
Abstract
This work presents a facile method of growing zinc-doped α-hematite (Zn-doped α-Fe2O3) nanostructures via thermal oxidation of Fe sheet in the presence of Zn2+ mist. Both undoped and Zn-doped α-Fe2O3 nanostructures exhibit blade-like morphology mixed with some nanowires. In general, smaller yet denser nanostructures are formed at higher oxidation temperatures. On the other hand, misting (water vapor) enhances the oxidation rate, leading to larger nanoblades. Raman and energy dispersive X-ray spectroscopy reveal the successful incorporation of Zn in the α-Fe2O3 lattice. However, excessive Zn2+ (0.01 M) promotes the formation of large Zn hydroxide chloride particles on top of the α-Fe2O3 nanoblades. The undoped α-Fe2O3 nanostructures prepared at 650 °C in water vapor effectively adsorb hexavalent chromium [Cr(VI)] in aqueous solution with about 95% removal efficiency. The sample oxidized in 0.005 M Zn2+ mist is also efficient in the Fenton-assisted photodegradation of methyl orange with > 90% removal even after five degradation cycles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Bodies of water near industrial facilities are constantly laden with various pollutants, both organic and inorganic, that could negatively affect the ecosystem if left untreated. Heavy metals, which exist in ionic forms, are one of the most common inorganic wastes that can be found in contaminated waters today. For example, hexavalent chromium, Cr(VI), is a strong carcinogenic and can lead to multiple complications, and even death, if ingested by humans. This contaminant is commonly found near textile, leather tanning, ore processing and refining, and electroplating facilities [1]. Due to its threat to human health, a maximum limit of 0.05 mg/L of Cr(VI) is set by the World Health Organization (WHO) for human consumption and fisheries [2]. Synthetic dyes are also produced in industrial scales as it gives color to almost any material, such as papers, textiles, and plastics [3]. Due to their inherent stability, dyes cannot be easily biodegraded and removed from solution upon discharge. This again poses risks to the environment if left untreated as synthetic dyes are hazardous compounds [4, 5]. In addition, colored wastewater can alter the ecosystem of a body of water as it impedes the efficient penetration of sunlight necessary for aquatic flora and fauna.
Common treatments for industrial effluents include ion exchange [6, 7], flotation [8], filtration [9], adsorption [10, 11] and coagulation and flocculation [12]. Among these methods, adsorption provides a direct and economical route for removing both organic and inorganic contaminants. Furthermore, adsorbent materials can potentially be reused through desorption/regeneration processes. Adsorption occurs because of van der Waals forces (physical adsorption) or from the formation of chemical bonds (chemical adsorption) between the adsorbent and adsorbate species [13]. Another promising way to remove organic pollutants is through advanced oxidation processes, specifically by photodegradation [14,15,16]. This process utilizes a semiconductor photocatalyst, such as transition metal oxides, which harnesses light energy for activation. Absorption of light energy greater than or equal to the band gap of the semiconductor results to photoexcitation, where electrons from the valence band are excited to the conduction band, leaving behind a hole in the conduction band. The production, and consequent utilization, of these charge carriers are integral in the redox processes producing the radical species that will then attack and degrade the target pollutants. Since its discovery, photocatalysis has been widely explored in its application for the degradation of organic pollutants, and even for the oxidation or reduction of some heavy metals to its less harmful or benign states. Since organic and inorganic pollutants necessitate different treatment approaches, the fabrication of multi-functional materials that can be utilized for both systems, i.e. for adsorption and photocatalysis, is therefore worth considering. Not only will it potentially reduce treatment costs, but it will provide a degree of flexibility by extending the applicability of the material for the treatment of different types of pollutants.
Hematite (α-Fe2O3) is a naturally occurring iron oxide abundant in rocks and soils [17]. Different synthesis methods for various nanostructured α-Fe2O3, such as combustion [18, 19], hydrothermal process [20,21,22], sol–gel [23, 24], precipitation [25,26,27], and thermal oxidation, [28,29,30] have already been explored for various applications. Several studies have investigated the utilization of nanostructured α-Fe2O3 for both adsorptive removal and photocatalytic degradation of pollutants [22, 23, 28, 31,32,33]. As a photocatalyst, however, α-Fe2O3 has several drawbacks including very fast photoexcited state lifetime due to its rapid recombination rate (10−12 s) [31] and short diffusion length (< 10 nm) [32, 34]. Thus, doping and nanoengineering the surface of α-Fe2O3 have been proposed to address these limitations.
In particular, Zn-doping has been employed by previous studies to improve the electronic properties of α-Fe2O3. Chu et al. [35] hydrothermally synthesized spindle-like, Zn-doped α-Fe2O3 nanostructures. It was further modified with tungsten sulfide (WS2) and were used as hybrid photocathode for hydrogen production. α-Fe2O3 microcubes were also hydrothermally produced in another study and used as electrode for acetone detection [36]. The amount of Zn2+ was also found to influence the size of resulting α-Fe2O3 microcubes. α-Fe2O3 thin films, on the other hand, were fabricated by Chen et al. [37] via spin coating and consequent thermal treatment for hydrogen production. Incorporation of dopant rendered the naturally n-type α-Fe2O3 to a p-type semiconductor and extended its absorption edge to enhance its photoactivity. These recent works suggest that Zn doping is beneficial in improving the properties of α-Fe2O3. While these techniques pose promising practical applications for removal of pollutants in solution, the production of equally efficient Zn-doped α-Fe2O3 nanostructures via a direct, one-step technique still comes with a challenge.
In this study, the fabrication of Zn-doped α-Fe2O3 nanoblades by thermal oxidation of iron (Fe) sheets in the presence of Zn2+ mist was described. Zn2+ was introduced into the α-Fe2O3 nanoblades by misting using a commercial nebulizer during thermal oxidation. To the best of our knowledge, this study is the first to demonstrate the growth of Zn-doped α-Fe2O3 nanostructures through this simple, direct process. At the same time, the effects of oxidizing temperature, time, and atmosphere on the morphology and phase composition of α-Fe2O3 nanostructures were investigated. The adsorptive removal of Cr(VI) using the α-Fe2O3 nanoblades was thoroughly studied by varying the initial concentration of Cr(VI), contact time and temperature. The photocatalytic properties of α-Fe2O3 nanoblades was also investigated by photodegrading methyl orange (MO) in the presence of minute amounts of hydrogen peroxide (H2O2). This work is also among the first to demonstrate the use of common, antiseptic grade H2O2 solution as activator of the Fenton reaction rather than concentrated solutions which are more expensive and difficult to handle. Finally, the reusability of the α-Fe2O3 nanoblades as a photocatalyst was also evaluated by performing several cycles of photodegradation of MO.
2 Methodology
2.1 Synthesis and characterization of Zn-doped α-Fe2O3 nanoblades by thermal oxidation
Iron (Fe) foils (99.99% purity, Nilaco Inc.) with thickness of 0.5 mm were cut to 1.0 × 1.0 cm2 dimensions. The Fe sheets were subjected to 1200 grit silicon carbide (SiC) paper wet grinding, followed by ultrasonic cleaning in acetone for 5 min. Grinding was done to ensure the surface of the foil is free from any native oxides. After cleaning, the Fe sheet was placed in an alumina crucible boat and positioned in a Carbolite MTF 12/38/250 tube furnace. Thermal oxidation was performed at 500, 650, and 800 °C in dry air and with water vapor for 180 min with a heating rate of 5 °C/min. Water vapor was introduced into the furnace using a compressor nebulizer (Omron NE-U17) with maximum air flow rate of 17 L/min and nebulization rate of approximately 0.3 mL/min at 1.7 MHz ultrasonic frequency. The samples were then furnace cooled to room temperature. The synthesis of Zn-doped α-Fe2O3 samples was performed at 500 °C for 3 h, while misting 100 mL of 0.005 or 0.010 M zinc chloride (ZnCl2, JT Baker) aqueous solution. Misting was done during ramp-up and soaking. After 3 h of misting, about 73 mL was left in the nebulizer. The morphology of both doped and undoped samples was observed under a field-emission scanning electron microscope (FE-SEM, Hitachi SU8230). Phase identification was performed by X-Ray diffractometry using Cu Kα radiation (XRD, Shimadzu XRD 7000 Maxima) and Raman spectroscopy (Horiba LabRAM HR Revolution) collected using a 50 × objective and a 632 nm He–Ne laser. Elemental analysis was carried out by energy dispersive X-ray spectroscopy (EDX, Horiba XMax).
2.2 Adsorptive removal of Cr(VI) by α-Fe2O3 nanoblades
Cr(VI) solutions were prepared by dissolving appropriate amounts of potassium dichromate (K2Cr2O7, Loba Chemie, 99.9%) in water. The pH of the solution was adjusted to 2 by adding sulfuric acid (18.4 M H2SO4, Sigma Aldrich). At this pH, Cr(VI) is present as HCrO4− anion. The α-Fe2O3 nanoblades were then immersed in the solution for 24 h. The effects of contact time, initial Cr(VI) concentration (30–210 mg/L), and temperature (25, 35, and 45 °C) on the adsorption capacity were investigated. The measurement of remaining Cr(VI) in solution was done through a standard colorimetry method in a UV–Visible spectrophotometer (Ocean Optics) at λ = 542 nm. About 2 drops of diphenyl-carbazide (C13H14N4O, DKL Laboratories), prepared by dissolving 0.5 mg of diphenyl-carbazide in 100 mL acetone (C3H6O, Fisher Chemical), were added to every 2.0 mL aliquots before UV–Vis testing.
2.3 Fenton-assisted photocatalytic degradation of methyl orange by α-Fe2O3 nanoblades
In a typical experiment, 2 pieces of 1.0 × 1.0 cm2 α-Fe2O3 foil were immersed in 30 mL 5 mg/L methyl orange (MO, C14H14N3NaO3S, Loba Chemie). Afterwards, 30.0 μL of 3 wt% hydrogen peroxide (antiseptic grade H2O2, Rhea) was added to initiate the heterogeneous Fenton reaction. Control set-ups were also included where MO alone, and MO with H2O2, was exposed to UV irradiation to account for their corresponding effects. Photocatalytic degradation was performed under two 10 W UV-C lamps (Sankyo Denki G10T8, λ = 253.7 nm) inside a blackbox set-up. Then, 2 mL aliquot samples were taken at set time intervals and tested by UV–Vis spectroscopy to monitor the change in the MO concentration with time. For the reusability studies, 2 pieces of 1.0 × 1.0 cm2 α-Fe2O3 foil were re-used in the photodegradation of 5 mg/L MO solution for five cycles. In each cycle, the catalysts were rinsed with deionized water twice before immersion into the MO solution and UV irradiation. Final concentration was then quantified using UV–Vis spectroscopy.
3 Results and discussion
3.1 Effects of temperature and atmosphere on the growth α-Fe2O3 nanoblades
Figure 1 shows the XRD patterns of the Fe sheets after thermal oxidation in air and water vapor at increasing temperature. The diffraction patterns suggest the formation of multilayered iron oxide phases after thermal oxidation. Peaks attributed to magnetite (Fe3O4) were determined at 2θ = 30.2, 43.2, 57.1, and 62.6° for the sample synthesized in air at 500 °C as seen in Fig. 1a. These peaks correspond to the reflections of the (220), (400), (511), and (440) Fe3O4 planes, respectively. The sharpest peak at 2θ = 35.8° could be an overlap between the (311) Fe3O4 and (110) Fe2O3 peaks. However, (311) is the strongest peak of Fe3O4, suggesting that the Fe oxide film could be predominantly Fe3O4. Other small peaks due to α-Fe2O3 (2θ = 33.2, 54.2°) and the Fe substrate (2θ = 45.0 and 65.3°) were also identified from the XRD pattern.

XRD pattern of thermally oxidized Fe sheets in dry air at (a) 500, (b) 650, (c) 800 °C and water vapor at (d) 500, (e) 650 and (f) 800 °C (Legend: I-Fe, H-α-Fe2O3, M-Fe3O4, W-FeO)
As the temperature was increased to 650 and 800 °C in dry air, the α-Fe2O3 peaks became more pronounced as shown in the XRD patterns in Fig. 1b, c. The peaks at 2θ = 24.2, 33.6, 40.9, 49.7, 57.6, and 64.1° are due to the reflections of the (012), (104), (113), (024), (511), and (300) planes of α-Fe2O3, respectively (PDF No 84-0307). These peaks became stronger as the temperature was raised. Then again, the relative intensity of the (110) α-Fe2O3, which coincides with the (311) Fe3O4 peak, was reduced after oxidation at 800 °C. This can be attributed to the decrease in the relative amount of Fe3O4. At 800 °C, the relative intensities of the (104) and (110) α-Fe2O3 also follow more closely the values from the standard diffraction pattern of α-Fe2O3. This further supports the growth of α-Fe2O3 at higher temperatures. On the other hand, the strong peak at around 2θ = 43.6° in Fig. 1c could be due to the (200) peak of wustite (FeO). Based on the Fe-oxygen phase diagram, FeO becomes stable at temperatures above 570 °C. It is known that the thickness of the FeO layer increases with temperature at the expense of the outer Fe3O4 and surface Fe2O3 layers [30]. This could explain the sharp FeO peak observed at 800 °C.
Similar XRD patterns were obtained for the samples prepared in the presence of water vapor. Like the samples oxidized in dry air, Fe3O4 peaks were more apparent at 500 °C, while the α-Fe2O3 peaks at 800 °C. This was also accompanied by a very sharp (200) FeO peak at 2θ = 43.5°. Then again, the XRD peaks of all samples were relatively sharper when water vapor was introduced. This possibly indicates minor crystal growth, which could be attributed to enhanced oxidation rate in the presence of water vapor.
Figure 2 shows the corresponding FE-SEM images of the as-synthesized α-Fe2O3 nanoblades. In all samples, the α-Fe2O3 nanostructures have blade-like appearance, with a broad base and narrow tip. Nanoblades have thicknesses not more than 80 nm. Short nanowires were also present particularly in the samples synthesized in dry air at 500 and 650 °C as seen in Fig. 2a, b. Denser nanosheet coverage was also apparent at 800 °C as in Fig. 2c. On the other hand, it is obvious in Fig. 2d–f that the nanoblades were larger when water vapor was present. The broadest blades were generated at 650 °C, which have an average width taken at half-height of around 830 nm. This is about 215% larger compared to the nanoblades produced in dry air at the same temperature.

FESEM images of α-Fe2O3 nanostructures synthesized in air at a 500, b 650, c 800 °C and in water vapor at d 500, e 650, f 800 °C. Inset shows images of samples tilted at about 45°
It has been reported that the formation of α-Fe2O3 nanostructures is mainly driven by the ‘relief’ of compressive stresses formed at the Fe3O4|α-Fe2O3 interface due to solid-state transformations [25, 28, 30]. The Pilling-Bedworth (P-B) ratio of Fe3O4 and α-Fe2O3 were reported to be 2.10 and 2.14, respectively [30]. This slight specific volume mismatch in the oxide layers is said to be sufficient to induce interfacial compressive stresses, which then triggers the nanostructure growth at the surface of the α-Fe2O3 layer. At the beginning, α-Fe2O3 nanowires possibly formed at the surface, which then laterally grew to nanoblades via surface diffusion driven by concentration gradient [30, 38]. Outward diffusion of Fe atoms from the substrate is easier at the bottom of the blades than at the tips, which explains the broader dimensions at the bottom of the nanostructures. Consequently, transport of Fe atoms from the substrate to the tips of the nanostructures is slow, ultimately forming the tapered, blade-like morphology.
Higher nanowire density and smaller α-Fe2O3 grain sizes were observed at elevated temperature in other works [30, 38, 39]. This indicates enhanced nucleation, which might have limited the subsequent lateral growth of the nanowires. This explains the denser and narrower nanoblades formed at 800 °C as seen in Fig. 2f, which also corresponds to the stronger α-Fe2O3 peaks in the XRD patterns. On the other hand, the presence of water vapor probably enhanced the oxidation rate by accelerating the inward diffusion of oxygen towards the Fe foil. This led to the faster growth of the nanostructures as evident from the SEM images and XRD patterns.
3.2 Effect of oxidation time
Figure 3 shows the development of the α-Fe2O3 nanostructures during thermal oxidation of Fe sheet at 650 °C for 30–180 min in the presence of water vapor. As seen in Fig. 3a, nanoblades of different sizes were formed after 30 min oxidation. The variety of nanoblade sizes implies that some structures may have formed earlier than others. Nanoblade widths range from as small as ~ 90 nm to about 840 nm. It is also apparent that several nanowires (shown in arrows) of around 65 nm in diameter were present and homogeneously distributed among the nanoblades. At 90 min oxidation time, structures were larger yet more uniform. Extending the oxidation time up to 180 min led to even bigger and denser nanoblades as shown in Fig. 3c. Prolonged oxidation allows more time for surface diffusion and atomic transport, which promotes growth of nanoblades. Figure 3d shows the tilted edge of the Fe substrate after oxidation at 650 °C in water vapor for 180 min. Layers with varying thickness and morphology can be observed in Fig. 3d. The upper layer measures about 1.7 µm in thickness while the lower layer, which is composed of columnar grains, has an average thickness of around 5.6 µm. These two layers likely correspond to the Fe3O4 + α-Fe2O3 and FeO phases, respectively [28, 30, 38].

SEM Images of α-Fe2O3 nanostructures formed in water vapor at 650 °C for a 30, b 90, c 180 min; d edge view SEM image of the as-synthesized sample at 650 °C in water vapor showing the different Fe oxide layer. Inset shows preferential nanowire growth at the edge
Depending on the oxidation temperature, different Fe oxide layers may form on the metal substrate [28, 30]. It is well established that at temperatures below 570 °C, a multilayer of Fe|Fe3O4|α-Fe2O3 film is formed. On the other hand, at oxidation temperatures higher than 570 °C, Fe|FeO|Fe3O4|α-Fe2O3 layers are produced [28, 30, 38]. According to the Fe–O phase diagram, FeO only forms at temperatures above 570 °C. The columnar habit of FeO, which was observed in previous works [30, 40], is also apparent in Fig. 3d. Oxidation of the topmost layer is most favorable since it is the most accessible to oxygen, leading to faster oxidation rate. However, the thickness of the columnar FeO inner layer was observed to be approximately three times larger than the upper Fe3O4 + α-Fe2O3 layer. The presence of the much thicker FeO layer suggests that the lower oxides were formed at the expense of the oxide above it as evident from the XRD patterns in Fig. 1. Growth of FeO comes from the degradation of the oxygen-richer Fe3O4 above it. Similarly, Fe3O4 continually forms by obtaining oxygen from the α-Fe2O3 layer [30]. The ease to which FeO forms the thickest layer at 650 °C may also be explained by the more efficient diffusion of oxygen (O) in FeO than in the more established Fe3O4 and α-Fe2O3 phases. Given enough energy, the direction of O diffusion is more favorable towards the phase with the lower O concentration, hence the degradation of the higher oxides. As a result, the FeO phase is more easily grown than the higher oxides above it.
It is also interesting to note that nanowires preferentially grow at the edges of the substrate during oxidation as seen in the inset of Fig. 3d. The edges are areas of high surface plastic deformation brought about by mechanical cutting of the parent Fe sheet. In a similar work, Fe sheet was oxidized at 600 °C for 1 h in the presence of 300 mbar O2. Prior to oxidation, sandblasting was done in the surface of the sheets to investigate its effect on the morphology of the oxide nanostructures. Sandblasting for 5 s drastically increased the density of the resulting nanowires relative to the untreated sample. However, extended sandblasting time to 9 s resulted in the formation of dense nanoblades. The results suggest that there is a certain degree of surface roughening that promotes nanowire growth [41]. Another work was also able to produce similar results. Etched Fe sheets were first induced with mechanical deformation via conventional hammering and were then oxidized in air at 640 °C for 1 h. As a result, densely packed nanowires were formed after oxidation. Other morphologies such as nanoflakes were hardly observed [42]. In the same manner, the mechanical cutting of the substrates in this study, which effectively induced plastic deformation and surface roughness along the edges, also possibly resulted in the formation of nanowires.
3.3 Effect of Zn doping on the morphology and structure of α-Fe2O3 nanoblades
The structural and compositional analysis of the Zn-doped α-Fe2O3 films oxidized at 500 °C in the presence of varying ZnCl2 concentration is shown in Fig. 4a. Upon introduction of Zn dopant, α-Fe2O3, Fe3O4, and Fe XRD peaks were still indexed from the XRD patterns. This suggests that the phase composition of the Fe oxides was preserved. However, the peak at 2θ = 35.8° was shifted to the left relative to the undoped sample as seen in the inset of Fig. 5a. This can be attributed to the Zn atoms being successfully incorporated in the α-Fe2O3 lattice. The ionic radius of Zn2+ (0.073 nm) [36] is larger than Fe3+ (0.064 nm) [32, 43] resulting to the shift to the left in the XRD pattern. However, an unknown peak at 2θ = 38.02° was identified in the diffraction patterns of the Zn-doped samples, which might be due to a Zn hydroxide chloride phase.

a XRD patterns (Legend: I-Fe, H-α-Fe2O3, M-Fe3O4, *zinc hydroxide chloride) and b Raman spectra of undoped and Zn-doped α-Fe2O3 samples synthesized at 500 °C for 3 h

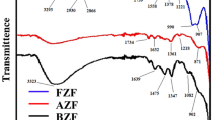
FTIR spectra of the undoped and Zn-doped α-Fe2O3 (0.005 M)
Raman spectroscopy was also employed to confirm the successful incorporation of Zn into α-Fe2O3 as shown in Fig. 4b. Raman peaks at 223 and 496 cm−1 are assigned to the A1g mode of α-Fe2O3, while the peaks at 244, 290, 404, and 610 cm−1 are attributed to the Eg mode of α-Fe2O3 [37, 43, 44]. All samples exhibit the same vibration pattern, specific only to α-Fe2O3, which suggests that it is the dominant phase at the surface of the samples. Noticeably, a new Raman band at about 655 cm−1 emerged after the introduction of dopant as seen in the inset of Fig. 5b. This peak is assigned to the Eu longitudinal optical (LO) mode that is attributed to structural defects brought about by Zn doping [37, 43,44,45].
FTIR analysis of the undoped and Zn-doped sample misted with 0.005 M ZnCl2 is shown in Fig. 5. The spectra for both the undoped and Zn-doped α-Fe2O3 samples are similar except for a pronounced peak centered at around 1079 cm−1 in the doped sample which can be assigned to Fe–OH hydroxo-complex [46]. Another notable difference is the apparent shift to higher wavelengths of the peak associated with the Fe–O stretching vibration at 500–560 cm−1 of the Zn-doped sample relative to pure α-Fe2O3. This may be ascribed to the effect of Zn-doping wherein the lattice parameters of α-Fe2O3 are altered due to the variation in the cation-oxygen bond length by substituting Fe3+ with Zn2+, consequently affecting band positions in the FTIR spectra [45]. This agrees well with the results of the XRD and Raman spectroscopy measurements indicating that Zn was successfully incorporated.
The morphology of the undoped and doped samples is compared in the FE-SEM images in Fig. 6. By misting with 0.005 M ZnCl2, the blade-like morphology of the α-Fe2O3 was still evident from the FE-SEM images. However, they appear less uniform relative to the undoped sample. The presence of chlorine ions (Cl−) in the oxidizing atmosphere possibly hindered the oxidation process and slowed down the formation of the nanostructures. Increasing the concentration to 0.010 M ZnCl2 resulted in non-uniform, slender nanoblades mixed with hexagonal plate-like particles. These particles are possibly the Zn chloride hydroxide detected from the XRD patterns in Fig. 5a.

Top-view SEM images of a, b undoped, and Zn-doped using c, d 0.005 and e, f 0.010 M Zn2+. Images on the right show tilted-view images
Elemental mapping of the doped samples in Fig. 7 shows that Fe, O, and Zn are present in the samples. By misting with 0.005 M ZnCl2, about 22.35 wt% Zn was incorporated in the α-Fe2O3 lattice as shown in Fig. 7a. The elemental maps suggest that Zn is uniformly distributed within the nanoblades, which further implies successful doping of Zn in the α-Fe2O3 lattice. The elemental map for the sample doped with 0.010 M ZnCl2 in Fig. 7b confirms that the hexagonal plate-like particles are composed of Zn. The total wt% of Zn was about 25.32%, which include both the atoms in the α-Fe2O3 lattice and the distinct hexagonal plate-like particles. These findings suggest that there is a maximum amount of Zn that can be introduced in the lattice. Excessive dopant concentration leads to the formation of a secondary phase.

Elemental map of α-Fe2O3 nanoblades doped with a 0.005 M and b 0.010 M Zn2+
3.4 Adsorptive removal of Cr(VI) by α-Fe2O3 nanoblades
The α-Fe2O3 nanoblades prepared by thermal oxidation at 180 min at increasing temperature with and without water vapor were used for the adsorptive removal of Cr(VI) ions in aqueous solution. Figure 8a summarizes the removal efficiency for Cr(VI) for all the samples after adsorption of 200 mg/L Cr(VI) solution for 24 h. Removal efficiency for all the samples ranges from around 75–95%. The sample synthesized at 650 °C in water vapor yielded the highest % removal of about 95%. This condition may have likely produced a good combination of nanowires and nanoblades on the surface of the Fe foil as seen in Fig. 2e. It is also evident from the inset of Fig. 2e that the nanoblades are wide, indicating a large surface area. This possibly results to more adsorption sites for Cr(VI). Thus, this same sample was used in the experiments for the effect of contact time, Cr(VI) initial concentration, and adsorption temperature.

a Equilibrium removal efficiencies of the α-Fe2O3 adsorbents for Cr(VI), b percent removal of Cr(VI) in solution with time with initial [Cr6+] = 15 mg/L, pH = 2, and area of adsorbent = 1.0 cm2, c effect of initial Cr(VI) concentration on the removal efficiency, d effect of adsorption temperature on the removal efficiency
Figure 8b presents the change in the percent removal of Cr(VI) with time after contact with α-Fe2O3 nanoblades. The solution has an initial concentration, volume, and pH of 15 mg/L, 75 mL, and 2, respectively. The area of adsorbate and agitation rate was set at 1.0 cm2 and 100 rpm, respectively. Initially, uptake of Cr(VI) was rapid. In fact, about half of the initial amount of Cr(VI) was removed after 10 min. Prolonging the adsorption led to the gradual increase in the Cr(VI) uptake until a plateau was reached, wherein almost 100% removal was attained after 50 min. As the amount of Cr(VI) in solution was reduced, the driving force for adsorption was also decreased, which explains the slower removal rates at longer adsorption time.
The effect of initial Cr(VI) concentration on the removal efficiency of the α-Fe2O3 films is shown in Fig. 8c. The total volume of the solution and area of adsorbate was 75 mL and 1.0 cm2, respectively. The pH was set at 2, while contact time was 24 h. The removal efficiency progressively decreased with increasing initial concentration, which suggests that there is a finite amount of adsorption sites available on the surface of the α-Fe2O3 nanoblades. Decrease in the removal efficiency can be due to the saturation of the adsorption sites of the α-Fe2O3 nanoblades. On the other hand, Fig. 8d reveals the enhancement in the adsorption efficiency with increasing temperature. Adsorption experiments were conducted at 25, 35, and 45 °C at an initial concentration of 210 mg/L, while keeping the pH, volume, agitation rate, and area of adsorbate constant. The removal efficiency was improved from 80.9 to 85.4% when the temperature was raised from 25 to 45 °C. The enhanced efficiency at elevated temperature implies that the adsorption process is highly likely to be endothermic in nature. The system is able to absorb energy at higher temperature to promote the adsorption of Cr(VI) onto the active sites of the adsorbent.
Figure 9 shows the FTIR spectra of the undoped α-Fe2O3 before and after adsorption with Cr(VI). The characteristic Fe-O stretching mode is evident at about 500–560 cm−1 [46,47,48,49,50]. The wide band found at 3211 cm−1 and at 1630 cm−1 are each assigned to the O–H stretch and H–O–H bending vibration modes respectively due to adsorbed water on the surface [46,47,48,49,50]. These peaks are pronounced only with the sample immersed in Cr(VI) solution. Another prominent peak found at about 1090 cm−1 is previously ascribed to Fe-OH hydroxo-complexes [46].

FTIR spectra of the undoped α-Fe2O3 before and after adsorption of Cr(VI)
The adsorption of Cr(VI) onto the α-Fe2O3 nanostructures is primarily driven by the electrostatic interactions between the adsorbent and adsorbate [28, 46, 47, 49, 50]. According to its speciation diagram, Cr in solution exists primarily as HCrO4− anions at pH = 2 [51, 52]. On the other hand, the point of zero charge (PZC) of α-Fe2O3 is previously reported at around pH = 8.3–8.8 [53, 54], rendering the adsorbent with a positive surface charge at acidic conditions. In aqueous medium, α-Fe2O3 is covered with hydroxyl groups (S-OH) which are protonated at acidic conditions (Eq. 1) [55]. This charge difference drives the negatively charged HCrO4− ions to be attracted onto the positively charged α-Fe2O3 surface, leading to the loss of Cr(VI) in solution (Eq. 2). This may be described by the following chemical reactions [46, 55]:
Related studies agree that the adsorption of HCrO4− onto α-Fe2O3 involves the formation of a surface complex [47, 52, 56, 57]. It was suggested that HCrO4− may have undergone ligand exchange with the surface hydroxyl groups of α-Fe2O3, ultimately forming an inner sphere surface complex [52], as represented in Eq. 2. This may be associated with the pronounced peak centered at about 1090 cm−1 of the FTIR spectrum of α-Fe2O3 after adsorption which was previously assigned to Fe-OH hydroxo-complexes as seen in Fig. 9. The highly acidic environment was able to increase the degree of protonation of the adsorbent thereby increasing adsorption sites, maximizing the high surface area of the α-Fe2O3 nanostructures to efficiently remove Cr(VI) in solution.
3.5 Fenton-assisted photodegradation of methyl orange by undoped and Zn-doped α-Fe2O3 nanoblades
Figure 10 shows the MO percent removal by photodegradation using the α-Fe2O3 nanoblades with increasing irradiation time. Pure MO was found to be highly stable even under UV-C irradiation since percent removal due to photolysis was only around 5.6%. On the other hand, the change in the concentration of MO after contacting with the α-Fe2O3 nanoblades without H2O2 was minimal with only about 6% removal efficiency after 5 h. One end of the structure of MO consists of a sulfonate group attached to sodium (Na). In aqueous solutions, Na exists as a dissolved cation, leaving behind the sulfonate group of MO with a negative charge. This negatively charged end of MO is electrostatically attracted towards the protonated surface hydroxyl groups of α-Fe2O3, potentially leading to the removal of MO in solution [58]. However, the PZC of α-Fe2O3 is earlier stated at around pH = 8.3–8.8 [53, 54]. The actual MO solution, on the other hand, is at neutral pH. Hence, α-Fe2O3 may have only formed a weak, positive surface charge, which resulted to fewer adsorption sites and small uptake of the anionic MO even after 5 h. The large size of the MO molecule further aggravates the adsorption process due to steric hindrance. Therefore, removal of MO via adsorption is not significant. Similarly, direct photocatalysis by illuminating both undoped and Zn-doped (0.01 M) α-Fe2O3 in MO was only able to remove around 5.3% and 5.6%, respectively.

Removal efficiencies of the undoped and Zn-doped α-Fe2O3 catalysts for the degradation of 5 mg/L MO
Meanwhile, adding 30 µL H2O2 in MO under UV-C light degraded a considerable amount of MO even in the absence of the α-Fe2O3 photocatalyst. The removal efficiency was around 50% after 5 h. When both α-Fe2O3 and H2O2 were present, photodegradation of MO was faster and more effective. In fact, around 72.8% of the initial MO concentration was removed after 5 h in the presence of both undoped α-Fe2O3 and H2O2. The Zn-doped α-Fe2O3 samples, together with H2O2, achieved even faster MO degradation. Removal percentages of approximately 99.8 and 92.4% were obtained for the α-Fe2O3 misted with 0.005 M and 0.010 M ZnCl2, respectively.
It is shown from the results that H2O2 alone, in the presence of UV light, can degrade a substantial amount of MO. Thus, the following chemical reaction plays a role in the degradation of MO [59, 60]:
The enhancement of MO degradation in the presence of α-Fe2O3 can be described based on (1) direct charge separation via photocatalysis or (2) dye sensitization that triggers the heterogeneous Fenton reaction. Direct charge separation via photocatalysis can be described by the following reactions:
Upon absorption of light energy greater than the bandgap of α-Fe2O3, photoexcitation produces electron–hole pairs which migrate to the surface of the material (Eq. 5). The photogenerated holes then react with H2O to produce OH· (Eq. 6) that will degrade MO (Eq. 4). The generated conduction band electrons may also react with dissolved dioxygen in the solution to produce a superoxide anion radical \({\text{O}}_{2}^{ \cdot - }\) which could also potentially contribute to the degradation of MO (Eq. 7).
On the other hand, degradation via dye sensitization and Fenton reaction commences via the following chemical reactions [61,62,63]:
MO attains an excited state (MO*) upon absorption of energy from UV light (Eq. 8). At the surface of α-Fe2O3, MO* reduces Fe3+ to Fe2+ (Eq. 9) which consequently reacts with H2O2 and triggers the heterogeneous Fenton reaction (Eq. 10). Likewise, the generated OH· then attacks MO resulting to its degradation (Eq. 4).
Based on experimental observations, the first mechanism is highly unlikely since the removal efficiency of direct photocatalysis is very low due to the fast recombination rate and slow hole mobility of α-Fe2O3 [31, 61]. This further suggests that the production of radical species in Eqs. 6 and 7 are highly improbable. As observed from the degradation results, set-ups with both UV illumination and H2O2 resulted in the highest percent removal. In fact, the undoped sample was even able to degrade substantial amount of MO, when illuminated in the presence of H2O2. In contrast, contacting MO with undoped α-Fe2O3 even in the presence of H2O2 without UV illumination resulted in removal of about 9%. Therefore, UV illumination and H2O2 are both necessary in the efficient degradation of MO and the second mechanism is the primary degradation route in this work.
Further, Zn-doping of α-Fe2O3 has further improved its photocatalytic performance. The introduction of Zn2+ in the lattice may have created impurity level/trap state in the band gap of α-Fe2O3 which possibly inhibits recombination of photoexcited electron–hole pairs to allow surface migration and reaction at the surface [31, 45]. However, this is not supported in the degradation results since direct illumination of Zn-doped α-Fe2O3 (0.01 M) without the presence of H2O2 did not exhibit any improvement relative to the undoped sample. Hence, extending separation lifetime of the photogenerated charge carriers is not the enhancing effect of Zn. Doping α-Fe2O3 with Zn2+ has been reported elsewhere to cause faster interfacial charge transfer as compared to pure α-Fe2O3 [37, 64]. The low interfacial charge transfer resistance is critical in the reaction rate of the Fenton reaction (Reaction 8) since an electron has to transfer from Fe2+ to the adsorbed H2O2 [65]. A more efficient electron transfer at the surface of α-Fe2O3 would also result to a more effective redox cycle between Fe2+ and Fe3+ ultimately leading to enhanced production of OH· that will finally degrade MO. Meanwhile, the apparent decrease in efficiency of the Zn-doped α-Fe2O3 (0.01 M) sample relative to Zn-doped α-Fe2O3 (0.005 M) can be ascribed to the fewer number of α-Fe2O3 nanoblades formed as evident in Fig. 6e, f. The higher concentration of Cl in 0.01 M ZnCl2 mist may have significantly slowed down oxidation rates which therefore led to visibly smaller and less dense α-Fe2O3 nanoblades. Furthermore, the presence of the large Zn hydroxide chloride platelets on top of the nanoblades might have hindered the adsorption of MO. Consequently, there are fewer active sites where pertinent chemical reactions can occur. Despite the less dense α-Fe2O3 nanoblades, the Zn-doped α-Fe2O3 (0.01 M) sample still exhibited better photocatalytic performance than the undoped sample. This clearly indicates the significant role of interfacial charge transfer rate on enhancing the Fenton reaction.
The performance of the Zn-doped α-Fe2O3 (0.005 M) sample was also subjected to five cycles of reusability as shown in Fig. 11. Almost complete MO removal was achieved after the first three cycles of degradation. However, a slight decrease in efficiency to about 92% was attained in the 4th and 5th cycles. The slight decrease in efficiency may possibly be attributed to adsorbed molecules on the catalyst surface thereby rendering some degradation sites inactive. Nevertheless, results show that the Zn-doped α-Fe2O3 films exhibit excellent photostability and may be reused for several number of cycles.

Removal efficiency of Zn-doped α-Fe2O3 nanoblades after five degradation cycles. (Zn2+ = 0.005 M)
4 Conclusion
In summary, α-Fe2O3 nanostructures were formed by thermal oxidation at varying temperature and atmosphere. Oxidation in the presence of water vapor produced larger nanoblades, which were effective (99%) adsorbent for Cr(VI). Removal efficiency was found to decrease with increasing Cr(VI) initial concentration, while it slightly improved at higher temperature. Zn-doped α-Fe2O3 nanoblades were also successfully prepared by misting 0.005 M ZnCl2 during thermal oxidation. At [ZnCl2] > 0.005 M, a secondary Zn-based phase was formed. Fenton-assisted degradation of methyl orange showed almost complete removal using the Zn-doped sample (0.005 M). Reusability studies showed that the α-Fe2O3 nanoblades in the presence of H2O2 can be reused up to five degradation cycles with a removal efficiency > 90%. The enhancement of the photocatalytic property of pure α-Fe2O3 may be ascribed to the enhanced interfacial charge transfer kinetics brought about by the successful introduction of Zn. This approach provides a new facile method in designing reusable and easily retrievable doped-metal oxide nanostructures for environmental applications.
Data availability
Available upon request to authors.
References
Yu D et al (2008) Agency for toxic substances and disease registry case studies in environmental medicine (CSEM): chromium toxicity
WHO (2011) Guidelines for drinking-water quality. World Health Organization
Cheremisinoff N, Rosenfeld P, Davletshin A (2008) Responsible care—a new strategy for pollution prevention and waste reduction through environmental management. Gulf Publishing Company, TX
Bazrafshan E, Zarei A, Nadi H, Zazouli M (2014) Adsorptive removal of methyl orange and reactive red 198 dyes by Moringa peregrina ash. Indian J Chem Technol 21:105
Zaman A, Das P, Banerjee P (2016) In: Rathoure A (ed) Biosorption of dye molecules, toxicity and waste management using bioremediation. IGI Global, Pennsylvania, pp 51–74
Da̧browski A, Hubicki Z, Podkościelny P, Robens E (2004) Selective removal of the heavy metal ions from waters and industrial wastewaters by ion-exchange method. Chemosphere 56(2):91–106
Abdel-Aziz M, Amin N, El-Ashtoukhy E (2013) Removal of heavy metals from aqueous solutions by liquid cation exchanger in a jet loop contactor. Hydrometallurgy 137:126–132
Ali S, Fazaelipoor M (2016) Evaluation of rhamnolipid (RL) as a biosurfactant for the removal of chromium from aqueous solutions by precipitate flotation. J Environ Manag 165:184
Huang Y, Wu D, Wang X, Huang W et al (2016) Removal of heavy metals from water using polyvinylamine by polymer-enhanced ultrafiltration and flocculation. Sep Purif Technol 158:124
Burakov A, Galunin E, Burakova I, Kucherova A et al (2018) Adsorption of heavy metals on conventional and nanostructured materials for wastewater treatment purposes: a review. Ecotox Environ Safe 148:702
Kobayashi Y, Ogata F, Nakamura T, Kawasaki N (2020) Synthesis of novel zeolites produced from fly ash by hydrothermal treatment in alkaline solution and its evaluation as an adsorbent for heavy metal removal. J Environ Chem Eng 8(2):103687
Pang F, Kumar P, Teng T, Omar A, Kailas L (2011) Wastewater. Removal of lead, zinc and iron by coagulation–flocculation. J Taiwan Inst Chem E 42(5):809
Dabrowski A (2001) Adsorption-from theory to practice. Adv Colloid Interface 93:135
Al-Mamun M, Kader S, Islam M, Khan M (2019) Photocatalytic activity improvement and application of UV–TiO2 photocatalysis in textile wastewater treatment: a review. J Environ Chem Eng 7(5):103248
Zhao L, Deng J, Sun P, Liu J et al (2018) Nanomaterials for treating emerging contaminants in water by adsorption and photocatalysis: systematic review and bibliometric analysis. Sci Total Environ 627:1253
Zhu D, Zhou Q (2019) Action and mechanism of semiconductor photocatalysis on degradation of organic pollutants in water treatment: a review. Environ Nanotechnol Monit Manag 12:100255
Wang T et al (2013) Influence of sodium halides (NaF, NaCl, NaBr, NaI) on the photocatalytic performance of hydrothermally synthesized hematite photoanodes. ACS Appl Mater Inter 5(16):7937–7949
Cao Z, Qin M, Jia B, Gu Y et al (2015) One pot solution combustion synthesis of highly mesoporous hematite for photocatalysis. Ceram Int 41(2):2806
Kopanja L, Milosevic I, Panjan M, Damnjanovic V, Tadic M (2016) Sol–gel combustion synthesis, particle shape analysis and magnetic properties of hematite (α-Fe2O3) nanoparticles embedded in an amorphous silica matrix. Appl Surf Sci 362:380
Tadic M, Trpkov D, Kopanja L, Vojnovic S, Panjan M (2019) Hydrothermal synthesis of hematite (α-Fe2O3) nanoparticle forms: synthesis conditions, structure, particle shape analysis, cytotoxicity and magnetic properties. J Alloys Compd 792:599
Trpkov D, Panjan M, Kopanja L, Tadić M (2018) Hydrothermal synthesis, morphology, magnetic properties and self-assembly of hierarchical α-Fe2O3 (hematite) mushroom-, cube- and sphere-like superstructures. Appl Surf Sci 457:427
Rapadas N, Balela M (2017) Hydrothermal synthesis of hierarchical hematite (α-Fe2O3) microstructures for photocatalytic degradation of methyl orange. Philipp J Sci 146(4):396
Demirci S, Yurddaskal M, Dikici T, Sarıoğlu (2018) Fabrication and characterization of novel iodine doped hollow and mesoporous hematite (Fe2O3) particles derived from sol–gel method and their photocatalytic performances. J Hazard Mater 345:27
Lian X, Yang X, Liu S, Xu Y, Jiang C, Chen J, Wang R (2012) Enhanced photoelectrochemical performance of Ti-doped hematite thin films prepared by the sol–gel method. Appl Surf Sci 258(7):2307
Fouad D, Zhang C, El-Didamony H, Yingnan L, Mekuria T, Shah A (2019) Improved size, morphology, and crystallinity of hematite (α-Fe2O3) nanoparticles synthesized via the precipitation route using ferric sulfate precursor. Results Phys 12:1253
Lassoued A, Lassoued M, Dkhil B, Gadri A, Ammar S (2017) Synthesis, structural, optical, and morphological characterization of hematite through the precipitation method: effect of varying the nature of the base. J Mol Struct 1141:99
Supattarasakda K, Petcharoen K, Permpool T, Sirivat A, Lerdwijitjarud W (2013) Control of hematite nanoparticle size and shape by the chemical precipitation method. Powder Technol 249:353
Budiman F, Kian T, Razak K, Matsuda A, Lockman Z (2016) The assessment of Cr(VI) removal by iron oxide nanosheets and nanowires synthesized by thermal oxidation of iron in water vapour. Procedia Chem 19:586
Canaria M, Ramos J, Sayson C, Balela M (2017) Growth of hematite nanostructures in iron foil for environmental cleaning. Solid State Phenom 266:101
Yuan L, Wang Y, Cai R, Jiang Q, Wang J, Li B, Sharma A, Zhou G (2012) The origin of hematite nanowire growth during the thermal oxidation of iron. Mater Sci Eng B Adv 177(3):327
Cao Z, Qin M, Gu Y, Jia B, Chen P, Qu X (2016) Synthesis and characterization of Sn-doped hematite as visible light photocatalyst. Mater Res Bull 77:41
Satheesh R, Vignesh K, Suganthi A, Rajarajan M (2014) Visible light responsive photocatalytic applications of transition metal (M = Cu, Ni and Co) doped α-Fe2O3 nanoparticles. J Environ Chem Eng 2(4):1956
Rahmat S, Rozana M, Tan W et al (2018) One-dimensional α-Fe2O3 nanowires formation by high temperature oxidation of iron and their potential use to remove Cr(VI) ions. In: Lockman Z (ed) 1-Dimensional metal oxide nanostructures: growth, properties, devices. CRC Press, Boca Raton, pp 115–136
Tsege E, Atabaev T, Hossain M, Lee D, Kim H, Hwang Y (2016) Cu-doped flower-like hematite nanostructures for efficient water splitting applications. J Phys Chem Solids 98:283
Chu D, Li K, Liu A, Huang J, Zhang C, Yang P, Du Y et al (2018) Zn-doped hematite modified by graphene-like WS2: a p-type semiconductor hybrid photocathode for water splitting to produce hydrogen. Int J Hydrog Energy 43(15):7307
Song H, Sun Y, Jia X (2015) Hydrothermal synthesis, growth mechanism and gas sensing properties of Zn-doped α-Fe2O3 microcubes. Ceram Int 41(10):13224
Chen Y, Kuo C, Hsu Y (2018) Facile preparation of Zn-doped hematite thin film as photocathode for solar hydrogen generation. J Alloys Compd 768:810
Hiralal P, Unalan H, Wijayantha K, Kursumovic A, Jefferson D, MacManus-Driscoll J, Amaratunga G (2008) Growth and process conditions of aligned and patternable films of iron(III) oxide nanowires by thermal oxidation of iron. Nanotechnology 19(45):455608
Bertrand N, Desgranges C, Poquillon D, Lafont M, Monceau D (2009) Iron oxidation at low temperature (260–500 °C) in air and the effect of water vapor. Oxid Met 73(1–2):139
Grigorescu S, Lee C, Lee K, Albu S, Paramasivam I, Demetrescu I, Schmuki P (2012) Thermal air oxidation of Fe: rapid hematite nanowire growth and photoelectrochemical water splitting performance. Electrochem Commun 23:59
Yuan L, Cai R, Jang J, Zhu W, Wang C, Wang Y, Zhou G (2013) Morphological transformation of hematite nanostructures during oxidation of iron. Nanoscale 5(16):7581
Dlugosch T, Chnani A, Muralidhar P, Schirmer A, Biskupek K, Strehle S (2017) Thermal oxidation synthesis of crystalline iron-oxide nanowires on low-cost steel substrates for solar water splitting. Sci Technol 32(8):084001
Mansour H, Bargougui R, Autret-Lambert C, Gadri A, Ammar S (2018) Co-precipitation synthesis and characterization of tin-doped α-Fe2O3 nanoparticles with enhanced photocatalytic activities. J Phys Chem Solids 114:1
Cha H, Noh H, Kang M, Kang Y (2013) Photocatalysis: progress using manganese-doped hematite nanocrystals. New J Chem 37(12):4004
Suman S, Kumar A, Kumar P (2020) Zn doped α-Fe2O3: an efficient material for UV driven photocatalysis and electrical conductivity. Crystals 10(4):273
Adegoke H, Adekola F, Fatoki O, Ximba B (2013) Adsorption of Cr(VI) on synthetic hematite (α-Fe2O3) nanoparticles of different morphologies. Korean J Chem Eng 31(1):142
Xiao Q, Sun Y, Zhang J, Li Q (2015) Size-dependent of chromium (VI) adsorption on nano α-Fe2O3 surface. Appl Surf Sci 356:18
Debnath A, Bera A, Chattopadhyay K, Saha B (2017) Facile additive-free synthesis of hematite nanoparticles for enhanced adsorption of hexavalent chromium from aqueous media: kinetic, isotherm, and thermodynamic study. Inorg Nano-Met Chem 47(12):1605
Gallo-Cordova A, Morales M, Mazarío E (2019) Effect of the surface charge on the adsorption capacity of chromium (VI) of iron oxide magnetic nanoparticles prepared by microwave-assisted synthesis. Water 11(11):2372
Trang V, Tam L, Van Quy N et al (2020) Enhanced adsorption efficiency of inorganic chromium(VI) ions by using carbon-encapsulated hematite nanocubes. J Sci Adv Mater Dev 5(3):392
Park H, Tavlarides L (2008) Adsorption of chromium (VI) from aqueous solutions using an imidazole functionalized adsorbent. Ind Eng Chem Res 47:3401
Huang X, Hou X, Song F, Zhao J, Zhang L (2016) Facet-dependent Cr(VI) adsorption of hematite nanocrystals. Environ Sci Technol 50(4):1964
Parks G, Bruyn P (1962) The zero point charge of oxides. J Phys Chem 66(6):967
Chatman S, Zarzycki P, Rosso K (2013) Surface potentials of (001), (012), (113) hematite (α-Fe2O3) crystal faces in aqueous solution. Phys Chem Chem Phy 15(33):13911
Singh D, Gupta G, Prasad G, Rupainwar D (1993) The use of hematite for chromium(VI) removal. J Environ Sci Health A 8:1813
Dzieniszewska A, Kyziol-Komosinska J, Pająk M (2020) Adsorption and bonding strength of chromium species by ferrihydrite from acidic aqueous solutions. PeerJ 8:e9324
Johnston C, Chrysochoou M (2014) Mechanisms of chromate adsorption on hematite. Geochim Cosmochim Acta 138:146
Grégoire B, Bantignies J, Le-Parc R et al (2019) Multiscale mechanistic study of the adsorption of methyl orange on the external surface of layered double hydroxide. J Phys Chem C 123(36):22212
Moon B, Kim T, Kim M, Choi J, Zoh K (2017) Degradation mechanisms of Microcystin-LR during UV-B photolysis and UV/H2O2 processes: byproducts and pathways. Chemosphere 185:1039
Starling M, Souza P, Le Person A, Amorim C, Criquet J (2019) Intensification of UV-C treatment to remove emerging contaminants by UV-C/H2O2 and UV-C/S2O82−: susceptibility to photolysis and investigation of acute toxicity. Chem Eng J 376:120856
Chan J, Ang S, Ye E, Sullivan M, Zhang J, Lin M (2015) Heterogeneous photo-Fenton reaction on hematite (α-Fe2O3){104}, 113 and 001 surface facets. Phys Chem Chem Phys 17(38):25333
Huang X, Chen Y, Walter E et al (2019) Facet-specific photocatalytic degradation of organics by heterogeneous Fenton chemistry on hematite nanoparticles. Environ Sci Technol 53(17):10197
Huang X, Zhao Q, Young R et al (2020) Photo-production of reactive oxygen species and degradation of dissolved organic matter by hematite nanoplates functionalized by adsorbed oxalate. Environ Sci Nano 7(8):2278
Mirbagheri N, Wang D, Peng C et al (2014) Visible light driven photoelectrochemical water oxidation by Zn- and Ti-doped hematite nanostructures. ACS Catal 4(6):2006
Zhang Y, Dong K, Liu Z et al (2017) Sulfurized hematite for photo-Fenton catalysis. Prog Nat Sci 27(4):443
Acknowledgements
The authors would like to acknowledge the Engineering Research and Development for Technology (ERDT), the Department of Science and Technology (DOST) through the National Academy of Science and Technology (NAST) in its Grants for Outstanding Achievements in Science entitled “Hydrothermal Synthesis of Hierarchical Hematite (α-Fe2O3) Nanostructures for Environmental Cleaning”, and the OVCRD Outright Research Grant with the title “Formation of Hematite Nanostructures on Iron Foil for the Adsorption of Cr(VI) Ions” for the financial support. The authors would also like to extend its gratitude to CHED-PCARI Projects nanoQuench (IIID-2016-007) and VERSe (IIID-2017-22) for the use of Raman equipment and the Active Nanomaterials Synthesis and Devices Laboratory (ANSyD) through Dr. Candy C. Mercado for the use of the UV–Vis spectrometer.
Funding
Funding was secured from the following agencies/institutions: DOST-Engineering Research and Development for Technology (ERDT); National Academy of Science and Technology (NAST)-Grants for Outstanding Achievements in Science; UP Diliman OVCRD Outright Research Grant.
Author information
Authors and Affiliations
Contributions
Christian Laurence E. Aquino: Conceptualization, Methodology, Investigation, Writing—original draft; Mary Donnabelle L. Balela: Conceptualization, Resources, Supervision, Writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aquino, C.L.E., Balela, M.D.L. Thermally grown Zn-doped hematite (α-Fe2O3) nanostructures for efficient adsorption of Cr(VI) and Fenton-assisted degradation of methyl orange. SN Appl. Sci. 2, 2099 (2020). https://doi.org/10.1007/s42452-020-03950-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-03950-1