electrochem 49:767–777. https://doi.org/10.1007/s10800-019-01325-y
Kamran U, Heo Y-J, Min B-G et al (2020) Effect of nickel ion doping in MnO2/reduced graphene oxide nanocomposites for lithium adsorption and recovery from aqueous media. RSC Adv 10:9245–9257. https://doi.org/10.1039/C9RA10277A
Wang X, Liu J, Qu R et al (2017) The laccase-like reactivity of manganese oxide nanomaterials for pollutant conversion: rate analysis and cyclic voltammetry. Sci Rep 7:1–10. https://doi.org/10.1038/s41598-017-07913-2
Lambert TN, Vigil JA, White SE et al (2017) Understanding the effects of cationic dopants on α-MnO2 oxygen reduction reaction electrocatalysis. J Phys Chem C 121:2789–2797. https://doi.org/10.1021/acs.jpcc.6b11252
Li X, Xu N, Li H et al (2017) 3D hollow sphere Co3O4/MnO2-CNTs: its high-performance bi-functional cathode catalysis and application in rechargeable zinc-air battery. Green Energy Environ 2:316–328. https://doi.org/10.1016/j.gee.2017.02.004
Worku AK, Ayele DW, Habtu NG et al (2021) Enhancing oxygen reduction reaction activity of ε-MnO2 nanoparticles via iron doping. J Phys Chem Solids 157:110207. https://doi.org/10.1016/j.jpcs.2021.110207
Gowda JI, Nandibewoor ST (2014) Electrochemical behavior of paclitaxel and its determination at glassy carbon electrode. Asian J Pharm Sci 9:42–49. https://doi.org/10.1016/j.ajps.2013.11.007
Davis DJ, Lambert TN, Vigil JA et al (2014) Role of Cu-Ion doping in Cu-α-MnO2 nanowire electrocatalysts for the oxygen reduction reaction. J Phys Chem C 118:17342–17350. https://doi.org/10.1021/jp5039865
Li J, Ren Y, Wang S et al (2016) Transition metal doped MnO2 nanosheets grown on internal surface of macroporous carbon for supercapacitors and oxygen reduction reaction electrocatalysts. Appl Mater Today 3:63–72. https://doi.org/10.1016/j.apmt.2016.03.003
Acknowledgements
The authors acknowledge Bahir Dar University, Bahir Institute of Technology, Ethiopia, for providing financial aid.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest regarding to the publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Worku, A.K., Ayele, D.W. & Habtu, N.G. Influence of nickel doping on MnO2 nanoflowers as electrocatalyst for oxygen reduction reaction. SN Appl. Sci. 3, 764 (2021). https://doi.org/10.1007/s42452-021-04746-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04746-7