
Abstract
Essential oils (EOs) are complex mixtures of compounds derived from plants that exhibit antimicrobial activity. Several studies have demonstrated their antifungal activity in food matrices or in vitro via vapor phase or direct addition. Recently, researchers are focusing on elucidating the target site or the mechanism of action of various EOs. Past research has suggested evidence of how EOs act in the fungal cells via assays assessed from cell wall alterations or gene expression modifications. However, no previous reports have summarized most methods for finding the target site of the mechanism of action for EOs. Therefore, this review presents the methods and assays used to discover the target site or the mechanism of action of EOs against fungal cells. Researchers commonly analyze the plasma membrane integrity using various techniques as well as the changes in cell morphology. Meanwhile, the quantification of the activity of the mitochondrial enzymes, ROS species, and gene expression are less assayed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Fungi are eukaryotic microorganisms able to attack both live or dead tissues, including fresh food and foodstuffs, and they also cause serious diseases in plants and animals, including humans. It is well-known that several fungi are resistant to various antifungals. Essential oils (EOs) play a fundamental role in inhibiting resistant fungi due to their broad spectrum of action arising from their varied chemical structures that are synthesized to protect plants from microbial pathogens. EO compounds include monoterpenes, sesquiterpenes, aldehydes, alcohols, esters, phenolics, ethers, and ketones [1].
EOs are generally complex mixtures of components with antibacterial and antifungal activities that are used as natural additives in foods and food products. The International Organization for Standardization [2] defines EO as the product produced via distillation (water or steam), mechanical processing, or dry distillation of natural material. EOs are liquid, volatile, limpid, colored mixtures of several aromatic compounds soluble in lipid and organic solvents. EOs can be obtained from various plant parts (buds, flower, leaves, stems, twigs, seed, fruits, roots, wood, or back), and are primarily extracted from herbs and spices, as they are stored in secretory cells, cavities, canals, epidermis cells, or glandular trichomes [1]. The antimicrobial properties of EOs, primarily depend on their chemical constituents, such as the quantity of the dominant compounds, and whether direct or vapor phase is utilized.
The diversity of EOs and their constituents provides a wide range of options to attack the fungal cells. Thus, researchers have proposed several techniques to elucidate the mechanism used via EOs to inhibit or inactivate the fungal cells. Techniques for studying the main cellular components initially focused on the cell wall, plasma membrane, mitochondria, and currently focus on gene modifications. Several articles have proposed methods to treat fungal cells, including yeast and molds. However, no previous report has compiled most of these techniques into one article. This review presents the methods and assays used to elucidate the target site or the mechanism of action of EOs against fungal cells.
2 Materials and methods
Original research papers (mainly peer review papers) and reviews were used for this literature review. Books from PubMed were also consulted to include concepts or fundaments. The search was conducted from January to June 2020. The search of previous studies about the antimicrobial activity effect of EOs and their individual compounds on the fungal cells was performed on library databases such as PubMed and Google Scholar. The keywords to realize the search in databases were “mechanism of action of EOs on molds, mechanism of action of EOs on yeasts, mode of action of antifungal EOs, antimicrobial effect of EOs on fungal cells”.
3 EO mechanism of action against fungi
The antifungal activity of EOs must be demonstrated before a target site, or mechanism of action can be established. Furthermore, it is important to recognize if a specific concentration of EO can kill or inhibits the fungal cells. Moreover, the assay tests should assist with identifying how a compound or EO interacts with the cell to cause inhibition or death. It is important to distinguish between tests that identify the target site from those that elucidate the mode of action. The site of action refers to the part of the cell that interacts with the compounds, such as the cell membrane, cell wall, intracellular proteins, enzymes, nucleic acids, or metabolites. However, the mode of action refers to the specific biochemical interaction that occurs via the application of EOs to fungi that produces inhibition or killing effects [3]. Moreover, it involves identifying the specific molecular targets that EO binds, such as an enzyme or receptor.
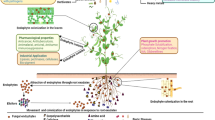
EOs effect fungi via inhibition of sporulation or producing cell damage, which leads to irreversible coagulation or denaturation of the cellular components. The EOs are capable of penetrating and disrupting the fungal cell wall and cytoplasmic membranes via utilizing a permeabilization process, that can reach the mitochondrial membranes. The changes in the fluidity of the plasma membrane may cause the electrolytes or cellular contents to leak, altering protein metabolism, and calcium ion concentration. Thus, EOs may damage the lipid, protein, and nucleic acid content of the cells. EOs also disrupt the depolarization of the mitochondrial membranes by affecting the proton pump and the ATP (Adenosine Triphosphate) pools, causing a decrease in membrane potential. Furthermore, the permeabilization of the inner and outer mitochondrial membranes hinders the cytochrome c pathways resulting in cell apoptosis or necrosis leading to cell death [1, 4, 5]. Since EOs can affect different cell components, several methods have been proposed to identify their targets of action and their mechanisms of action. Figure 1 depicts the main methods used to identify the target of action or to elucidate the mechanism of action.

Main test to determine the essential oils target site or mechanisms of action on fungal cells. TEM Transmission electron microscopy, SOD Superoxide dismutase, GSH Reduced tripeptide glutathione, GPx Glutathione peroxidase, SDH Succinate dehydrogenase, MDH Malate dehydrogenase, RT-PCR Real-time polymerase chain reaction, RT-FQ-PCR Real-time fluorescence quantitative polymerase chain reaction, ROS Reactive oxygen species, NO Nitric oxide, LPO Lipid peroxidation
4 Methods to determine the target of action of EOs and compounds on fungal cells
4.1 Assays available to identify cell wall alteration
The rigid fungal cell wall, which is primarily composed of chitin (microfibrils), glucans, mannans, proteins, lipids, inorganic salts, and pigments, is essential for adhesion, viability, and fungal pathogenesis [6, 7] The cell wall also acts as a protective barrier, limiting the access of molecules to the plasma membrane and is generally considered a therapeutic target [6, 7]. The action of EOs in molds cause a thinning and distortion of the hyphal wall and subsequent cell wall disruption. Furthermore, a final transformation flattens and empties the hyphal tips that are then bifurcated into bud-like structures.
The following sections describe the techniques used to measure the antifungal activity of EOs, which are directly related to the cell wall. The method used routinely to identify the cell wall as the target site is the sorbitol test due to the ease and cost compared to other methods.
4.1.1 Alkaline phosphatase activity measurement
The enzyme alkaline phosphatase (AKP EC 3.1.3.1) is produced in the cytoplasm, then secreted into the periplasmic space. It is associated with catalyzing the cleavage of the mono-phosphate groups from organic backbones. Damage to the external cell wall layer causes the release of AKP from the cell, which is considered as marker of microbial activity. This activity has also been suggested as an indicator of the proportion of hyphae involved in phosphorus metabolism, and fungal AKP activity may be sensitive to some fungicides [8, 9]. Shao et al. [9] conducted a study to discover the effect of tea tree EO (1 mL/L) on the cell wall of Botrytis cinerea via an assay kit (Nanjing, Jiangsu Institute of Bioengineering, China) which evaluated the activity of AKP from the supernatant. The results were recorded in U/100 mL. Increased AKP activity was observed in the culture medium following tea tree EO treatment for 4 h, which indicated early destruction of the B. cinerea cell wall. The AKP values increased for 36 h during the tea tree EO treatment. Furthermore, OuYang et al. [10] applied the AKP test to determine the cinnamaldehyde effect on Geotrichum citri-aurantii and the extracellular AKP activity detected after 60 or 120 min of treatment was also found to be related to the disruption of the cell wall.
4.1.2 Sorbitol protection assay
Sorbitol is a sugar alcohol, is commonly used as a humectant and osmotic protectant to stabilize fungal protoplasts. This protectant is important, as the minimal inhibitory concentration (MIC) of a compound will shift (because of damage to the cell wall) to a much higher value in the presence of osmotic support [11, 12]. Therefore, cell wall damage from EO may be reversed via the addition of sorbitol in the medium as it can stabilize the fungal structure. De Oliveira Pereira et al. [13] and De Oliveira Pereira et al. [14] also assessed the effect of sorbitol (0.8 mol/L) on the MIC values obtained for eugenol, geraniol, and citronellol against Trichophyton rubrum. They reported increases in the geraniol MIC by 64-fold, citronellol MIC by 32-fold, and the eugenol MIC by 4-fold when adding sorbitol; these findings suggested that compounds can interfere with the cell wall. Danielli et al. [15] evaluated the effect of sorbitol (0.8 mol/L) on dermatophyte strains (Microsporum canis, M. gypseum, T. rubrum, and T. mentagrophytes,) treated with Schinus lentiscifolius EO. The addition of sorbitol increased the MIC by 2 to 8 times in six of the strains [15]. These results also suggested that this EO acted on the fungal cell wall.
4.1.3 Test of calcofluor white
OuYang et al. [10] tested the effect of cinnamaldehyde on the cell wall integrity of G. citri-aurantii. The cells were analyzed by calcofluor white staining coupled with fluorescence microscopy. The fluorochrome stain calcofluor binds preferentially to areas containing chitin and is widely used to determine cell wall integrity. The 2-day-old mycelia were collected from 50 mL PDB containing ½ MIC (0.25 µL/mL) of cinnamaldehyde and centrifuged at 4000 × g for 10 min, the collected mycelia were stained with 10 µL of calcofluor white stain after the addition of 10 µL KOH (10%), this addition was suggested by the manufacturer. Samples were observed with a fluorescence microscope. The authors reported a weaker, uneven fluorescence in the cell walls, particularly in the septa, which indicated that the chitin content in the cell walls was affected by the cinnamaldehyde treatment.
4.2 Assays related to the integrity of the plasma membrane
The fungal plasma membrane plays a vital role in maintaining a homeostatic environment, exchanging material, and transferring energy and information in the cell to keep cells healthy and alive. The plasma membrane primarily contains lipids and protein with small quantities of carbohydrates. The main lipids are the amphipathic phospholipids and sphingolipids, which form the lipid layer. The principal sterol is ergosterol, necessary for the growth and normal function of the cell membrane, provides structure, modulates membrane fluidity, and may control some of the physiological events [16]. Antifungal agents interfere with fungal activity by binding directly to ergosterol (causing the formation of pores in the membrane) or by disrupting the biosynthetic pathway. Thus, EOs have also been tested to identify their role in ergosterol. The plasma membrane is also one of the main targets of antifungal EOs; hence several techniques have been developed to identify membrane damage or alterations.
4.2.1 Plasma membrane integrity analysis using fluorescence
Propidium iodide (PI) can determine if lesions in the plasma membrane exist in the fungal cells. PI is a nucleic acid-binding fluorescent dye used to study the effect of drugs on the cell membranes [17]. It only penetrates cells with severe membrane lesions; the lesions show increased red fluorescence. After the fungal cells are stained, their fluorescence can be measured with different instruments, such as confocal scanning laser microscopy (green helium–neon laser, 543 nm; [18]) epifluorescence microscope [19], and an optical microscope equipped with an individual fluorescein rhodamine filter set [20,21,22]. A fluorescence spectrophotometer [10], or cytometer analyzes treated and untreated cells and quantifies the percentage of stained cells that are directly related to the plasma membrane damage. In addition, if a microscope is used, micrographs can be obtained, showing the red fluorescence in the inner membrane of the fungal cells. Wang et al. [20] used these tests to examine damage to the conidia treated with eugenol; however, PI could not penetrate the spores.
4.2.2 Flow cytometry analysis
Flow cytometry analysis also uses PI to visualize lesions on the plasma membrane via screening for increased red fluorescence. This analysis involves the following, yeast cells or spores at levels of 106 are placed in PBS solution with 2% d-glucose and the desirable EO concentrations. The mixtures are incubated for periods that ranged from 3 to 12 h at an optimal temperature for the fungal cells. After the cells are rinsed, 0.5 mL PBS were used to re-suspend the cells and stained with 1 µg/mL PI as a final concentration. The samples are incubated for 10–30 min and analyzed to evaluate morphological changes. A flow cytometer is utilized to visualize the percentage of PI-positive cells. The results are examined using software [23,24,25]. This method has been used to identify the effect of EOs on molds and yeasts. Selected studies that have used flow cytometry are presented in Table 1. The flow cytometry analysis is widely used to show losses of plasma membrane integrity when fungal cells are treated with EOs. The level of stained cells is directly related to the unviable cells.
4.2.3 Methylene blue dye exclusion assay
Methylene blue aqueous dispersion can be used to stain dead yeast cells. This occurs because the methylene blue reductase is located in the plasma membrane of yeast cells; thus, if the integrity is lost, the enzyme is not active. Moreover, live cells reduced the dye due to enzymatic activity [32]. Hammer et al. [33] tested the permeability of the cell membrane using Candida albicans treated with tea tree EO and components, using methylene blue dye. First, C. albicans was grown at levels of ~ 107 CFU/mL, collected, washed, and resuspended in sterile distilled water. The desirable EO concentrations were added to the cells. Samples were incubated at 35 °C with shaking, and aliquots were taken at different times. Each sample of 80 µL was added with 20 µL of 0.05% methylene blue (w/v, prepared in sterile distilled water), mixed and then left for 5 min at room temperature. Cells were examined microscopically using a final magnification of 400X. One hundred cells (minimum) in consecutive visual fields were examined, and the percentage of stained cells was calculated. This method helped to show that tea tree EO or the components increased the yeast cell permeability. The cells that were colored had suffered damage to their cell membrane [33].
4.2.4 Extracellular pH and conductivity measurements
Permeability of the cell membrane can be expressed in terms of electrical conductivity and extracellular pH values. Rachitha et al. [34] measured the extracellular pH of Fusarium sporotrichioides following Mentha piperita EO treatment. The fungal cell suspension (105 spores/mL) was added to 20 mL of the liquid medium and then incubated at 28 °C for 20 min. Following incubation, it was centrifuged (4000 × g, 20 min), washed twice with water, and re-suspended in 20 mL of the sterilized double-distilled water. Then the EO was added, and the pH was determined. The pH of the suspensions of mold treated with the EO (500 or 1000 μg/mL) was significantly lower than the suspensions without EO; the ΔpH ranged from 0.95 to 1.2 at different contact times (30- to 90 min). The pH drops were related to the H+ losses and the plasma membrane integrity [34]. The electric conductivity for the supernatant of the cell culture was determined to explain the changes in membrane permeabilization and the release of cellular material, such as the leakage of ions.
An example of this in yeast cultures was trialed in Saccharomyces cerevisiae from an overnight culture obtained by centrifugation and re-suspended in a liquid medium to expose the culture to the EO. Conductivity was measured using a conductivity meter [35]. Increases in the conductivity values indicated leakage of ions to the extracellular medium due to loss of integrity in the cellular membrane. This effect was observed for thyme (0.5–1.0 μL/mL), oregano (0.2–1.0 μL/mL), and turpentine (0.01–0.2 μL/mL) EOs on S. cerevisiae. This test was also applied to molds. Yu et al. [21] tested the effect of tea tree EO on B. cinerea mycelium, and the observed values were significantly different between the treated or untreated cells. Similar results were reported for cinnamon EO (0.05 or 0.1 μL/mL, after 12 h) against Rhizopus nigricans [36]; geraniol (0.02% v/v) against Aspergillus flavus [37]; and Litsea cubeba EO (0.5 and 1.0%) against B. cinerea [38].
4.2.5 Cellular leakage assay (leakage of cytoplasmic content)
The cellular leakage assay assumes that a disruption in the membrane will cause a release of the intracellular components from the fungal cell. The cellular components, which absorb at 260 nm, represent one class of leakage components, the primary nucleotides were uracil-containing compounds that exhibited the strongest absorbance [11]. An assay was used to determine the effect of different EOs or their components on yeast cells such as Debaryomyces hansenii, C. albicans, or C. glabrata at different incubation times, from 1 to 6 h [19, 33]. A high number of cells (107 – 109 CFU/mL) were suspended in PBS, treated with EO, incubated, centrifuged, filtered, and the absorbance of the supernatant at 260 nm was measured. Hammer et al. [33] reported significant incremental increases (p < 0.05) in the absorbance of the supernatants of the C. albicans and C. glabrata treated with tea tree EO (0.25% to 1.0%) after the 6 h. Furthermore, an increase in the absorbance was observed from the supernatant of D. hansenii at 128 and 256 μg/mL of carvacrol or thymol after 6 h [19]. Similar results have been reported for eugenol (50 to 200 μg/mL) against B. cinerea [20]; eugenol (32 μg/mL), or geraniol (32 and 64 μg/mL), against T. rubrum [13, 14]; cinnamon EO (0.05 or 0.1 μL/mL) against R. nigricans [36]; M. piperita EO (500 or 1000 μL/mL) against F. sporotrichioides [34]; cinnamaldehyde (128 μg/mL) and eugenol (256 μg/mL) against C. glabrata [39]; and trans-cinnamaldehyde (0.313 µg/mL) against Penicillium italicum [22].
Rajkowska et al. [40] conducted this test and expressed the results for DNA concentration (μg/mL) using the formula:
where A is the absorbance of the supernatant at 260 nm, and d.f. is a dilution factor of 1. The cells of C. albicans ATCC 10231 were treated with clove (0.5% v/v), tea tree (0.5% v/v) or thyme (0.25% v/v) EOs, then the DNA concentrations of the supernatants were determined to be 8.11, 15.38, and 42.75 μg/mL, respectively; after 3 h of treatment. These results revealed leakage of the intracellular compounds and indicated an increase in cell membrane permeability.
Furthermore, the release of cellular proteins was determined from the supernatant readings at an absorbance of 750 nm following the Lowry method or 595 nm using the Bradford method. For both assays, a standard curve for bovine serum albumin (BSA) should be used to calculate the protein concentration [38, 40]. In this regard, the protein concentrations in the supernatants obtained from the C. albicans ATCC 1021 treated with clove (0.5% v/v), thyme (0.25% v/v) or tea tree (0.5% v/v) EOs, were 1251.96, 70.36, and 15.98 μg/mL, respectively; following 3 h of treatment. These results indicated that proteins were released from the cells due to a loss of integrity in the cell membrane. Similar results were reported for cinnamon EO (0.05 or 0.1 μL/mL) against R. nigricans [36]; trans-cinnamaldehyde (0.157–0.626 µg/mL) against P. italicum [22]; and L. cubeba EO (1.0%, exposure time 6 h) against B. cinerea [38].
4.2.6 Ergosterol binding assay
This method considers the exposure of a test compound to exogenous sterols, as an affinity for sterols will lead to the rapid formation of a complex, thereby impeding complexation with the sterols of the cell membrane resulting in a MIC increase [41]. If the EO binds to ergosterol, it will rapidly form a complex, preventing the cellular membrane from interacting with the ergosterol, and the observed MIC for EO increases [12, 40, 42].
Freires et al. [43] examined whether the coriander (Coriandrum sativum L.) EO interacts with ergosterol. First, the MIC of the EO against Candida spp. was obtained by the microdilution method in the absence or presence of exogenous ergosterol at concentrations of 100, 200, and 400 µg/mL. Plates were incubated at 35 °C for 24 h and read via spectrophotometer. According to their results, the MIC values of the EO increased between 8 and 16 times while the exogenous ergosterol concentration had also increased, indicating that the EO seemed to bind to the membrane ergosterol, which increased ionic permeability, ultimately causing cell death. Miron et al. [42] evaluated the ability to bind the ergosterol of five monoterpenes (common compounds of EOs) against Trichosporon asahii TAH10 via determining the MIC by adding exogenous ergosterol. The MIC of citral, geraniol, and nerol had increased fivefold in the presence of exogenous ergosterol (200 or 250 μg/mL). Similar results were observed for the MIC results as they increased by fourfold for carvacrol, or by eightfold for thymol on C. albicans ATCC 14053 and C. albicans CBS 562, respectively; when exogenous ergosterol were added (250–300 μg/mL or 312.5 μg/mL) [44, 45]. Rajkowska et al. [40] reported an increase in the MIC against C. albicans ATCC 10231 by eightfold and 32-fold for thyme and clove EO, respectively; when 400 μg/mL was added. De Oliveira Pereira et al. [13] also reported a fourfold MIC for T. rubrum treated with eugenol when exogenous ergosterol was added (400 μg/mL).
The binding assay was used because ergosterol was the main sterol in the plasma membrane, and it is only present in fungi. Furthermore, the assay exhibited low toxicity to humans and was easy and inexpensive compared to the ergosterol quantification, which required extraction steps and analysis with high-performance liquid chromatography (HPLC) or a spectrophotometer.
4.2.7 Ca2+ concentration quantification using a dependent aequorin luminescence
The test is based on the capability of the aequorin-coelenterazine complex to bind Ca2+ which emits light, the resulting luminescence intensity can be quantified and correlated to the Ca2+ concentration [46]. Rao et al. [46] investigated the effect of carvacrol on the cytosolic Ca2+ levels in the S. cerevisiae strain BY4742 (a mutant yeast which expressed the protein aequorin after the addition of the cofactor coelenterazine). The mutant was constructed via transferring the plasmid pEVP11-Aeq-89 responsible for the expression of the aequorin. The yeast cells were cultivated in synthetic complete (SC) medium overnight, adjusted the optical density (OD)600 to 1.0 per micro-centrifuge tube, and were spun down. After centrifugation, the pellets were suspended in 1 mL of glucose (2%), spun, and poured; then SC medium (50 µL) was aggregated, the cells were vortexed, and coelenterazine was aggregated (6 µL from 12.5 mg/mL in ethanol, stored at − 20 °C). Following the incubation (2 h at 30 °C), the cells were spun down and transferred into 2 mL of SC medium and mixed (vortexing), then 150 µL were placed in each well of a white 96-well microtiter plate. Additionally, the OD was measured to cells (150 µL) transferred to another microtiter plate. Carvacrol or eugenol were blended (0.0125, 0.25, 0.5, and 0.1%), and luminescence was determined on a microplate reader. The readings were taken every second, normalized regarding OD, and averaged. Carvacrol at concentrations of 0.0125 to 0.05% increased in a dose-dependent manner Ca2+ levels, returned to the baseline after 1 to 2 min of treatment. The spike was consistent with a rapid influx of Ca2+ from the extracellular medium, the vacuole, and other intracellular stores. This was followed by sequestration into stores or efflux from the cells and concomitant desensitization of the channels. The effect was recognized as Ca2+ bursts.
4.2.8 Potassium ion efflux analysis
Efflux of the potassium-free medium is a common cell response to the presence of some compounds such as EO. Gucwa et al. [12] evaluated the potassium efflux in cells of C. albicans ATCC 10231 exposed to different EOs. First, cells were obtained from a liquid medium which was grown overnight by centrifugation and washed twice with Milli-Q water. Optical density (OD660) was adjusted to 1.0. Cells were treated with EOs (thyme, lemon, geranium, cinnamon, basil, and clove) at different concentrations. Solutions of EOs of the same concentration were prepared as the controls for potassium ion content in the EOs. Cell suspensions were shaken energetically at room temperature for 10 min. The samples were centrifuged (3000 rpm, 5 min), and the supernatant was transferred to new tubes. The ion potassium concentration was measured with a flame ionizing detector BWB-1. According to these results, three of the tested EOs (thyme, lemon, and clove oil) influenced the potassium ion efflux and were directly related to the cell membrane disintegration.
Huang et al. [22] used a 2-day-old mycelium culture from a liquid medium of P. italicum that was collected by vacuum filtration. The mycelium was suspended in ultrapure water and treated with trans-cinnamaldehyde. The concentration of free potassium ions was measured after 0, 30, 60, 120 min, by a photometric procedure using the flame atomic absorption spectroscopy. Following 30 min of treatment, a high level of potassium ion efflux indicated that trans-cinnamaldehyde (0.157–0.626 µg/mL) had severely damaged the K+ transport of the cell membrane. Similar results were reported by Wang et al. [20] for eugenol (100 µg/mL) against B. cinerea after 3 min of treatment.
4.3 Changes in inner cell morphology
Microscopy techniques assist with the observation of the effect of EOs on cell structure and identify changes in the structure when the treated and untreated cells are compared. The effect of EOs on the hyphal morphology can be observed via Scanning Electron Microscopy (SEM) and the cell ultrastructure via Transmission Electron Microscopy (TEM) [21].
SEM observations, including the mycelium at 2- to 10-days-old is used depending on the type of fungal cell. Mycelium with and without EO should be prepared. Preparation includes the following equal amounts (wet weight) of mycelium are re-suspended in an equal volume of sterile water that contains Tween-80 and the EO. Mixtures are incubated, and then each sample is fixed, dehydrated with ethanol, washed with tert butyl alcohol, dried in a freeze dryer, gold-coated, and observed at a specific magnification (2000X) [21]. Tea tree EO (1.5 μL/mL) severely altered the morphology of B. cinerea, SEM revealed that the hyphae lost linearity, formed warty surfaces, and had flattened [21]. Table 2 exhibits selected studies that used SEM to show the effect of EOs on the fungal cell morphology. This test also analyzed the antifungal effect of EO in a vapor phase, as shown by Shao et al. [9] and Hua et al. [47]. SEM analysis was also applied to assess the effect of EOs on the integrity of the yeast biofilm. C. sativum EO ranged from 156 to 625 μg/mL was tested on the oral Candida species biofilm. This EO caused disruption of the cells, and the biofilm exhibited a withered appearance [43].
TEM observations of the mycelium preparation were similar to those previously described for SEM. Subsequently, the mycelium samples were fixed and dehydrated with the application of a series of ethanol and acetone. Afterwards, the samples are treated with epoxy resin until the resin concentration was adequate. Samples are incubated in pure epoxy (37 °C, 12 h) and polymerized (60 °C, 36 h). Then sections are treated with uranyl acetate and lead citrate; a diamond knife was used to cut the ultrathin sections for each sample (70 nm); then, the samples are examined by TEM [21]. In the hyphal ultrastructure of B. cinerea treated with tea tree EO, it was observed that the intracellular constituents were seriously damaged, plasmolysis occurred, and the plasmalemma ruptured and detached from the cell wall [21]. Table 3 presents the selected studies which use TEM to show the effect of EOs on the fungal cell ultrastructure.
4.4 Alterations of the inner fungal cell
4.4.1 Intracellular pH determination
The determination of the intracellular pH (cytoplasmic or vacuolar) can only be qualitative in the yeast cells. Rao et al. [46] used pHluorin to measure cytoplasmic pH. S. cerevisiae BY4742 was previously transformed with a plasmid pZR4.1. First, yeast cells were cultured until the mid-logarithmic phase, treated with carvacrol, and read the fluorescence using the dual excitation at 410 and 485 nm and emission at 520 nm. The BCECF/AM (2ʹ,7ʹ-bis-(Carboxyethyl)-5(6ʹ)-carboxyfluorescein Acetoxymethyl Ester, 2ʹ,7ʹ-bis-(Carboxyethyl)-5(6ʹ)-carboxyfluorescein Acetoxymethyl Ester, a pH-sensitive fluorophore) were added to cells and poured into a microplate to measure the vacuolar pH. The effect of carvacrol was determined by reading the fluorescence at dual excitation at 450 and 485 nm and emission at 520 nm. The addition of carvacrol (0.05%) increased 0.5 units of the vacuolar pH, which had persisted the alkalization. Consequently, carvacrol (0.05%) induced immediate acidification of the cytosol; this was shown by the drop of cytoplasmic pH (~ 0.5 pH units) [46].
4.4.2 Cytosolic Ca2+ measurement
This test establishes if the biological effect of EO in the mycelia is related to changes in the internal Ca2+ concentration, and the concentration of the free cytosolic Ca2+. Wang et al. [20] assessed the effect of eugenol (100 μg/mL) in the mycelia of B. cinerea using the fluorescent Ca+2 indicator Fluo3-AM. The final concentration of Fluo3-AM was adjusted to 5 µmol/L and prepared from a 5 mmol/L dimethyl sulfoxide (DMSO) stock solution. The buffer used was 10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.4), and the samples were observed with a Confocal Laser Scanning Microscope. Following treatment, the concentration of Ca2+ increased and had reached the highest level at 30 min, suggesting that eugenol either directly or indirectly changed the cytosolic Ca2+ concentration and was related to the biological effects. This Ca2+ increment in the cytosol was potentially due to the Ca2+ released from internal stores (such as the vacuoles or the endoplasmic reticulum) [20].
4.4.3 Reactive oxygen species (ROS) analysis
ROS, such as the peroxide anion (O2−), hydrogen peroxide (H2O2), nitric oxide (NO), and the hydroxyl radical (OH·), consisted of radical and non-radical oxygen species formed by the partial reduction of oxygen. ROS are derived from the end products of aerobic cellular metabolism. The main ROS producers are peroxisomes, NADPH oxidases, uncoupled nitric oxide synthase (NOS), cytochrome c, and the electron transport chain in mitochondria. An excessive amount of ROS in the mitochondria leads to hyperpolarization that results in apoptosis. Other cellular effects are morphological changes, nuclear fragmentation, chromatin condensation, cell swelling, enzyme inactivation, membrane disruption, and phosphatidylserine externalization that ultimately results in cell death. Therefore, ROS quantification is closely related to cell viability [58, 59]. EOs stimulate the production of ROS in abundance, causing damage to the proteins, lipids, and DNA, leading finally to cell death.
The endogenous ROS measurement in mold and yeast cells exposed to EO is analyzed by fluorescence intensity using a flow cytometer. The assay uses fluorescent dye 2′,7′-dichlorofluorescein diacetate (DCFH-DA), which is well established for use in the quantification of ROS production to investigate the changes in the intracellular ROS levels of fungal cells. After, DCFH-DA (hydrophobic and non-fluorescence) reaches the interior of the cell, it is hydrolyzed via intracellular esterase enzymes to generate DCFH, and following oxidation steps lead to DCF (fluorescence molecule) [54]. The intensity of fluorescence is directly proportional to the level of the ROS molecules. Tian et al. [25] and Chen et al. [29] measured the ROS formation in A. flavus spores and C. albicans cells, respectively, when they were treated with dill seeds EO using the DCFH-DA. The spores (4 × 106 spore/mL) or exponentially growing yeast cells were suspended in sterile PBS. Yeast cell analysis included the addition of adequate amounts of EO to the yeast cell suspensions and then incubated them for 12 h at 30 °C. Afterwards, the yeast cells were harvested by centrifugation, washed twice, and re-suspended in PBS. Thus, for yeast cells, a 10 µmol/L DCFH-DA concentration was achieved using a ROS kit. Mold cell analysis included the following DCFH-DA was directly added to the spore mixture with a final concentration of 10 µmol/L. Subsequently, the incubation step was 4 h at 28 °C for mold and 30 min at 30 °C for the yeast cells, then both kinds of cells were collected via centrifugation, washed twice, and re-suspended in PBS. ROS were expressed as the fluorescence intensity of the DCFH-DA using a flow cytometer (excitation and emission wavelengths of 485 and 535 nm). Dill seeds of the EO increased the fluorescence intensity of the mold spores by 4 to 12 times for 0.25 or 2.0 µL/mL, respectively, which resulted in an increase of ROS-positive cells [25]. Chen et al. [29] found that dill seeds EO caused a dose-dependent increase in the fluorescence intensity of the C. albicans cells. Therefore, the ROS production by the EO caused losses in the cell’s viability. Similar results were reported for the Coreopsis tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31]; thymol (100 or 200 μg/mL) against A. flavus spores [52]; eugenol (256 μg/mL) against C. glabrata [39]; Cymbopogon martinii EO (100 to 600 μL/L) against Fusarium graminearum [54], and geraniol (0.02% v/v) against A. flavus [37].
The ROS accumulation measurement was conducted in P. italicum and treated with trans-cinnamaldehyde using the luminol chemiluminescence method [22]. Mycelium from a 2-day-old was collected by vacuum filtration from a liquid medium and ground into powder with liquid nitrogen. Following centrifugation of the sample, 50 µL of the supernatant and 830 µL of HEPES buffer (0.02 mol/L, pH 7.4) were injected into a glass container. MPI-B Chemiluminescence analysis test system was started immediately when 20 µL horseradish peroxidase (3 g/mL) was added under absolutely dark conditions. Trans-cinnamaldehyde (0.626 µg/mL) triggered the production of ROS, and thus, accumulated, as a potential danger to the mitochondrial membrane.
4.4.4 Detection of H2O2 in hyphae
H2O2 is an unavoidable by-product from all living organisms that depend on the respiration for energy production. The mitochondria are the main site of H2O2 production and are involved in several oxidase reactions. The cytotoxicity of H2O2 is generally attributable to the damage inflicted on the macromolecules, including lipids, DNA, and proteins. H2O2 is less toxic compared to other ROS; however, it can diffuse away from the original production site before reaching a target [60]. Due to the short life and reactivity of the ROS, a way to measure them is through the quantification of H2O2 as it is more stable than the rest of the ROS and will degrade into dangerous substances for the cells.
Wang et al. [20] evaluated the ability of eugenol to generate H2O2 in B. cinerea. Discs of (6 mm) mycelial from mold were taken from 3-day-old colonies and placed in flasks with a liquid medium and incubated at 25 °C with gentle shaking, the mycelia were centrifuged (10,000 × g, 10 min), and washed three times in 20 mL of PBS buffer (0.05 mol/L, pH 7.0). 0.5 g of mycelia were ground into a fine powder with a mortar and pestle using liquid nitrogen and then re-suspended with 4 mL of PBS buffer. The blend was centrifuged (10,000 × g, 4 °C, 10 min), and the H2O2 concentration was measured in the supernatant. 100 µL of the supernatant was added to 100 µL of the assay reagent (500 µmol/L ferrous ammonium sulfate, 50 µmol/L H2SO4, 200 µmol/L xylenol orange, and 200 mmol/L sorbitol). Then incubated for 30 min at room temperature, the peroxide-mediated oxidation of Fe2+ to Fe3+ was determined by measuring the absorbance at 570 nm of the Fe+3-xylenol orange complex. Following treatment of the cells with eugenol, the accumulation of H2O2 had increased by 1000% at 4 h.
4.4.5 Nitric oxide (NO) assay
NO synthesis in the fungal cells has not yet been completely elucidated. However, it is considered to be related to several vital functions, such as homeostasis, pathogenesis, morphogenesis, and reproduction [61]. Therefore, NO must be maintained at adequate physiological levels to correct cell function. NO in spores has been measured using DAF-FM DA (3-amino,4-aminoethyl-2′,7′-difluorencein diacetate) utilizing an inverted fluorescence microscope in the range of excitation of 488 nm and emission of 525 nm. Shen et al. [52] tested the effect of thymol on the A. flavus spores using this assay. Adequate spores/mL were incubated in a liquid medium for 30 min at 37 °C prior to treatment with thymol. Afterwards, the spores were treated with different concentrations of thymol and transferred to 20 mmol/L HEPES–NaOH (pH 7.5) buffer solution containing 15 µmol/L specific NO fluorescent probe DAF-FM DA, and then incubated in darkness for 15 min [62]. The spores were removed and washed with saline solution. 100 µL of spore suspension in saline solution were added into the wells of a 96-well plate directly for fluorescence photography. Thymol (100 or 150 μg/mL) stimulated the NO production of the spores in a dose-dependent manner. NO contributed ROS to the spores causing loss of viability.
4.4.6 Quantification of the primary scavenging enzymes of ROS
The superoxide dismutase (SOD) rapidly disrupted the univalent reduced oxygen, the superoxide anion radical, and converted it to H2O2. Catalase is an enzyme belonging to the category of the oxidoreductases, that catalyzes the reaction by which H2O2 is decomposed to water and oxygen. Detoxification of H2O2 is a fundamental aspect of the cellular antioxidant responses in which catalase plays a major role. Catalase prevents the accumulation of H2O2 and protects cellular organelles and tissues from damage by H2O2, which is continuously produced by numerous metabolic reactions. The induction of SOD and the catalase activities are responses to enhanced levels of ROS; these enzymes have key roles in the defense mechanisms against the free radicals in aerobic organisms [63]. Moreover, the glutathione peroxidase (Gpx), catalase, and reduced tripeptide glutathione (GSH) are the cell antioxidant armor that counters the peroxides and hydroxiperoxides to prevent cell damage. GSH is particularly vital for normal cellular function and constitutes one of the physiologically important mechanisms used to curtail the progression of cell damage [17]. Consequently, they are the primary scavenging enzymes of ROS. Therefore, the evaluation of their activity relates to ROS production and cell defense against EOs.
Before the enzyme activity measurements take place, a cell-free extract must be prepared. Treated and untreated cells of yeast (1 g wet weight) collected from the mid-exponential phase of growth in a liquid medium were suspended in 2 mL of grinding medium (250 mmol/L sucrose, 10 mmol/L Tris HCl, pH 7.5, 1 mmol/L PMSF) and 2 g of glass beads (0.45 mm). The suspension was disrupted mechanically in a cell homogenizer by agitating in cycles of seconds at 4000 vibrations/min. The homogenate was collected, and centrifuged (1000 × g, 5 min, 4 °C) to remove the unbroken cells and glass beads. In order to obtain an adequate supernatant, another centrifugation step is required at 15000 × g for 40 min at 4 °C [17].
Superoxide dismutase (SOD) activity measurement Khan et al. [17] measured SOD activity from C. albicans exposed to eugenol and estragole (0–100 μg/mL) by the inhibition of pyrogallol autoxidation. The assay solutions should be freshly prepared at the time of the analysis. In order to create the test sample, 0.1 mL of the cell-free extract was added to pyragallol and Tris buffer (50 mmol/L, pH 8.5), then an induction period of 90 s was required. The pyrogallol inhibition was monitored every 30 s for 3 min at a wavelength of 420 nm. For each sample, control was taken in which 2.9 mL Tris buffer was added to 0.1 mL pyragallol (20 mmol/L). Pyrogallol autoxidation per 3 mL of the assay mixture was calculated as follows:
where A is the difference in absorbance per 1 min in the control and B the difference in absorbance per 1 min in the test samples. Results are expressed in U/mg protein. The authors found that the SOD activity increased significantly in the yeast cells treated with eugenol and estragole.
Catalase activity assay Khan et al. [17] assessed the catalase activity in C. albicans cells exposed to different concentrations of eugenol and estragole (0–100 µg/mL). Once the cell-free extract was prepared, 10 µL of the extract was added to 1.99 mL phosphate buffer (0.05 mol/L, pH 7.0) and 1.0 mL of H2O2 (0.0019 mol/L) to a total volume of 3.0 mL in a quartz cuvette. The decrease in absorbance due to the disappearance of H2O2 was recorded at 30 s intervals up to 3 min at 230 nm at 25 °C. Catalase activity was calculated in terms of the µmol of H2O2 consumed/min mg protein using an extinction coefficient of 0.081 × 10−1/mol/L cm. The authors reported significant increases in catalase activity in cells treated with 50 or 100 µL of eugenol or estragole. The increase in catalase activity was related to the peroxide formation induced by this treatment.
Reduced tripeptide glutathione (GSH) activity quantification Cells of C. albicans were treated with different amounts of eugenol and estragole (0–100 µg/mL), and after the cell-free extract was prepared, the cytosolic reduction of GSH activity was assayed [17]. A 1.0 mL aliquot of the cell-free extract is precipitated with 1 mL of 4% sulphosalicylic acid, kept for 1 h at 4 °C and then centrifuged at 1200 × g for 15 min at 4 °C. Following centrifugation, 0.1 mL of the supernatant was mixed with 2.7 mL of phosphate buffer (0.01 mol/L, pH 7.4) and 0.2 mL of the freshly prepared 5,5′-dithiobis-2-nitrobenzene (40 mg in 10 mL of 0.1 mol/L phosphate buffer pH 7.4) for a total volume of 3 mL. The yellow color develops due to the formation of 5-thio-2-nitrobenzoate and is measured at 412 nm at 25 °C. Results are expressed in µmol GSH/mg protein. Significant amounts of GSH depletion were observed by the authors for all the concentrations of the tested compounds.
Glutathione peroxidase (GPx) activity measurement Cells of C. albicans treated with different amounts of eugenol and estragole (0–100 µg/mL), and after the cell-free extract was prepared, the GPx activity was assayed [17]. First, the reaction mixture is prepared with 1.53 mL of phosphate buffer (0.05 mol/L, pH 7.0), 0.1 mL of 1 mmol/L EDTA, 0.1 mL of 1 mmol/L NaN3, 0.1 mL of 1 mmol/L GSH, 0.1 mL of 0.2 mmol/L NADPH, and 0.01 mL of 0.25 mmol/L H2O2. 100 µL of the cell-free extract was added to the reaction mixture for a final volume of 2.0 mL. The activity was measured in terms of a decrease in absorbance at 340 nm suggestive of the disappearance of NADPH at 30 s intervals for 3 min at 25 °C. The enzyme activity was calculated as µmol of NADPH oxidized/min mg protein using a molar extinction coefficient of 6.22 × 103/mol/L cm. The author’s findings were an increase of GPx activity occurred in the cells treated with 10 µg/mL for both compounds, followed by a significant decrease resulting from the increasing concentration of eugenol or estragole.
4.5 Dysfunction of the fungal mitochondria
Mitochondria are the site of most energy-yielding oxidative reactions and the coupled synthesis of ATP through the tricarboxylic cycle acid (TCA). Although the TCA is central to energy-yielding metabolism, the role of this cycle is not limited to energy conservation. For instance, four- and five-carbon intermediates of the cycle serve as precursors for a wide variety of products. In eukaryotes, the entire set of reactions of the TCA takes place in the mitochondria. Isolated mitochondria were found to contain all of the enzymes and coenzymes required for the TCA, and also all of the enzymes and proteins necessary for the last stage of respiration-electron transfer and ATP synthesis via oxidative phosphorylation. Mitochondrial dehydrogenases are the responsible enzymes to catalyze the biosynthesis of ATP. Lactate dehydrogenase (LDH), malate dehydrogenase (MDH), and succinate dehydrogenase (SDH), are very important enzymes in the biosynthesis of ATP that participate in TCA. LDH catalyzes the conversion of lactate into pyruvate, it is the last step in aerobic glycolysis, and a vital step in the energy production of cells. MDH catalyzes the interconversion of malate to oxaloacetate in the TCA. The oxidation of succinate to fumarate is catalyzed via SDH in the TCA [30]. EOs can affect mitochondrial effectiveness by inhibiting the action of mitochondrial dehydrogenases, consequently disturbing the TCA and inhibiting ATP synthesis [64].
4.5.1 Mitochondrial dehydrogenase activity assay
Total dehydrogenase activity determines the effect of the antifungal substance. This assay examines the activity of various dehydrogenases in the mitochondrion, such as LDH, MDH, and SDH. Viable and metabolically active cells are capable to reduce the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-(2H)-tetrazolium-5-carboxanilide) as a substrate to an orange water-soluble formazan product via mitochondrial dehydrogenase, thus this activity is using to develop a colorimetric method which assess the enzymes activity. This method has utilized by Tian et al. [25] and Chen et al. [29] to assess the effect of dill seeds EO on the fungal cells. 200 µL of 2 × 106 spore/mL were added to 96-well flat-bottom microplates and incubated with different concentrations of EO. Following 24 h of incubation (28 °C), 50 µL aliquots of XTT with menadione were added to the wells in order to obtain a final concentration of 50 µg/mL XTT and 25 µM menadione. The OD450 was read directly using a 96-well scanner, following 2 h of exposure to XTT [25]. The dill seeds EO significantly reduced the mitochondrial dehydrogenase activity of A. flavus and ranged from 16 to 68% for 0.0625 to 4.0 µL/mL, respectively [25]; resulting in the inhibition of ATP synthesis. Chen et al. [29] reported a drop in the activities of the mitochondrial dehydrogenases of C. albicans treated with dill seeds EO varied from 13 to 82% for 0.312 to 10 µL/mL, respectively. Similar results were reported for C. tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31]; and Acorus calamus EO (0.2 to 1.0 μg/mL) against Aspergillus niger [65].
4.5.2 Assessment of the mitochondrial reductase enzyme activity
This assay determines the metabolic activity through the formazan product obtained by the action of the mitochondrial reductases on the thiazolyl blue tetrazolium bromide [MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide]. Pinto et al. [66] assessed the effect of Ferulago capillaris EO on the mitochondrial reductase activity of C. albicans ATCC 10231 using MTT. Yeast cell suspensions were prepared in NaCl 0.85%, and the turbidity was adjusted to a 0.5 McFarland standard. Dilutions 1:50, followed by a 1:20, were performed in an RPMI (Roswell Park Memorial Institute) culture medium. 500 µL of RPMI were added to the same volume of the previous cell suspension into a 12-well plate and incubated overnight (18–24 h at 37 °C). Following incubation, the cells were homogenized, centrifuged (10,000 rpm, 5 min), the supernatant was removed, and 1 mL of the EO was added to each tube in the required concentration. The mixture was homogenized, transferred to the 12-well plate, and incubated for 1 h at 37 °C. Later, the cell suspensions were centrifuged, the supernatant was removed, and 500 µL of MTT solution (0.5 mg/mL prepared in RPMI) were added to each well and incubated for 30 min at 37 °C. The yellow tetrazolium salt MTT was converted by mitochondrial dehydrogenase of the metabolically active cells to an insoluble purple formazan product and was then solubilized with 300 µL of DMSO. The extent of the reduction to formazan within the cells was quantified by measuring the absorbance at 510 nm in a plate reader. F. capillaris EO reduced the activity of the mitochondrial reductase enzymes by 50% in concentrations that ranged from 0.08 μL/mL (1/8 MIC) to 0.64 μL/mL (1 MIC).
4.6 Effect on gene expression
4.6.1 Analysis of the transcriptional response
The EOs may cause entire cell changes that are analyzed via the whole assay of gene expression during treatment. These changes in the cell are examined using the transcriptional response. In order to determine the time of cell exposition to EO and analyze the transcriptional effect, additional tests are required since the test should be subinhibitory. Rao et al. [46] studied the early-log phase cultures (OD600 of 0.1) of S. cerevisiae BY4742 to evaluate the transcriptional effect of carvacrol (0.005% and 0.01%) after the direct contact for 15 min. Untreated cells were also tested as a control. The RNA was isolated from both samples, treated and untreated cells. The purity and the quality of the RNA samples was established via polyacrylamide gel electrophoresis. cDNA synthesis, labeling, and hybridization, image scanning and processing took place in adequate facilities. Following the design of microarray data, the information was introduced to software (Partek GS) to normalize and analyze. To the hierarchical clustering analyses, the data set was introduced to Gene Cluster 3.0 and compared along with previously published DNA microarray data for these genes in response to thymol, calcium chloride (CaCl2), rapamycin, amiodarone, nitrogen depletion, and grown in YPD, diauxic shift, and four classes of antifungals. Java Tree View software was used to display the results and edited with adequate software. The server of the Munich Information Center for Protein Sequences (MIPS) Functional Catalogue was used to analyze and enhance the gene set.
Cells treated with carvacrol (0.01%) had upregulated 800 genes and downregulated 603 genes by regulating ≥ twofold. Genes related to alternative metabolic and energy-handling pathways (glucogen and trehalose biosynthesis, polyamine degradation, and fatty acid transport and oxidation), stress response (sporulation, oxygen, and free radical detoxification), heat shock proteins and chaperones, autophagy, and vacuolar degradation mechanisms were overexpressed mainly. Meanwhile, the repressed genes were related to nucleic acid and RNA synthesis, processing, and modifications leading to growth inhibition [46].
Wang et al. [67] also extracted total RNA to sequence the RNA and prepared cDNA libraries of treated and untreated conidia from A. flavus YC-15 to evaluate the effects of cinnamaldehyde on the mold. For the total RNA using a Fungal RNA kit and cDNA libraries were made using an Illumina® TruSeq™ RNA Sample Preparation kit and the Illumina® HiSeq™ 4000 system. Afterwards, they were mapped to the A. flavus genome, the EST sequencing, rRNA sequencing, and then assembled using programs TopHat 1.31, Bowtie, and Cufflinks, respectively. The FPKM values were counted to calculate and normalize the transcription levels of genes in A. flavus. The mRNA data revealed 1032 genes that were significantly differentially transcribed from A. flavus treated or untreated cells, and 427 genes were upregulated, and 605 genes were downregulated. The upregulated genes were related to biological processes (mainly to biological regulation, cellular process, localization, metabolic process), cellular components (cell and macromolecular complex), and molecular functions (binding and catalytic activity).
5 Methods to assess the mechanism of action of EOs and their components on fungal cells
5.1 Assay to identify cell wall changes
5.1.1 Determination of the chitin content
Chitin is a β-(1,4)-linked polymer of N-acetylglucosamine, and an important structural component in cell walls; that is found in considerable quantities in filamentous fungi (10–20%) playing a critical role in fungi development and pathogenicity [7]. However, chitin is only present in very small amounts in yeast (1–2%) [7].
OuYang et al. [10] evaluated the chitin content of G. citri-aurantii cells treated with different amounts of cinnamaldehyde in potato dextrose broth (PDB). The samples were dried and ground into a powder. Then, the powder (W1) was treated with saturated potassium hydroxide (KOH) solution at 160 °C for at least 15 min until the products became a transparent thin film. Later, the products were poured onto filter paper and rinsed slowly with distiller water. The extracts were successively dehydrated with 95% and 100% alcohol and weighed again (W2). The chitin content was calculated via the following formula:
where 1.26 is the conversion factor.
Following quantification of the chitin content, the G. citri-aurantii mycelia rapidly decreased from the cinnamaldehyde treatment after 60 min of exposure, which suggested that cinnamaldehyde may exhibit antifungal activity against the mold by interfering with the construction of the cell wall.
5.2 Methods to determine the functionality of the plasma membrane
5.2.1 Ergosterol quantification assay
EOs or their components can reduce or inhibit ergosterol biosynthesis. Absence or reduced content of ergosterol in the fungal membranes results in osmotic and metabolic instability of the fungal cell, compromising reproduction, and infectious activity [64].
The two main analysis quantified the ergosterol content in the sample, based on the ability to absorb UV light at 282 nm, spectrophotometry (UV–Vis), and an HPLC system equipped with a UV–Vis detection component. Furthermore, gas chromatography coupled to mass spectrometry (GC–MS) also was used by Essid et al. [68] to quantify the sterols (including ergosterol). The effect of EOs on the ergosterol content of the fungal cells was quantified via the following steps. The fungi were cultivated in a liquid medium supplemented with different levels of EO and incubated for long periods, 4–14 days for molds and 16 to 24 h for yeasts, at an optimal fungi growth temperature (25–28 °C for molds and 17 to 35 °C for yeasts). Treated and untreated cells were assayed. Afterwards, the biomass was separated via either filtering for molds or centrifugation for yeasts, washed, and weighed. Then the lipids were extracted with KOH solution under favorable conditions (temperatures 70–85 °C, 40 min–4 h, with shaking). Later, sterols were extracted from the sample using mixtures of n-heptane or n-hexane, followed by dilution or concentration of the extract until they were ready to analyze [28, 31, 53, 54]. If the spectrophotometry technique was used, the spectrum between 230 and 300 nm was scanned. A characteristic four-peaked curve showed the presence of ergosterol at 232 nm and the late sterol intermediate 24,28-dehydroergosterol (DHE). The absence of detectable ergosterol in the extract was indicated by a flat line. Ergosterol content was calculated as a percentage of the wet weight of the cells, via the equation reported by Ahmad et al. [28]:
where F is the factor dilution, and 290 and 518 are the E values (as percentages/cm) determined for crystalline ergosterol and 24,28-DHE, respectively. Table 4 presents the effect of selected EOs or their components on the ergosterol content of fungal cells. Most studies used UV/VIS spectrophotometry technique likely because it is easy and inexpensive. The ergosterol content reduction is considered EO concentration dose-dependent. However, the EO, the microorganism, and the range of tested concentrations determine the effect on the fungal cell ergosterol content. Based on information from Table 4, the ergosterol content reduction in fungal cells ranged from 18 to 100%. Despite the fact that most studies utilized liquid mediums (commonly culture broths) to evaluate the EO effect on the ergosterol content of the fungal cells; recently, Perczak et al. [69] reported the quantification of ergosterol in wheat grains treated with eight different EOs.
5.2.2 Determination of the total lipid content
Lipids are important cell components that are widely distributed in the organelles. Phospholipids, sterols, and sphingolipids are amphipathic molecules which are the main components of biological membranes including fungal cells. Phospholipids and sphingolipids include fatty acids in their structural backbones [72]. Researchers are using the quantification of fatty acids to assess the effect of EOs on the fungal cell membranes.
Shao et al. [9] evaluated the effect of tea tree EO on the cell membrane by quantifying the total lipids. First, B. cinerea was inoculated into the liquid medium and incubated for 2 days (25 °C with shaking), then 1 mL/L EO was added and incubated for 36 h. Following incubation, the cells were collected by centrifugation (4000 × g, 10 min) and washed twice in sterile water. Total lipids were extracted as described by Ruess et al. [73], via saponification conducted in sodium hydroxide-methanol solution at 100 °C for 30 min (liberating the fatty acids from cellular lipids), followed by acid methanolysis in hydrochloric acid (HCl)-methanol (80 °C, 10 min). The fatty acid methyl esters were extracted into hexane-methyl tertiary butyl ether and washed with aqueous sodium hydroxide (NaOH). Fatty acids were separated and quantified by CG-MS. Then 20 µL of the sample solution was injected to conduct the analysis; N2 was used as the gas carrier, and the capillary column used was Spb-50 silica. The unsaturated/saturated fatty acid ratio was calculated using the formula:
The analysis revealed important increments of palmitic acid (16:0), stearic acid (18:0), oleic acid (18:1), and linolenic acid (18:3). While the linoleic acid (18:2) content dropped ~ 59%. In addition, the ratio of the unsaturated/saturated fatty acids was 1.17 or 0.66 for the untreated and treated cells. A high degree of saturated fatty acids in the membrane lipid bilayer was related to a decrease in the membrane fluidity [9]. Essid et al. [68] reported a decline in the total amount of fatty acids in C. albicans ATCC 10231 cells following Pelargonium graveolens (500 μg/mL) treatment, this reduction was more pronounced for oleic acid (from 41.29 to 11.58%) than the other fatty acids. This imbalance in the fatty acid composition could result in changes to the membrane fluidity of C. albicans.
OuYang et al. [10] determined the total lipid content for the G. citri-aurantii spores with 0.25 μg/mL cinnamaldehyde treatment in PDB at different incubation times (30 to 120 min), using the phosphovanillin method. Following treatment, the samples were freeze-dried (4 h) and the dry mycelia (~ 0.1 g) were mixed and ground with liquid nitrogen. Subsequent, for the extraction step, 4.0 mL of the methanol/chloroform/water mixture (2:1:0.8 v/v/v) were used shaking vigorously for 30 min, then samples were centrifuged (4000 × g, 10 min). Lipids were contained in the lower phase thus was carefully mixed with a saline solution (0.2 mL) and centrifuged (4000 × g,10 min). A lipid mixture was shifted to a clean tube added 0.2 mL chloroform and 0.5 mL sulfuric acid (H2SO4) and heated for 10 min in a boiling water bath. Finally, phosphovanillin (3 mL) was aggregated, energetically shaken, incubated at room temperature (10 min), and read the absorbance at 520 nm. To calculate the total lipid content, the absorbance from the standard calibration curve was used (cholesterol was the standard). Cinnamaldehyde induced a massive accumulation of the total lipid content after 60 min of exposure. This might lead to the thickening of cell membranes. Rachitha et al. [34] found that the lipid content of F. sporotrichioides decreased (25 to 50%) following treatment with M. piperita EO (500 or 1000 μL/mL), which was related to a decrease in membrane stability.
5.2.3 ATPase activity and ion efflux in the cell membrane
Efflux pumps are a part of all living cells, and their function is to transport toxic substances out of the cell. Fungal cells often include the transport of accumulated drugs out of the cell [74]. H+-ATPases have important physiological functions in fungal cells since they are responsible to carry out the transportation of protons across the cell membrane to maintain the gradient transmembrane electrochemical balance for nutrient uptake. Electrochemical balance is evolved with the regulation of the intracellular pH, cell growth, fungal pathogenicity, and medium acidification. Thus, alterations in the H+-ATPases functionality derives in intracellular acidification and inhibition of the cell growth or related to cell death [64, 70].
The K+ and Ca2+ efflux pumps of the cell membrane of the fungi also provide a tool that allows the cells to control their entry and exit. The permeability barrier is essential to support various cellular functions related to the regulation of energy and cell metabolism, transduction processes coupled to the plasma membrane, cell integrity (turgor pressure), and solute transport [75].
Determination of the acidification of the external medium (proton efflux measurement) The proton pumping activity of the fungal cells is detected by monitoring the glucose-induced acidification of the external medium via measurement of the pH. Glucose-induced cells exhibited a decrease in the extracellular pH values, which results in a maximal pH gradient across the cell membrane. Tian et al. [25] assessed the effect of dill seeds EO on the A. flavus cells. First, the spore suspension (107) was inoculated into the PBS with 2% glucose and then incubated for 48 h at 28 °C. Fungal cells were separated using filter paper and rinsed twice with distilled water. For glucose starvation, the wet mycelia (~ 1.0 g) was re-suspended in 50 mM potassium chloride (KCl) solution (40 mL) and incubated (18 h at 4 °C). EO at different concentrations was added to the solution. Then the volume was adjusted to 45 mL with 50 mM KCl solution, the mixtures were incubated for 10 min at room temperature, and the mycelia were filtered using filter paper. To induce medium acidification, the mycelia was combined with 20 mL solution of glucose (10%). The external pH was determined using a pH meter at various times (0 to 60 min). Dill EO (0.25, 0.5, and 1.0 μL/mL) significantly inhibited the medium acidification after 10 min, and 60 min, the ΔpH ranged from 0.25 to 0.68 for the control. In contrast, 2.0 μL/mL completely inhibited the acidification after 30 min. This result was related to the plasma membrane ATPase dysfunction. Konuk and Ergüden [35] also observed inhibition of the ability of S. cerevisiae to acidify the external medium when it was treated with EOs (1.0 μL/mL oregano or 0.2 μL/mL turpentine).
5.3 Methods to evidence changes in the inner fungal cell
5.3.1 Lipid peroxidation (LPO) analysis
One of the main predictors of cell death is oxidative stress, when it is related to high levels of ROS, which is the most important cause for generating lipid peroxidation (LPO). LPO occurs primary in unsaturated lipids to produce polar lipid hydroperoxides that disrupt hydrophobic phospholipids and lead to increased membrane fluidity [17]. The LPO alters the integrity and function of the membrane integrity and produces the aldehyde by-product malondialdehyde (MDA), which is considered an indicator of lipid oxidation. The MDA reacts with DNA and produces propane adduct with 2′-deoxyguanosine, which has an extreme influence on the physiological cell functions, including cell signaling, cell proliferation, differentiation, and apoptosis [76].
Khan et al. [17] tested the formation of MDA due to peroxidation; it was measured using thiobarbituric acid. Prior to the LPO analysis, the plasma membrane must be prepared. In this case, cells of C. albicans were grown in a liquid medium with different concentrations of eugenol and estragole (0–100 μg/mL) at 30 °C with shaking at 200 rpm up to OD600 ≈ 1.25 (mid-log phase). The cells were disrupted using a cell homogenizer. The homogenizing buffer contained 2 mmol/L of 2,2′,2″,2‴-(Ethane-1,2-diyldinitrilo)-tetraacetic acid (EDTA), 20% glycerol (v/v), 1 mmol/L phenylmethylsulphonyl fluoride (PMSF), and 50 mmol/L Tris, pH 7.5. Following, the mixture was centrifuged (1957 × g, 10 min) and rinsed twice; then the plasma membrane fractions were pelleted from the supernatant by ultracentrifugation (54 782 × g, 45 min), then the pellet was suspended in 20% glycerol, 0.5 mmol/L EDTA, 0.5 mmol/L PMSF, and 10 mmol/L HEPES, pH 7.0. A 0.2 mL aliquot of the plasma membrane was added to 2 mL of the reaction mixture containing 1.8 mL of the phosphate buffer (0.1 mol/L, pH7.4), the mixture was incubated (37 °C in a water-bath shaker for 1 h), and the reaction was terminated by adding 0.1 mL of trichloroacetic acid (10%) followed by the addition of 1.0 mL of 0.67% thiobarbituric acid. Samples were kept in a boiling water bath for 20 min and subsequently cooled on ice and centrifuged (2500 × g, 10 min). The absorbance of the resultant supernatant, containing TBARS, was measured at 432 nm against a reagent blank at 25 °C. The results were expressed as nmol TBARS formed/mg protein using a molar extinction coefficient of 1.56 × 105/mol/L cm. Following treatment, the authors reported increases of LPO in the cells by 2.7–3.8-fold in the presence of 10–100 µg/mL estragole, while cells treated with eugenol reached an LPO of 1.76–4.93-fold compared with the control cells.
In addition, LPO could be measured by determining the MDA content in the cells. Kalagatur et al. [54] inoculated 10 µL of the fungal spore suspension of F. graminearum in 100 mL of the liquid medium with different amounts of C. martinii EO and incubated the suspension for 7 days at 28 °C. Following incubation, the mycelia were separated through filter paper. The amount of MDA present in the fungal sample was analyzed. The quantification of MDA was conducted using a spectrometric dependent lipid-peroxidation assay kit following the manufacturer’s instructions. The MDA levels in the fungal cells increased with the EO treatment and ranged from 3.5 to 39.0 nmol/L/mg of protein for 100 to 600 μL/L.
5.4 Assays related to mitochondrial functionality
As mentioned in Sect. 4.5, mitochondrion is the central energy-yielding source for cells. Especially, enzymes related to TCA and electron transport that are vital to maintain the cell alive.
5.4.1 Determination of the mitochondrial succinate dehydrogenase (SDH) activity
Li et al. [36] investigated the SDH activity in R. nigricans treated with cinnamon EO. ~ 2 g wet mycelium were re-suspended in 10 mL of phosphate buffer (PBS, pH 7.4), broken with ultrasound in an ice bath for 10 min, and centrifuged at 5000 rpm for 5 min at 4 °C to remove the mycelia fragments and conidia. The supernatant was assayed using an SDH assay kit purchased from Nanjing Jiancheng, Institute of Bioengineering (Nanjing, Jiangsu, China) following the manufacturer’s instructions. The activity of SDH was expressed as 1 U/mg prot = ΔA600/(0.01 min mg protein). The activity of SDH dropped constantly for 12 h and almost disappeared after 24 h in the treated cells with cinnamon EO (0.1 μL/mL). The loss of SDH activity involved a loss of metabolic energy in the cell that resulted in the loss of viability. Other EOs reportedly reduce SDH activity, such as fennel seeds EO (0.018–0.078 µL/mL [30]) against T. rubrum; and C. tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31].
5.4.2 Analysis of the malate dehydrogenase (MDH) activity
Li et al. [36] assessed the effect of cinnamon EO on the activity of MDH from R. nigricans. ~ 2 g wet mycelium were re-suspended in 10 mL PBS (pH 7.4). The cell suspension was broken with ultrasound in an ice bath for 10 min and then centrifuged at 10,000 rpm for 30 min at 4 °C. The supernatant was collected and stored on ice for the MDH measurements. The activity of MDH was tested using the MDH assay kit purchased from the Nanjing, Jiancheng Institute of Bioengineering (Nanjing, Jiangsu, China), according to the manufacturer’s instructions. The activity of MDH was expressed as:
During the first 3 h of treatment with cinnamon EO (0.1 μL/mL), the activity of MDH dropped sharply, and gradually decreased for up to 24 h. Other EOs reportedly reduced the MDH activity such as fennel seeds EO (0.018–0.078 µL/mL [30]) against T. rubrum; and C. tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31].
5.4.3 Determination of the mitochondrial ATPase activity (proton efflux measurement)
Mitochondrial ATPase activity is also affected by EOs. Tian et al. [25] and Zeng et al. [30] used a micro-ATPase assay kit to analyze the effects of dill EO (0.25–1.0 µL/mL) on A. flavus or fennel EO (0.018–0.078 µL/mL) on T. rubrum, respectively. Jianchen Institute of Biological Engineering of Nanjing (Jiangsu, China) provided the kit. The measure was performed following the manufacturer’s protocol to determine the OD636 after the 5 min reaction, using a 20 nmol/L inorganic phosphate as the control. One unit of ATPase activity was defined as 1 µmol inorganic phosphorus catalyzed by 1 mg enzyme for 1 h [µmol Pi/mg protein/h]. Dill EO at 0.25, 0.5, 0.75, and 1.0 µL/mL reduced the ATPase activity of A. flavus that ranged from 7% to 70% [25]. Zeng et al. [30] found that the ATPase activity of T. rubrum was significantly reduced following treatment with fennel EO at all of the concentrations tested. Similar results were reported for the C. tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31].
5.4.4 Cytochrome c assay
Cytochrome (Cyt) c is a multi-functional enzyme essential for mitochondrial electron transport and is involved in the life and death decisions of the cell (apoptosis). Under healthy conditions, Cyt c function is regulated by the mitochondrial membrane potential (MMP) and acts as a ROS scavenger. In contrast, under stressed conditions (apoptotic stimuli or excessive calcium) Cyt c conducts a maximal electron transport chain resulting in hyperpolarization (MMP > 140 mV) which triggers excessive ROS and leads to Cyt c dissociation and release from the mitochondria causes the apoptosis [77].
Gupta et al. [39] evaluated the effect of cinnamaldehyde and eugenol in the C. glabrata cells and elucidated the specific changes that occurred with the concentration of Cyt c in the cytoplasm and the mitochondria. Cells in the early stationary phase were diluted in PBS (1 × 107 cells/mL) and added to 3 mL of yeast extract-peptone-dextrose (YPD) broth containing cinnamaldehyde (128 μg/mL) and eugenol (256 μg/mL), and then incubated for 24 h at 30 °C. Cells were centrifuged, washed twice with PBS, resuspended in the homogenization medium (50 mmol/L Tris (pH 7.5), 2 mmol/L EDTA, 2% glucose, and 1 mmol/L phenylmethylsulfonyl fluoride). Then, the cells were mixed in the vortex and centrifuged (5000 rpm, 10 min), the supernatant was again centrifuged at 20 000 rpm for 45 min. The supernatant was quantified for Cyt c released from the mitochondria to cytoplasm. To quantify the Cyt c remained in the mitochondria, the pellet was suspended in 50 mmol/L Tris (pH 5.0) with 2 mmol/L EDTA and then incubated at 37 °C for 5 min, the pellet was suspended again, and centrifuged at 10 000 rpm for 30 s. Protein content in the supernatant and the pellet was estimated by the Bradford method, using BSA as the standard. Both samples were reduced with 500 mg/L of ascorbic acid at room temperature for 5 min, and the Cyt c was quantified at 550 nm. The amount of Cyt c released from mitochondria increased by 120% and 70% when it was treated with cinnamaldehyde and eugenol, respectively [39].
5.4.5 Mitochondrial membrane potential (MMP) measurement
MMP represents the transmembrane electrochemical potential across the mitochondrial membrane that is regulated via proton pumps. It is in the transmembrane space where ATP is generated as an energy source for cellular functions. Under stable conditions or normal physiological activity, the levels of MMP and ATP in cells remain relatively stable (with small fluctuations). Under stress conditions, changes in both factors can cause cell damage. In addition, if there are long-term drastic drops or increases in MMP, cell death occurs. MMP also regulates anion and cation homeostasis in mitochondria, apoptosis, and active transport protein; among other functions necessary to maintain healthy mitochondria [59].
Tian et al. [25] and Chen et al. [29] measured the MMP to assess the effect of dill seeds EO on A. flavus or C. albicans, respectively. The MMP was examined by flow cytometry using rhodamine 123 (Rh 123). Rh123 is a cationic and lipophilic dye that permeates the negatively charged mitochondria and reflects the mitochondrial membrane potential. The ability of Rh123 to permeate microbial cells depended on the type of cell, for instance, yeast cells require a shorter time (< 1 h) whereas mold requires a longer time (> 4 h), this might occur as there are fewer lipophilic components in the cell membrane or cell wall of the filamentous fungus [25]. The fungal cells were exposed to various doses of EO. Yeast cell analysis included the following, the incubation period with EO (12 h, 30 °C) occurred before the Rh123 was added, then the cells were centrifuged, washed twice, and resuspended at 6x106 CFU/mL in PBS. Rh123 was added to a final concentration of 0.1 mg/mL and incubated for 30 min in the dark at 30 °C. Conversely, mold analysis involved 100 ng/L of Rh 123 and was incubated for 12 h at 28 °C in the dark, centrifuged (5000 × g, 5 min) and washed twice with PBS. A flow cytometer was used to visualize the fluorescence intensity of Rh123 and correlate it with the MMP. A. flavus cells exposed to different amounts of dill EO (0.25, 0.5, 1.0, and 2.0 μL/mL) caused an increment in MMP in a concentration-dependent manner showing significant hyperpolarization [25]. Exposure to different concentrations of EO (0.312 to 1.25 μL/mL) showed significant hyperpolarization of the C. albicans MMP in a dose-dependent manner, suggesting mitochondrial dysfunction [29]. Similar results were reported for Ferulago capillaris EO (0.01 to 0.64 μL/mL) against C. albicans [66]; C. tinctorial flowers EO (0.391 to 1.563 μL/mL) against C. neoformans [31]; and cinnamaldehyde (128 μg/mL) and eugenol (256 μg/mL) against C. glabrata [39].
5.5 Methods to evidence gene expression changes
5.5.1 Quantitative real-time PCR analysis
QRT-PCR analysis assesses the gene expression levels of specific genes in the cells under EO treatment at sublethal doses. The expression patterns can show downregulation or overexpression of the genes quantified. For example, the gene ERG3 (a sterol Δ5,6-desaturase gene) downregulated in C. albicans cells treated with different concentrations of carvacrol was consistent with the reduction of ergosterol content in the treated cells [45]. Gupta et al. [39] used QRT-PCR to study the effect of subinhibitory concentration of cinnamaldehyde (32 μg/mL) and eugenol (64 μg/mL) on C. glabrata. Total RNA was extracted from the log-phase cells and treated using an RNAeasy kit, RNA was quantified and used to synthetize cDNA using a kit. Adequate primers were designed and synthesized by IDT. SYBR green mix was used in a Mastercycler for the QRT-PCR. The expression level of the sterol importer (AUS1), GPI-anchored cell wall protein (KRE1), and 1,3-β-glucan synthase (FKS1) genes were significantly downregulated upon treatment.
Darvishi et al. [78] used RT-PCR to quantify the expression of EST2 (Telomerase reverse transcriptase) and TLC1 (RNA component of telomerase enzyme) and showed the effect of thymol on S. cerevisiae BY4741 (haploid yeast strain). Cells were treated at a subinhibitory concentration (60 μg/mL) of thymol for 24 h. Total RNA was extracted using an extraction kit, DNase was treated, quantified, and cDNA was synthesized using 1 μg of total extracted RNA. Oligonucleotide primers were designed from the Saccharomyces Genome Database. Following RT-PCR, the relative values of gene expression were calculated with the untreated samples as controls and then normalized using the phosphoglycerate kinase (PGK1) expression as a standard. Finally, the PCR products were examined by electrophoresis in 1.2% agarose gels. EST2 gene was significantly downregulated in the presence of thymol by a factor of 2.2 compared to the control. However, TLC1 remained unchanged after treatment. These results suggested that the thymol probably interfered with the biological pathway related to the transcription of the EST2 gene, reducing the telomerase activity during the cell cycle.
Shen et al. [52] used this analysis to show the expression of nitric oxide synthase (NOS) and nitrate reductase (NR) following spore treatment (A. flavus) with thymol (200 μg/mL). First, the treated spores were ground in liquid nitrogen. The total RNA was extracted using an RNA pure Plant kit following the manufacturer’s instructions, treated with DNase I, and then reverse-transcribed to form the first-strand cDNA. cDNA was amplified for the semi-quantitative analysis. The primers of NOS and NR were designed by Primer-blast in NCBI, and a 18S rRNA gene was used as a standard. The RT-PCR was achieved, and the products were separated in 2% agarose gels and stained with ethidium bromide. Densitometric scanning was used for image analysis and quantified the signal intensity of each band. The RT-PCR results confirmed that NOS and NR genes were upregulated in response to the thymol treatment; thus, it induced NOS- and NR-dependent NO generation in the spores of A. flavus.
This test also has been used to analyze the effects of EOs on mycotoxin production in molds (Aspergillus and Penicillium genus) since one concern about mold growing on food is their mycotoxins. Hence the importance of measuring the genes regulated the mycotoxin production. In this regard 1.0 mmol/L of cinnamaldehyde downregulated the level of pks (98%), nrps (96%), laeA (84%), veA (76%), and velB (74%) genes which were ochratoxin biosynthetic and regulatory in Aspergillus ochraceous. These reductions in gene expression resulted in a drop of 82% of ochratoxin production following 7 days of treatment [55].
5.5.2 Real-time fluorescence quantitative PCR (RTFQ-PCR) analysis
The RTFQ PCR also elucidates the effect of EO on specific gene expression. OuYang et al. [10] tested RTFQ-PCR analysis on G. citri-aurantti spores treated with 0.25 μg/mL (1/2 MIC) cinnamaldehyde for 30- and 60-min. RNA was collected from the G. citri-aurantii cells using the Trizol reagent following the manufacturer’s instructions. 2 μg of the RNA free DNA were used for reverse transcription using the oligo dT18. RTFQ-PCR was performed in a BIO-RAD CFX Connect Thermal Cycler using the FastStart Universal SYBR Green Master. The primers for the test genes were designed based on RNA-Seq data for G. citri-aurantii. RTFQ-PCR was programmed as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The 2−Δ ΔCT method was used to quantify the value of each sample using the r-actin gene as an internal reference. Following 30 min of exposure to cinnamaldehyde, the expression levels of CHS2, chsA, chsB, chsG, and UAP1 involved in chitin biosynthesis were significantly lower than that of the control samples. Moreover, the expression level of CHI1 related to chitin decomposition was significantly higher in the spores treated with cinnamaldehyde than in the control. Therefore, cinnamaldehyde inhibited chitin synthesis and induced the hydrolysis of chitins affecting the formation of the cell wall and destroying the cell wall integrity.
6 Other techniques to determine the target site
Surface-Enhanced Raman Spectroscopy (SERS) has shown a strong correlation between nucleic acid release, protein leakage, and lipid losses in the P. italicum treated with trans-cinnamaldehyde. This suggests that it would be an alternative to analyzing the antifungal mechanisms of EOs on the fungus, due to the SERS offered ultrasensitive fingerprint information [22]. SDS-PAGE electrophoresis also was proposed to evaluate the effect of L. cubeba EO on the B. cinerea internal proteins; three bands were clearly fainter than the untreated fungal cells [38]. However, the proteins should be identified and quantified to elucidate their function or role. Another approach was analyzed the length of the telomere restriction fragment which was based on previous observations of the thymol effects on the haploid S. cerevisiae cells [78]. The test involved the extraction of DNA, digestion, hybridization with a telomeric probe, visualization of bands, and calculation of the telomere sizes using internal control fragments. However, observations should be completed with RT-PCR for the specific telomerase gene to conclude the analysis.
7 Concluding remarks and future trends
Several methods have been proposed to elucidate mainly the target site or the mechanism of action of the EOs in fungal cells. These methods have evolved from the initial testing of the cell wall components to the gene modifications of today. Therefore, they vary in complexity, time consumption, and cost. Researchers commonly test the plasma membrane integrity using dye techniques, ergosterol binding assay, ergosterol quantification, and changes in the cell morphology. Less used techniques are the quantification of the activity of the mitochondrial enzymes, ROS species, and gene expression. Most methods reported are focused on target site. In the future, it is necessary to develop or adapt new methods to elucidate the mechanisms of action of EOs and their components on fungal cells. In addition, fungal cells treated with EOs should be analyze by different methods that allow integrate the results and glimpse the changes triggered. An integral analysis can determine the sequence steps in the mode of action of EOs.
References
Akthar MS, Degaga B, Azam T (2014) Antimicrobial activity of essential oils extracted from medicinal plants against the pathogenic microorganisms: a review. Issues Biol Sci Pharm Res 2(1):1–7
The International Organization for Standardization (2013) International Standard ISO 9235:2013 (E/F), Aromatic natural raw materials—vocabulary, 2nd edn. ISO, Geneva
Hyldgaard M, Mygind T, Meyer RL (2012) Essentials oils in food preservation: mode of action, synergies, and interactions with food matrix components. Front Microbiol 3:12. https://doi.org/10.3389/fmicb.2012.00012
Saad NY, Muller CD, Lobstein A (2013) Major bioactivities and mechanism of action of essential oils and their components. Flavour Fragr J 28(5):269–279. https://doi.org/10.1002/ffj.3165
Swamy MK, Akhtar MS, Sinniah UR (2016) Antimicrobial properties of plant essential oils against human pathogens and their mode of action: an updated review. Evid Based Complement Alternat Med 2016:3012462. https://doi.org/10.1155/2016/3012462
Frost DJ, Brandt KD, Cugier D, Goldman R (1995) A whole-cell Candida albicans assay for the detection of inhibition towards fungal cell wall synthesis and assembly. J Antibiot 48(4):306–310. https://doi.org/10.7164/antibiotics.48.306
Scorzoni L, de Paula e Silva ACA, Marcos CM, Assato PA, de Melo WCMA, de Oliveira HC, Costa-Orlandi CB, Mendes-Giannini MJS, Fusco-Almeida AM (2017) Antifungal therapy: new advances in the understanding and treatment of mycosis. Front Microbiol 8:36. https://doi.org/10.3389/fmicb.2017.00036
Kjøller R, Rosendahl S (2000) Effects of fungicides on arbuscular mycorrhizal fungi: differential responses in alkaline phosphatase activity of external and internal hyphae. Biol Fertil Soils 31:361–365. https://doi.org/10.1007/s003749900180
Shao X, Cheng S, Wang H, Yu D, Mungai C (2013) The possible mechanism of antifungal action of tea tree oil on Botrytis cinerea. J Appl Microbiol 114:1642–1649. https://doi.org/10.1111/jam.12193
OuYang Q, Duan X, Li L, Tao N (2019) Cinnamaldehyde exerts its antifungal activity by disrupting the cells wall integrity of Geotrichum citri-aurantii. Front Microbiol 10:15. https://doi.org/10.3389/fmicb.2019.00055
Carrasco H, Raimondi M, Svetaz L, Di Liberto M, Rodriguez MV, Espinoza L, Madrid A, Zacchino S (2012) Antifungal activity of eugenol analogues. Influence of different substituents and studies on mechanism of action. Molecules 17(1):1002–1024. https://doi.org/10.3390/molecules17011002
Gucwa K, Milewski S, Dymerski T, Szweda P (2018) Investigation of the antifungal activity and mode of action of Thymus vulgaris, Citrus limonum, Pelargonium graveolens, Cinnamomum cassia, Ocimum basilicum, and Eugenia caryophyllus essential oils. Molecules 23:1116. https://doi.org/10.3390/molecules23051116
De Oliveira PF, Mendes JM, de Oliveira LE (2013) Investigation on mechanism of antifungal activity of eugenol against Trichophyton rubrum. Med Mycol 51:504–513. https://doi.org/10.3109/13693786.2012.742966
De Oliveira PF, Mendes JM, Lima IO, Mota KSDL, de Oliveira WA, de Oliveira Lima E (2015) Antifungal activity of geraniol and citronellol, two monoterpenes alcohols, against Trichophyton rubrum involves inhibition of ergosterol biosynthesis. Pharm Biol 53(2):228–234. https://doi.org/10.3109/13880209.2014.913299
Danielli LJ, Pippi B, Duarte JA, Maciel AJ, Lopes W, Machado MM, Oliveira LFS, Vainstein MH, Teixeira ML, Bordignon SAL, Fuentefria AM, Apel MA (2018) Antifungal mechanism of action of Schinus lentiscifolius Marchand essential oil ant its synergistic effect in vitro with terbinafine and ciclopirox against dermatophytes. J Pharm Pharmacol 70:1216–1227. https://doi.org/10.1111/jphp.12949
McGinnis MR, Tyring SK (1996) Introduction to mycology. In: Baron S (ed) Medical microbiology, 4th edn. University of Texas Medical Branch at Galveston, Galveston
Khan A, Ahmad A, Akhtar F, Yousuf S, Xess I, Khan LA, Manzoor N (2011) Induction of oxidative stress as a possible mechanism of the antifungal action of three phenylpropanoids. FEMS Yeast Res 11:114–122. https://doi.org/10.1111/j.1567-1364.2010.00697.x
Khan A, Ahmad A, Akhtar F, Yousuf S, Xess I, Khan LA, Manzoor N (2010) Ocimum sanctum oil and its active principles exert their antifungal activity by disrupting biosynthesis and membrane integrity. Res Microbiol 161:816–823. https://doi.org/10.1016/j.resmic.2010.09.008
Chavan PS, Tupe GS (2014) Antifungal activity and mechanism of action of carvacrol and thymol against vineyard and wine spoilage yeasts. Food Control 46:115–120. https://doi.org/10.1016/j.foodcont.2014.05.007
Wang C, Zhang J, Chen H, Fan Y, Shi Z (2010) Antifungal activity of eugenol against Botrytis cinerea. Trop Plant Pathol 35(3):137–143. https://doi.org/10.1590/S1982-56762010000300001
Yu D, Wang J, Shao X, Xu F, Wang H (2015) Antifungal modes of action of tea tree oil and its two characteristic components against Botrytis cinerea. J Appl Microbiol 119:1253–1262. https://doi.org/10.1111/jam.12939
Huang F, Kong J, Ju J, Zhang Y, Guo Y, Cheng Y, Qian H, Xie Y, Yao W (2019) Membrane damage mechanism contributes to inhibition of trans-cinnamaldehyde on Penicillium italicum using surface-enhanced Raman spectroscopy (SERS). Sci Rep 9:490. https://doi.org/10.1038/s41598-018-36989-7
Pinto E, Pina-Vaz C, Salgueiro L, Gonçalves MJ, Costa-de-Oliveira S, Cavaleiro C, Palmeira A, Rodrigues A, Martinez-de-Oliveira J (2006) Antifungal activity of the essential oil of Thymus pulegioides on Candida, Aspergillus and dermatophyte species. J Med Microbiol 55:1367–1373. https://doi.org/10.1099/jmm.0.46443-0
Zuzarte M, Gonçalves MJ, Cavaleiro C, Canhoto J, Vale-Silva L, Silva MJ, Pinto E, Salgueiro L (2011) Chemical composition and antifungal activity of the essential oils of Lavandula viridis L’Hér. J Med Microbiol 60:612–618. https://doi.org/10.1099/jmm.0.027748-0
Tian J, Ban X, Zeng H, He J, Chen Y, Wang Y (2012) The mechanism of antifungal action of essential oil from dill (Anethum graveolens L.) on Aspergillus flavus. PLoS ONE 7(1):e30147. https://doi.org/10.1371/journal.pone.0030147
Pina-Vaz C, Gonçalves-Rodríguez A, Pinto E, Costa-de-Oliveira S, Tavares C, Salgueiro L, Cavaleiro C, Gonçalves MJ, Martinez-de-Oliveira J (2004) Antifungal activity of Thymus oils and their major compounds. J Eur Acad Dermatol Venereol 18(1):73–78. https://doi.org/10.1111/j.1468-3083.2004.00886.x
Pinto E, Vale-Silva L, Cavaleiro C, Salgueiro L (2009) Antifungal activity of the clove essential oil from Syzygium aromaticum on Candida, Aspergillus and dermatophyte species. J Med Microbiol 58:1454–1462. https://doi.org/10.1099/jmm.0.010538-0
Ahmad A, Khan A, Kumar P, Bhatt RP, Manzoor N (2011) Antifungal activity of Coriaria nepalensis essential oil by disrupting ergosterol biosynthesis and membrane integrity against Candida. Yeast 28:611–617. https://doi.org/10.1002/yea.1890
Chen Y, Zeng H, Tian J, Ban X, Ma B, Wang Y (2013) Antifungal mechanism of essential oil from Anethum graveolens seeds against Candida albicans. J Med Microbiol 62:1175–1183. https://doi.org/10.1099/jmm.0.055467-0
Zeng H, Chen X, Liang J (2015) In vitro antifungal activity and mechanism of essential oil from fennel (Foeniculum vulgare L.) on dermatophyte species. J Med Microbiol 64:93–103. https://doi.org/10.1099/jmm.0.077768-0
Zeng H, Li T, Ding H, Tian J (2016) Anti-cryptococcus activity and mechanism of essential oil from Coreopsis tinctoria flowering on Cryptococcus. Fungal Genom Biol 6(1):132. https://doi.org/10.4172/2165-8056.1000132
Krishnamoorthy JR, Ranganathan S, Gokul Shankar S, Ranjith MS (2006) Dano: a herbal solution for dandruff. Afr J Biotechnol 5(10):960–962
Hammer KA, Carson CF, Riley TV (2004) Antifungal effects of Melaleuca alternifolia (tea tree) oil and its components on Candida albicans, Candida glabrata and Saccharomyces cerevisiae. J Antimicrob Chemother 53:1081–1085. https://doi.org/10.1093/jac/dkh243
Rachitha P, Krupashree K, Jayashree GV, Gopalan N, Khanum F (2017) Growth inhibition and morphological alteration of Fusarium sporotrichioides by Mentha piperita essential oil. Pharmacogn Res 9:74–79. https://doi.org/10.4103/0974-8490.199771
Konuk HB, Ergüden B (2017) Antifungal activity of various essential oils against Saccharomyces cerevisiae depends on disruption of cell membrane integrity. Biocell 41(1):13–18
Li Y, Nie Y, Zhou L, Li S, Tang X, Ding Y, Li S (2014) The possible mechanism of antifungal activity of cinnamon oil against Rhizopus nigricans. J Chem Pharm Res 6(5):12–20
Tang X, Shao Y-L, Tang Y-J, Zhou W-W (2018) Antifungal activity of essential oil compounds (geraniol and citral) and inhibitory mechanisms on grain pathogens (Aspergillus flavus and Aspergillus ochraceus). Molecules 23:2018. https://doi.org/10.3390/molecules23092108
Wang L, Hu W, Deng J, Liu X, Zhou J, Li X (2019) Antibacterial activity of Litsea cubeba essential oil and its mechanism against Botrytis cinerea. RSC Adv 9:28987–28995. https://doi.org/10.1039/c9ra05338g
Gupta P, Gupta S, Sharma M, Kumar N, Pruthi V, Poluri KM (2018) Effectiveness of phytoactive molecules on transcriptional expression, biofilm matrix, and cell wall components of Candida glabrata and its clinical isolates. ACS Omega 3:12201–12214. https://doi.org/10.1021/acsomega.8b01856
Rajkowska K, Nowak A, Kunicka-Styczynska A, Siadura A (2016) Biological effects of various chemically characterized essential oils: investigation of the mode of action against Candida albicans and HeLa cells. RSC Adv 6:97199–97207. https://doi.org/10.1039/c6ra21108a
De Lira Mota KS, de Olivera Pereira F, de Olivera WA, Lima IO, de Oliviera Lima E (2012) Antifungal activity of Thymus vulgaris L. essential oil and its constituent phytochemicals against Rhizopus oryzae: interaction with ergosterol. Molecules 17(12):14418–14433. https://doi.org/10.3390/molecules171214418
Miron D, Battisti F, Silva FK, Lana AD, Pippi B, Casanova B, Gnoatto S, Fuentefria A, Mayorga P, Schapoval EES (2014) Antifungal activity and mechanism of action of monoterpenes against dermatophytes and yeasts. Rev Bras Farmacogn 24:660–667. https://doi.org/10.1016/j.bjp.2014.10.014
Freires IDA, Murata RM, Furletti VF, Sartoratto A, Alencar SMD, Figueira GM, Rodrigues JADO, Duarte MCT, Rosalen PL (2014) Coriandrum sativum L. (coriander) essential oil: antifungal activity and mode of action on Candida spp., and molecular targets affected in human whole-genome expression. PLoS ONE 9(6):e99086. https://doi.org/10.1371/journal.pone.0099086
De Castro RD, de Souza TMPA, Bezerra LMD, Ferreira GLS, de Brito Costa EMM, Cavalcanti AL (2015) Antifungal activity and mode of action of thymol and its synergism with nystatin against Candida species involved with infections in the oral cavity: an in vitro study. BMC Complement Altern Med 15:417. https://doi.org/10.1186/s12906-015-0947-2
Alizadeh F, Khodavandi A, Esfandyari S, Nouripour-SIsakht S (2018) Analysis of ergosterol and gene expression profiles of sterol Δ5,6-desaturase (ERG3) and lanosterol 14α-demethylase (ERG11) in Candida albicans treated with carvacrol. J Herbmed Pharmacol 7(2):79–87. https://doi.org/10.15171/jhp.2018.14
Rao A, Zhang Y, Muend S, Rao R (2010) Mechanism of antifungal activity of terpenoid phenols resembles calcium stress and inhibition of the TOR pathway. Antimicrob Agents Chemother 54(12):5062–5069. https://doi.org/10.1128/AAC.01050-10
Hua H, Xing F, Selvaraj JN, Wang Y, Zhao Y, Zhou L, Liu X, Liu Y (2014) Inhibitory effect of essential oils on Aspergillus ochraceus growth and ochratoxin A production. PLoS ONE 9(9):e108285. https://doi.org/10.1371/journal.pone.0108285
Bennis S, Chami F, Chami N, Bouchikhi T, Remmal A (2004) Surface alteration of Saccharomyces cerevisiae induced by thymol and eugenol. Lett Appl Microbiol 38:454–458. https://doi.org/10.1111/j.1472-765X.2004.01511.x
Xing F, Hua H, Selvaraj JN, Zhao Y, Zhou L, Liu X, Liu Y (2014) Growth inhibition and morphological alterations of Fusarium verticillioides by cinnamon oil and cinnamaldehyde. Food Control 46:343–350. https://doi.org/10.1016/j.foodcont.2014.04.037
Da Silva BN, Nakassugi LP, Oliveira JFP, Kohiyama CY, Mossini SAG, Grespan R, Nerilo SB, Mallmann CA, Filho BAA, Machinski M Jr (2015) Antifungal activity and inhibition of fumonisin production by Rosmarinus officinalis L. essential oil in Fusarium verticillioides (Sacc.) Nirenberg. Food Chem 166:330–336. https://doi.org/10.1016/j.foodchem.2014.06.019
Kohiyama CY, Ribeiro MMY, Mosinni SAG, Bando E, da Silva BN, Nerilo SB, Oliveira Rocha GH, Grespan R, Graton Mikcha JM, Machinski M Jr (2015) Antifungal properties and inhibitory effects upon aflatoxin production of Thymus vulgaris L. by Aspergillus flavus Link. Food Chem 173:1006–1010. https://doi.org/10.1016/j.foodchem.2014.10.135
Shen Q, Zhou W, Li H, Hu L, Mo H (2016) ROS involves the fungicidal action of thymol against spores of Aspergillus flavus via the induction of nitric oxide. PLoS ONE 11(5):e0155647. https://doi.org/10.1371/journal.pone.015564712(1):20-26
Avanço GB, Ferreira FD, Silva Bomfim N, Rodrigues dos Santos PADS, Peralta RM, Brugnari T, Mallmann CA, de Abreu Filho BA, Mikcha JMG, Machinski M Jr (2017) Curcuma longa L. essential oil composition, antioxidant effect, and effect on Fusarium verticillioides and fumonisin production. Food Control 73:806–813. https://doi.org/10.1016/j.foodcont.2016.09.032
Kalagatur NK, Nirmal Ghosh OS, Sundararaj N, Mudili V (2018) Antifungal activity of chitosan nanoparticles encapsulated with Cymbopogon martinii essential oil on plant pathogenic fungi Fusarium graminearum. Front Pharmacol 9:610. https://doi.org/10.3389/fphar.2018.00610
Wang L, Jin J, Liu X, Wang Y, Liu Y, Zhao Y, Xing F (2018) Effect of cinnamaldehyde on morphological alterations of Aspergillus ochraceus and expression of key genes involved in ochratoxin A biosynthesis. Toxins 10:340. https://doi.org/10.3390/toxins10090340
Tian J, Huang B, Luo X, Zeng H, Ban X, He J, Wang Y (2012) The control of Aspergillus flavus with Cinnamomum jensenianum Hand.-Mazz essential oil and its potential use as a food preservative. Food Chem 130:520–527. https://doi.org/10.1016/j.foodchem.2011.07.061
Cardoso NNR, Alviano CS, Blank AF, Romanos MTV, Fonseca BB, Rozental S, Rodrigues IA, Alviano DS (2016) Synergism effect of the essential oil from Ocimum basilicum var. Maria Bonita and its major components with fluconazole and its influence on ergosterol biosynthesis. Evid Based Complement Alternat Med 2016:5647182. https://doi.org/10.1155/2016/5647182
Pozniakovsky AI, Knorre DA, Markova OV, Hyman AA, Skulachev VP, Severin FF (2005) Role of mitochondria in the pheromone- and amiodarone-induced programmed death of yeast. J Cell Biol 168(2):257–269. https://doi.org/10.1083/jcb.200408145
Zorova LD, Popkov VA, Plotnikov EJ, Silachev DN, Pevzner IB, Jankauskas SS, Zorov SD, Babenko VA, Zorov DB (2018) Functional significance of the mitochondrial membrane potential. Biochem (Mosc) Suppl Ser A Membr Cell Biol 12(1):20–26. https://doi.org/10.1134/S1990747818010129
Li Q, McNeil B, Harvey LM (2008) Adaptive response to oxidative stress in the filamentous fungus Aspergillus niger B1-D. Free Radic Biol Med 44(3):394–402. https://doi.org/10.1016/j.freeradbiomed.2007.09.019
Cánovas D, Marcos JF, Marcos AT, Strauss J (2016) Nitric oxide in fungi: is there NO light at the end of the tunnel? Curr Genet 62:513–518. https://doi.org/10.1007/s00294-016-0574-6
Guo K, Xia K, Yang Z-M (2008) Regulation of tomato lateral root development by carbon monoxide and involvement in auxin and nitric oxide. J Exp Bot 59(12):3443–3452. https://doi.org/10.1093/jxb/ern194
Angelova MB, Pashova SB, Spasova BK, Vassilev SV, Slokoska LS (2005) Oxidative stress response of filamentous fungi induced by hydrogen peroxide and paraquat. Mycol Res 109(2):150–158. https://doi.org/10.1017/S0953756204001352
Nazzaro F, Fratianni F, Coppola R, De Feo V (2017) Essential oils and antifungal activity. Pharmaceuticals 10:86. https://doi.org/10.3390/ph10040086
Iswarya S, Subha TS, John Sundar V, Gnanamani A (2016) Microsphere formulations of essential oil of Acorus calamus L. controls Aspergillus niger growth in finished leather at an extended period of storage: featuring extraction, formulation and evaluation. Int J Pharmacognosy 3(7):295–305. https://doi.org/10.13040/IJPSR.0975-8232.IJP.3(7).295-05
Pinto E, Hrimpeng K, Lopes G, Vaz S, Gonçalves MJ, Cavaleiro C, Salgueiro L (2013) Antifungal activity of Ferulago capillaris essential oil against Candida, Cryptococcus, Aspergillus and dermatophyte species. Eur J Clin Microbiol Infect Dis 32:1311–1320. https://doi.org/10.1007/s10096-013-1881-1
Wang P, Ma L, Jin J, Zheng M, Pan L, Zhao Y, Sun X, Liu Y, Xing F (2019) The anti-aflatoxigenic mechanism of cinnamaldehyde in Aspergillus flavus. Sci Rep 9:10499. https://doi.org/10.1038/s41598-019-47003-z
Essid R, Hammami M, Gharbi D, Karbouch I, Hamouda TB, Elkahoui S, Limam F, Tabbene O (2017) Antifungal mechanism of the combination of Cinnamomum verum and Pelargonium graveolens essential oils with fluconazole against pathogenic Candida strains. Appl Microbiol Biotechnol 101:6993–7006. https://doi.org/10.1007/s00253-017-8442-y
Perczak A, Gwiazdowska D, Marchwinska K, Jus K, Gwiazdowski R, Waskiewicz A (2019) Antifungal activity of selected essential oils against Fusarium culmorum and F. graminearum and their secondary metabolites in wheat seeds. Arch Microbiol 201:1085–1097. https://doi.org/10.1007/s00203-019-01673-5
Khan A, Ahmad A, Manzoor N, Khan LA (2010) Antifungal activities of Ocimum sanctum essential oil and its lead molecules. Nat Prod Commun 5(2):345–349
Rajput SB, Karuppayil SM (2013) Small molecules inhibit growth, viability and ergosterol biosynthesis in Candida albicans. SpringerPlus 2:26. https://doi.org/10.1186/2193-1801-2-26
Klug L, Daum G (2014) Yeast lipid metabolism at a glance. FEMS Yeast Res 14:369–388. https://doi.org/10.1111/1567-1364.12141
Ruess L, Häggblom MM, Zapata EJG, Dighton J (2002) Fatty acids of fungi and nematodes—possible biomarkers in the soil food chain? Soil Biol Biochem 34(6):745–756. https://doi.org/10.1016/S0038-0717(01)00231-0
Freiesleben SH, Jäger AK (2014) Correlation between plant secondary metabolites and their antifungal mechanisms—a review. Med Aromat Plants 3(2):154. https://doi.org/10.4172/2167-0412.1000154
Cox SD, Mann CM, Markham JL, Bell HC, Gustafson JE, Warmington JR, Wyllie SG (2000) The mode of antimicrobial action of the essential oil of Melaleuca alternifolia (tea tree oil). J Appl Microbiol 88(1):170–175. https://doi.org/10.1046/j.1365-2672.2000.00943.x
Vasilaki AT, McMillan DC (2011) Lipid peroxidation. In: Schwab M (ed) Encyclopedia of cancer. Springer, Berlin, pp 2054–2055. https://doi.org/10.1007/978-3-642-16483-5_3373
Hüttermann M, Pecina P, Rainbolt M, Sanderson TH, Kagan VE, Samavati L, Doan JW, Lee I (2011) The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11(3):369–381. https://doi.org/10.1016/j.mito.2011.01.010
Darvishi E, Omidi M, Bushehri AA, Golshani A, Smith ML (2013) Thymol antifungal mode of action involves telomerase inhibition. Med Mycol 51:826–834. https://doi.org/10.3109/13693786.2013.795664
Acknowledgements
This work was supported by the National Council for Science and Technology (CONACyT) of Mexico [Grant CB-2016-01-283636] and Universidad de las Americas Puebla (UDLAP). Author Cortés-Zavaleta acknowledges financial support for her postdoctoral fellowship from CONACyT and UDLAP. The authors would like to thank Eduardo Quío-Velasco for Figure design.
Author information
Authors and Affiliations
Contributions
OC: Researched studies and wrote part of the manuscript. EM: completed the research studies, compiled data, interpreted results, and wrote the complete manuscript. AL: Conceptualized the idea of review, reviewed and edited the manuscript, and acquired the funding resources. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest declared associated with this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mani-López, E., Cortés-Zavaleta, O. & López-Malo, A. A review of the methods used to determine the target site or the mechanism of action of essential oils and their components against fungi. SN Appl. Sci. 3, 44 (2021). https://doi.org/10.1007/s42452-020-04102-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-04102-1