
Abstract
Several methods have been developed for the analysis and detection of mycotoxins in food; however, most do not make use of critical statistics and mathematical tools for precise optimization. This study developed, optimized, and validated a modified quick, easy, cheap, effective, rugged, and safe (QuEChERS) extraction procedure for the extraction of multiple mycotoxins in maize and subsequent validation using liquid chromatography tandem mass spectrometry (LC–MS/MS). Central composite design (CCD) was used to optimize extraction conditions. Data analysis of full factorial screening experiments revealed that MeCN (%), FA (%), and extraction time significantly affected the mycotoxins recovery. Assessment of the statistical significance of the generated model using analysis of variance (ANOVA), coefficient tables, and surface plots showed the relative interactions of factors and the adequacy of the model. Thus, P values from the lack of fit (LOF) test ranged from 0.137– 0.467 and a composite desirability function of 0.91 was obtained. Using the optimum extraction conditions of 0.1% (v/v) FA in 80.2% MeCN for 74 min, 10 mycotoxins were effectively extracted with satisfactory recoveries (85–114%), coefficients of regression (R2 > 0.98), coefficients of variation (CVs < 15%), limit of quantifications (LOQs) (0.33–60.45 µg/kg), and other associated method validation parameters. The method validation was carried out according to Commission Implementing Regulation 2021/808 and Commission Regulation (EC) No 401/2006 of 23 February 2006. Application of this method to 20 maize samples collected from markets in Botswana showed detectable mycotoxins in 13 samples, with 2 exceeding the European Union (EU) maximum aflatoxin B1 (AFB1) limit, suggesting potential exposure to high levels of toxic mycotoxins in Botswana.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Mycotoxin contamination of food and feed is a major issue around the world, with the Food and Agricultural Organization of the United Nations (FAO) reporting that it affects 25% of grain and oil crops annually [1]. Mycotoxins are produced by fungi that grow in field crops such as maize, sorghum, and groundnuts [2,3,4]. Although contamination can occur during plant growth, most of the contamination happens during storage and transportation of the produce when there are high temperatures and moisture. The International Agency for Research on Cancer (IARC) classified some of the important mycotoxins, aflatoxins (AFs), fumonisins (FBs), ochratoxin A (OTA), trichothecenes (T2 and HT2 toxin), and zearalenone (ZEA), as belonging to groups 1, 2, and 3 of toxic substances.
Mycotoxins are regulated due to their high toxicity [5, 6]. For instance, the EU has set their maximum levels (MLs) in food and feed (Table 1) [7]. Proper regulation and monitoring of these contaminants require sensitive, accurate, and quick analytical procedures, and so the development of analytical procedures with a few steps that may be applied to routine analysis of mycotoxins has become popular [8, 9]. Nevertheless, due to the numerous parameters, method development is not always a straightforward operation. The main challenge is optimizing the extraction process because of the quantity and physiochemical diversity of the compounds. The complexity rises with the variety of analyte-matrix interactions, which can result in analyte suppression or enhancement [10, 11].
Various sample preparation techniques have been developed for the extraction of multiple mycotoxins in food matrices. These include dispersive liquid–liquid micro-extraction (DLLME), solid–liquid extraction (SLE), accelerated solvent extraction (ASE), supercritical fluid extraction (SFE), microwave-assisted extraction (MAE), vortex-assisted low density solvent-microextraction (VALDS-ME), solid phase extraction (SPE), molecularly imprinted solid phase extraction (MISPE), and immunoaffinity columns (IACs) [12,13,14,15,16]. Multiple mycotoxins extraction using some of these techniques, such as ASE, MAE, and VALDE-ME is expensive, while others are relatively time-consuming and utilize large amounts of organic solvents. However, the QuEChERS sample preparation method has many advantages, such as easy operation, low cost, being highly effective, quick, and safe [17, 18]. Hence, it has been widely used to determine a variety of environmental contaminants, including pesticides, drug residues, and mycotoxins [19,20,21]. This technique relies on the dispersion of salts to achieve the extraction of a diverse array of analytes from complex matrices while also facilitating the cleanup of the resulting extract [22].
Although multiclass approaches are becoming more popular in the field of food safety, the diversity in the chemical characteristics of mycotoxins has slowed the development of methods that integrate them in analytical studies. In most prior studies, each factor was mostly optimized using a one-factor-at-a-time (OFAT) strategy, while all the other factors were fixed at a constant level. Sirhan et al. [23] developed a QuEChERS method for the extraction of AFs in various food samples by optimizing the extraction time, the type, and the amounts of salts using the OFAT technique. This approach to optimization is unable to discern the relative importance of each factor and identify the true optimum. In this case, statistical experimental approaches, such as factorial design and response surface methodology (RSM), could optimize the parameters collectively and get rid of the constraints of traditional optimization processes. Sirhan et al. [23] expanded their investigation by optimizing the influence of extraction solvent using the design of experiments (DOE) approach, which configures the RSM using statistical and mathematical techniques.
In DOE optimization, a two-level full factorial design is an effective way to screen for and identify the main factors among many variables and reduce the number of experiments needed for the actual optimization. The experiments take up an Lk format, where L represents levels for each factor, often two (low and high), and k represents the number of factors under evaluation. This sets up a range within which the optimum is deemed to be, leading to the total number of experiments (N), as shown by Eq. 1.
After the evaluation of significant factors, response optimization can then be used to identify the optimum points for each factor. CCD is one of the tools used for response optimization. CCD allows for estimation of the constant, linear terms, interaction terms, quadratic terms, and evaluation of curvature using the model as shown in Eq. 2.
where y is the estimated response, a0 is the constant, ai is the regression coefficient for the linear parameters, aij represents the regression coefficients of interactions, x is the input variable, and ɛ is the error or residual associated with the experiments. Gbashi et al. [10] used CCD to optimize a pressurized hot water extraction method for the simultaneous extraction of multiple mycotoxins from maize. Zhou et al. [24] optimized QuEChERS extraction of mycotoxins by RSM for application in egg and milk matrices using the Plackett–Burman design for screening and CCD for optimization of FA (%) in MeCN, sodium chloride (NaCl) amount, and the amount of dispersive solid phase extraction salts needed for extract clean-up.
The aims of this study were to (1) evaluate the effect of extraction solvent, extraction time, and salt addition on QuEChERS extraction of multiple mycotoxins in maize and (2) optimize the recovery of each mycotoxin via CCD. The established method is anticipated to contribute to the risk monitoring and investigation of contamination levels in maize.
2 Materials and Methods
2.1 Chemicals and Reagents
Mycotoxin standards (AFB1, AFB2, AFG1, AFG2, OTA, HT2-Toxin, T2-Toxin, FB1, and FB2) were purchased from Trigology Analytical Laboratory, Inc (USA), and ZEA was supplied by Sigma Aldrich (Austria). Lichrosolv chemicals and reagents were purchased from Merck, Supelco (Germany); pure water for chromatography (LC–MS grade), methanol (hyper-grade for LC–MS), acetonitrile (gradient grade for liquid chromatography). Formic acid (ACS, > 98%) was purchased from Carl Roth (Germany), and sodium chloride analytical reagent was obtained from Rochelle Chemicals (South Africa).
2.2 Sample Collection and Treatment
A blank maize samples was purchase from Choppies superstore in Botswana and determined to have no detectable mycotoxins. Twenty maize samples were purchased from Francistown and Gaborone city markets. About 1 kg of each sample was finely blended using a laboratory blender and stored at -20 °C in a tightly closed 500 mL plastic jar.
2.3 Preparation of the Multiple-mycotoxin Standard Solution
Preparation of the multi-mycotoxin standard solution was carried out according to Sect. 2.3 of our previously published work [11]. The standard solution contained; 10 µg/L of each of AFB1, AFB2, AFG1, AFG2; 250 µg/L of each of FB1, FB2, T2-toxin; 100 µg/L of each of H2-toxin, ZEA, and 5 µg/L of OTA. These were the concentrations used as MLs for each analyte. This solution was kept at -20 °C in an amber bottle when not in use.
2.4 Modified QuEChERS Extraction
In a 50 mL centrifuge tube, 2.0 ± 0.1 g of finely ground sample was extracted with 8 mL MeCN/H2O (80.2:19.8 v/v) containing 0.1% FA. The mixture was homogenized for 30 s with a probe homogenizer and then left to shake for 74 min for maximum extraction. After shaking was complete, 1 g of NaCl was added to the mixture, followed by a vortex for 1 min. The sample was then centrifuged for 10 min at 5000 rpm, 4 °C. The supernatant (≈5 ml) was collected into a glass tube and preconcentrated to 500 µL over a gentle stream of nitrogen (40 °C) using a turbo vap, then reconstituted with 500 µL MeOH/H2O (50:50 v/v). Finally, the extract was filtered and ready for injection into LC–MS/MS.
2.5 LC–MS/MS Parameters and Analysis
The LC–MS/MS parameters used in this study are the same as those used and elaborated in a Sect. 2.5 of our previously published work [11]. An ExionLC™ LC paired with a linear ion trap quadrupole (QTRAP 6500 +) mass spectrometer from AB Sciex with electrospray ionization (ESI) was used for the analysis. A Zorbax Eclipse Plus C18 (2.1 × 150 mm, 5 μm, Agilent, USA) column was used for separation. Mobile phase A was composed of H2O + 0.1% FA, while mobile phase B was composed of MeOH/MeCN (50/50 v/v) + 0.1% FA. The tabulated binary gradient is illustrated in Table S1. The flow rate was held constant at 0.5000 mL/min throughout the runs, with a 1-min equilibration time. All measurements were done in multiple reaction monitoring (MRM) mode.
2.6 CCD Optimization
Optimization using DOE is usually applied as a two-step process. The first step is screening, which tells us how important the effects of the factors are on the response. These preliminary tests were done to narrow down the number of factors so that there is less experimentation and more effectiveness in the next step [25]. In this study, screening was carried out via a two-level full factorial design to examine the significance of four factors on the recovery of selected mycotoxins. The factors subject to screening were the extraction solvents MeCN (%), FA (%), the extraction time (min), and the amount of NaCl (g) added during the extraction process. Table 2 shows the high and low levels for each factor.
The second step is the optimization phase, where significant factors are assessed to determine their optimum. The center levels for factors selected for optimization are shown in Table 2. Optimization was replicated to establish the measurement of reproducibility and model the lack of fit (LOF) [26]. The Minitab 21 statistical software (Minitab Inc., Pennsylvania, US) was used for all designs and statistical analysis.
2.7 Method Validation
The method developed was validated by assessment of linearity, limits of detection (LODs) LOQs, selectivity, precision (repeatability and reproducibility within-laboratory), recovery, and decision limit (CCα) as described in Sect. 2.6 of our previously published work [11]. Matrix-matched calibration curves were utilized for quantification of each mycotoxin. The curves were constructed from blank samples that were spiked at four concentrations that covered the analyte concentration range expected to be present in the experiments (Table S2). Linearity was evaluated visually and statistically using the R2 values. The LOD and LOQ were calculated using Eqs. 3 and 4. Where Sa is the standard deviation of the response and b is the slope of the calibration curve. In this study, Sa was estimated using the y-residuals (ŷ) of the response.
Recovery was evaluated using six replicates of each concentration: 0.25, 1, and 1.5 times the ML of each mycotoxin. The samples were then extracted, and their concentrations were determined. The recovery, standard deviation (SD), and CVs were calculated using Eqs. 5, 6, and 7, respectively.
The precision of this analytical method was determined as follows. Six replicate samples fortified with the multi-mycotoxin standard to yield concentrations equivalent to 0.25, 1, and 1.5 times the ML of each mycotoxin were prepared and analyzed (n = 6 for 3 different concentrations). The validation batch was made up of samples that had already been analyzed and found to be negative. These steps were repeated in three consecutive runs intra-day to evaluate the within-laboratory repeatability (CVr). The reproducibility (CVR) was determined in a similar manner to CVr, however the experiments were repeated in three consecutive weeks, and the method ruggedness was demonstrated through the use of 3 different operators to performed 3 QuEChERS extractions at the three spiking levels over the three consecutive weeks. The mean concentrations, SD, and CVs (%) of the fortified samples were then calculated. The total ion flow chart and MS/MS spectrum of each mycotoxin for the validation sample fortified with multi-mycotoxin standard at MLs are presented in Fig. S1 and Fig. S2 respectively.
CCα is defined as the limit at and above which it can be concluded with an error probability of α that a sample is non-compliant [27]. For substances with established MLs such as mycotoxins, α = 5%. CCα was determined by analyzing 20 blank samples fortified with the analytes at ML concentrations. The mean concentration at ML plus 1,64 times the corresponding standard measurement uncertainty at ML equals CCα (Eq. 8).
3 Results and Discussion
3.1 LC–MS/MS Optimization
ESI and MS/MS parameters were carefully optimized as elaborated in Sect. 3.1 of our previously published work [11] to obtain the ideal conditions for detection of the selected mycotoxins (Table S3).
3.2 Method Development and Optimization of the QuEChERS Extraction
A modified QuEChERS extraction procedure was developed for the extraction of 10 mycotoxins in maize. Four factors were screened via the two-level full factorial design. Data analysis was carried out using normal probability plots of standardized effects, pareto charts of standardized effects, and residual plots. Figure 1 shows the normal probability plots of standardized effects and pareto charts of standardized effects AFB1, while all plots for other mycotoxins are shown in Fig. S3, Fig. S4 and Fig. S5. Results show that the recovery (%) of all analytes is positively affected by the amount of MeCN (A) in the extraction solvent but negatively affected by the amount of FA (B) spiked into the extraction solvent. Except for T2-toxin, the extraction time (C) showed a positive effect on the recovery of all analytes. The main effects due to the addition of NaCl (D) were not significant for all analytes except T2-toxin. However, the contribution of factor D (17.5% weight) towards the recovery of T2-toxin was smaller compared to the contributions of factors A (95.5% weight) and C (89.0% weight). The normal probability plots of standardized effects and pareto charts also showed that there are significant contributions to the recoveries due to interactions between factors A, B, and C. Furthermore, the residual plots signified the absence of systematic errors and confirmed adequacy of the model (Fig. S5). Finally, the statistical significance of the screening model applied was evaluated using the p-value from the analysis of variance (ANOVA) at 95% confidence level and R2 values were used to assess the fitness of the model for the experimental data. All the model p-values were below 0.05, which indicated that the model is suitable for all analytes. R2 values range from 98.3% (HT2- toxin) to 99.9% (AFG2 and OTA), thus suggesting that the model fits adequately with the experimental data. According to Demirel and Kayan [28], a regression coefficient is suggested to be at least 80% for a good fit of a model.

(a) Normal probability plot of standardized effects AFB1 and (b) Pareto chart of standardized effects for AFB1
Therefore, factors A, B, and C (MeCN (%), FA (%) in the extraction solvent and the extraction time) were optimized using CCD to ensure an optimum performance of the modified QuEChERS method and thereby improve the recoveries of selected mycotoxins. Here, experiments were conducted according to a set of statistically predetermined conditions and a quadratic equation (Eq. 2) was fitted to the experimental data. The full quadratic model applied to the data obtained from extraction of AFB1 is represented by Eq. 9. Similar equations for the extraction of other mycotoxins can be drawn using Eq. 2 and the coefficients in the coded coefficient tables. In this equation, the factors being investigated are represented by x. Where, x1 is the MeCN (%), x2 is the FA (%), and x3 is the extraction time (min).
Statistical testing of experimental values was carried out using Fisher's test from ANOVA results. The results indicate that the response equations proved to be suitable for the CCD experiment. According to Miller and Miller [25], if the model has a very high degree of adequacy for predicting the experimental results, the computed F value should be greater than the tabulated F value at a level of significance α (0.05). Fmodel values ranging from 22.34 – 83.95 were observed, suggesting that the models are significant for the extraction of selected mycotoxins. Thus, the calculated Fmodel values were compared with the tabulated F value (F 0.05, df, (n − df + 1)) at a significance level of 0.05, with the df for the model being 12 and n = 20. It can be observed that the tabular F value (F 0.05, 12, 9 = 3.07) is less than the calculated Fmodel values. Additionally, Pmodel values less than 0.05 indicate that the model terms are significant. All P values for the models were observed to be 0.000.
The quality of the polynomial model was validated by the R2 and R2 (adj) values. These values ranged from 91–97% for R2 indicating that the model adequately explains variations in the response. Whereas, for R2 (adj) values ranged from 87–96% indicating that the models presented a good estimation of the true relationship between the dependent variable (i.e. recovery) and the control variables (i.e. MeCN (%), FA (%). An R2 value above 80% is considered high enough for model adequacy [28].
The fitness of the model was assessed using the LOF test. For the LOF to be considered insignificant the PLOF must be greater than α (0.05) [28]. An insignificant LOF means that the model accounts for errors brought about by the regressor-response relationship. In this study, PLOF values ranged from 0.137 – 0.467 indicating good fit of the model with experimental data. Multicollinearity between independent factors is assessed using the Variance Inflation Factor (VIF). This factor indicates how the much variance of a coefficient is inflated due to correlations among independent factors in the model [29]. A VIF = 1 indicates no correlation, 1 < VIF < 5 indicates moderate correlation, and a VIF > 5 indicates high correlation. Here, VIF values of 1.00 and 1.01 were observed for independent factors, thus indicating minimal difficulties in the interpretation of statistical significance.
3.3 Interactive Effects of Independent Factors
QuEChERS extraction dynamics and mechanism have already been extensively discussed [8, 19, 30]. However, the QuEChERS procedures developed for multiple mycotoxins extraction have been based on single-factor optimization. In this study, the focus was on understanding the regressor-response relationship better. The relationship between factors is represented in three-dimensional response surface plots. For example, the response surface plots of AFB1 are shown in Fig. 2. Response surface plots for other mycotoxins are shown in Fig. S6 to Fig. S14. Low recoveries were obtained at the extremities of both MeCN (%) and FA (%), possibly due to different polarities between the analytes and the extraction solvent. Higher recoveries are observed in regions towards the center of both MeCN (%) and FA (%) composition. The addition of FA increases the polarity and hydrophilicity of the extraction solvent; therefore, polar analytes such as AFB1 are extracted more at higher FA (%) and nonpolar analytes like ZEA are poorly extracted at higher FA (%). Figure 2 b shows that the mycotoxin recovery is low at lower MeCN (%) and shorter extraction time, as the analytes are more soluble in a polar organic solvent. The recovery increased with increasing extraction time because penetration of the complex matrix by the solved needs time.

Response surface plots of (a) AFB1 vs FA (%), MeCN (%), (b) AFB1 vs Extraction time(min), MeCN (%), and (c) AFB1 vs Extraction time(min), FA (%)
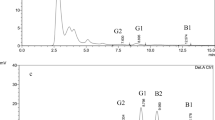
Although these response surface plots show how a combination of factors affects the response, they do not definitively give the optimal values. This is because the optimal values for each mycotoxin are localized in different regions, therefore it is difficult to find conditions that simultaneously satisfy all the responses, i.e., maximum extraction of all analytes. Thus, the multi-objective optimum solution for the dependent variables was computed using the multiple response optimizer. This tool considers all responses simultaneously in order to establish a compromised optimum [26]. A function commonly known as the desirability function (D) is used to assess this optimization [26, 31]. The scale of the individual desirability function spans from d = 0, which represents a wholly undesirable response, to d = 1, which represents a fully desirable response, above which no more changes are necessary [26]. A composite desirability of 0.91 was recorded, meaning that the fitted conditions are desirable. The optimum solution generated was extraction with 0.1% (v/v) FA in 80.2% MeCN for 74 min. Subsequent validation of these optimized conditions by laboratory experiments showed high recoveries for all mycotoxins ranging from 84–94% (Fig. 3).

QuEChERS extraction of multiple mycotoxins using CCD-optimized extraction conditions of MeCN (%), FA (%) in the extraction solvent, and the extraction time
3.4 Analytical Method Validation
After optimization, the method’s performance was validated according to Commission Implementing Regulation (EU) 2021/808 of March 22, 2021 [27] and Regulation (EC) no. 401/2006 [32]. This step was essential to authenticate the extraction procedure and to compare the findings with defined specifications for mycotoxin extraction and analysis. All the R2 values from the calibration curves were greater than 0.98, which suggests that the recorded signals can be strongly correlated with the real concentration of the mycotoxins. The computed values for LOD and LOQ ranged from 0.11–20.52 µg/kg and 0.33–60.45 µg/kg, respectively. Table 3 shows the linearity ranges, LODs, and LOQs for the mycotoxins in this study.
Results of recovery showed that analytes were determined with reasonable accuracy ranging from 95–104%, 85–94%, 94–112%, and 93–98% for AFs, FBs, T2&HT2 toxins, and ZEA respectively, which have recommended recovery levels by Commission Regulation (EC) No 401/2006 of 70–110%, 60–120%, 60–130%, and 60–120% respectively [32]. For OTA, all recoveries were within the recommended range of 70 – 110% except for one sample which as spiked at 0.25 ML that had a recovery of 114%. Overall, these are well within the recommended ranges. The CVr and CVR results for method precision are presented in Table 4. The method demonstrated good repeatability, as CVr values ranged from 4–14%. These values are all less than 15% as recommended by Commission Regulation (EC) No 401/2006 with specific recommendations as ≤ 20% for FBs and OTA, ≤ 25% for ZEA, and ≤ 30% for T2 toxin. The method also demonstrated good reproducibility, as CVR values ranged from 1–14% which are within recommended levels of ≤ 25% and ≤ 30% for mass fractions of 10–20 µg/kg and < 10 µg/kg. Although recommended levels for CVs are stated, Commission Implementing Regulation (EU) 2021/808 also states that these values serve as guidelines and should be as low as reasonably possible [27]. Our results concerning the performance criteria for this method are in line with other studies [8, 33], although Zachariasova et al. (2010) proved that using internal standards would improve the results [34].
CCα is also interpreted as the concentration at and above which, with an error probability of 1 – α, a decision can be made that if a signal is detected, it is not noise and the analyte detected is truly present or above the ML [11]. According to Commission Implementing Regulation (EU) 2021/808, CCα values shall be higher than but as close as possible to the ML. The CCα values obtained are listed in Table 4 and are all within the recommended guidelines.
3.5 Comparison of the Modified QuEChERS Method with Analytical Performance of other Methods Reported in Literature
Table 5 compares the findings of this study with the findings and analytical performance parameters of other methods reported in literature. The use of CCD for method development proves to be superior, with higher recoveries obtained. Furthermore, the QuEChERS method also proves to produce higher recoveries compared to other extraction techniques such as pressurized hot water extraction.
3.6 Application of Method to Real Samples
The developed method was used to analyze 20 maize samples meant for human consumption obtained in Gaborone and Francistown marketplaces in Botswana (Table S4). A total of 13 samples had detectable mycotoxins. The most frequently detected mycotoxins were the AFs, FBs, and ZEA. T2, HT2, and OTA were not detected in any of the samples. Co-occurrence of mycotoxins was observed in 3 samples. All detected concentrations except for AFB1 were below the maximum permitted levels by the EU. Two samples had levels of AFB1 (2.55 and 4.07 µg/kg) above the maximum permitted levels of 2 µg/kg in cereals intended for human consumption [27]. Thus, indicating possibility of exposure to high levels of AFB1. These findings reflected the possible contamination of Botswana’s maize. This was also reported by Masitha et al. [35], who identified mycotoxigenic fungi such as Aspergillus (flavus, niger), Fusarium (proliferatum, fujikuroi) and Alternaria in maize and sorghum samples from Botswana.
4 Conclusion
The use of RSM to develop a modified QuEChERS procedure for the extraction of multiple mycotoxins in maize is reported. CCD optimization helps to systematically determine the best extraction conditions, minimize experimental error, and maximize the recovery of mycotoxins from samples. Effective extraction of 10 mycotoxins was achieved with sensitive and accurate detection using LC–MS/MS. Chemometric method optimization improves method performance and paves the way for the development of methods that can determine hundreds of mycotoxins simultaneously. Several mycotoxins were detected in most of the samples analyzed. Therefore, stricter regulation and guideline levels of contamination with mycotoxins, particularly AFB1, in maize products are required in Botswana. Even though this method is promising, future research can focus on multiple matrix analysis using QuEChERS for a wider application.
Data Availability
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
References
Escrivá L, Font G, Manyes L, Berrada H. (2017) Studies on the presence of mycotoxins in biological samples: An overview. Toxins (Basel). 9(8). https://doi.org/10.3390/toxins9080251
James A, Zikankuba VL (2018) Mycotoxins contamination in maize alarms food safety in sub-Sahara Africa. Food Control 1(90):372–381
Wu F, Groopman J, Pestka J (2014) Public health impacts of foodborne mycotoxins. Annu Rev Food Sci Technol 5(1):351–72. https://pubmed.ncbi.nlm.nih.gov/24422587/. Accessed 1 Nov 2023
Agriopoulou S, Stamatelopoulou E, Varzakas T (2020) Advances in occurrence, importance, and mycotoxin control strategies: prevention and detoxification in foods. Foods 9:518. https://www.mdpi.com/journal/foods. Accessed 1 Nov 2023
Fletcher MT, Blaney BJ (2016) Mycotoxins. Encycl Food Grains Second Ed 2(4):290–296
Bennett JW, Klich M. (2003) Mycotoxins. Clin Microbiol Rev. 16(3):497–516. https://journals.asm.org/doi/abs/10.1128/CMR.16.3.497-516.2003
EUROPEAN COMMUNITIES (2023) Commission Regulation (EU) 2023/915 of 25 April 2023 on maximum levels for certain contaminants in food and repealing Regulation (EC) No 1881/2006 Off J Eur Union. L119:103–57. http://https//eur-lex.europa.eu/homepage.html. Accesssed 19 Mar 2024
Pantano L, La SL, Olibrio F, Galluzzo FG, Bongiorno C, Buscemi MD et al (2021) Quechers lc–ms/ms screening method for mycotoxin detection in cereal products and spices. Int J Environ Res Public Health 18(3774):1–17. https://doi.org/10.3390/ijerph18073774
Guan X, Feng Y, Suo D, Xiao Z, Wang S, Liang Y et al (2022) Simultaneous determination of 11 mycotoxins in maize via multiple-impurity adsorption combined with liquid chromatography–tandem mass spectrometry. Foods 11(22):3624. https://www.mdpi.com/2304-8158/11/22/3624/htm. Accessed 5 Jul 2023
Gbashi S, Njobeh PB, De Saeger S, De Boevre M, Madala NE (2020) Development, chemometric-assisted optimization and in-house validation of a modified pressurized hot water extraction methodology for multi-mycotoxins in maize. Food Chem 1:307
Mbisana M, Rebagamang T, Mogopodi D, Chibua I (2023) Development and validation of a QuEChERS-LC-MS/MS method for determination of multiple mycotoxins in maize and sorghum from Botswana. Front Fungal Biol 3(4):1141427
Salim SA, Sukor R, Ismail MN, Selamat J (2021) Dispersive Liquid-Liquid Microextraction (DLLME) and LC-MS/MS Analysis for Multi-Mycotoxin in Rice Bran: Method Development. Optimization and Validation Toxins (Basel) 13(280):1–21. https://doi.org/10.3390/toxins13040280
Gholizadeh S, Mirzaei H, Khandaghi J, Afshar Mogaddam MR, Javadi A (2022) Ultrasound–assisted solvent extraction combined with magnetic ionic liquid based-dispersive liquid–liquid microextraction for the extraction of mycotoxins from tea samples. J Food Compos Anal 1(114):104831
He T, Zhou T, Wan Y, Tan T (2020) A Simple Strategy Based on Deep Eutectic Solvent for Determination of Aflatoxins in Rice Samples. Food Anal Methods 13:542–550. https://doi.org/10.1007/s12161-019-01665-7
Facorro R, Llompart M, Dagnac T (2020) Combined (d)SPE-QuEChERS extraction of mycotoxins in mixed feed rations and analysis by high performance liquid chromatography-high-resolution mass spectrometry. Toxins 12(3):206. https://www.mdpi.com/2072-6651/12/3/206/htm. Accessed 27 Oct 2023
Somsubsin S, Seebunrueng K, Boonchiangma S, Srijaranai S (2018) A simple solvent based microextraction for high performance liquid chromatographic analysis of aflatoxins in rice samples. Talanta 176:172–177
González-Curbelo M, Socas-Rodríguez B, Herrera-Herrera AV, González-Sálamo J, Hernández-Borges J, Rodríguez-Delgado M (2015) Evolution and applications of the QuEChERS method. Trends Anal Chem 71(9):169–185. https://doi.org/10.1016/J.TRAC.2015.04.012
Perestrelo R, Silva P, Porto-Figueira P, Pereira JAM, Silva C, Medina S et al (2019) QuEChERS - Fundamentals, relevant improvements, applications and future trends. Anal Chim Acta 6(1070):1–28
Nakhjavan B, Ahmed NS, Khosravifard M (2020) Development of an improved method of sample extraction and quantitation of multi-mycotoxin in feed by LC-MS/MS. Toxins 12(7):462. https://www.mdpi.com/2072-6651/12/7/462/htm. Accessed 27 Jan 2022
Kresse M, Drinda H, Romanotto A, Speer K (2019) Simultaneous determination of pesticides, mycotoxins, and metabolites as well as other contaminants in cereals by LC-LC-MS/MS. J Chromatogr B 1(1117):86–102. https://doi.org/10.1016/J.JCHROMB.2019.04.013
Jo HW, Park MK, Heo HM, Jeon HJ, Choi SD, Lee SE et al (2021) Simultaneous determination of 13 mycotoxins in feedstuffs using QuEChERS extraction. Appl Biol Chem 64(34):1–12. https://doi.org/10.1186/s13765-021-00602-9
Anastassiades M, Lehotay SJ, Stajnbaher D (2023) Quick, Easy, Cheap, Effective, Rugged, and Safe (QuEChERS) approach for the determination of pesticide residues. In: 18th Annual Waste Testing and Quality Assurance Symposium, WTQA 2002 - Proceedings. https://www.researchgate.net/publication/283647077. Accessed 1 Nov 2023
Sirhan AY, Tan GH, Al-Shunnaq A, Abdulra’uf L, Wong RCS (2014) QuEChERS-HPLC method for aflatoxin detection of domestic and imported food in Jordan. J Liq Chromatogr Relat Technol. 37(3):321–42. https://doi.org/10.1080/10826076.2012.745138
Zhou J, Xu JJ, Cong JM, Cai ZX, Zhang JS, Wang JL et al (2018) Optimization for quick, easy, cheap, effective, rugged and safe extraction of mycotoxins and veterinary drugs by response surface methodology for application to egg and milk. J Chromatogr A 12(1532):20–29
Miller JN, Miller JC (2005) Statistics and chemometrics for analytical chemistry, 5th edn. Pearson Prentice Hall, Harlow, pp 181–190
Bezerra MA, Santelli RE, Oliveira EP, Villar LS, Escaleira LA (2008) Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta 76(5):965–977
EU (2021) Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the performance of analytical methods for residues of pharmacologically active substances used in food-producing animals and on the interpretation of results as well as on the methods to. Off J Eur Union 180(401):84–109. http://extwprlegs1.fao.org/docs/pdf/eur203999.pdf. Accessed 1 Jul 2021
Demirel M, Kayan B. (2012) Application of response surface methodology and central composite design for the optimization of textile dye degradation by wet air oxidation. Int J Ind Chem. 3(1):1–10. https://link.springer.com/article/10.1186/2228-5547-3-24
Wu S (2020) Multicollinearity in regression. Why it is a problem?. https://towardsdatascience.com/multi-collinearity-in-regression-fe7a2c1467ea. Accessed 29 Oct 2022
Seo H, Jang S, Jo H, Kim H, Lee S, Yun H, et al. (2021) Optimization of the QuEChERS-Based Analytical Method for Investigation of 11 Mycotoxin Residues in Feed Ingredients and Compound Feeds. Toxins (Basel). 13(11):767. https://www.mdpi.com/2072-6651/13/11/767/htm
Murphy TE, Tsui KL, Allen JK (2005) A review of robust design methods for multiple responses. Res Eng Des 15(4):201–215
EC (2006) Commission Regulation (EC) No 401/2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off J Eur Union 24:1–42. https://eur-lex.europa.eu/eli/reg/2006/401/oj. Accessed 2 Feb 2022
Leite M, Freitas A, Barbosa J, Ramos F (2023) Comprehensive assessment of different extraction methodologies for optimization and validation of an analytical multi-method for determination of emerging and regulated mycotoxins in maize by UHPLC-MS/MS. Food Chem Adv 1(2):100145
Zachariasova M, Lacina O, Malachova A, Kostelanska M, Poustka J, Godula M et al (2010) Novel approaches in analysis of Fusarium mycotoxins in cereals employing ultra performance liquid chromatography coupled with high resolution mass spectrometry. Anal Chim Acta 662(1):51–61. https://pubmed.ncbi.nlm.nih.gov/20152265/. Accessed 28 Dec 2023
Masitha B, Sereme-Mothobole B, Kabelo K (2019) Current status of mycotoxigenic fungi in cereal grains in the central region of botswana: a mini survey. J Food Saf Hyg 5(2):120–5. https://doi.org/10.18502/JFSH.V5I2.3950
Acknowledgements
We would like to thank Botswana National Veterinary Laboratory for allowing access to their analytical instruments and chemical reagents.
Funding
Open access funding provided by University of Botswana. This study was partly funded by the University of Botswana through the Office of Research and Development (ORD).
Author information
Authors and Affiliations
Contributions
MM: Conceptualization; Data curation; Formal analysis; Methodology; Validation; Visualization; writing—original draft; and Writing—review & editing. DM: Funding acquisition; Supervision; Writing—review & editing. TR: Methodology; Resources; Validation. IC and BN: Supervision.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mbisana, M., Mogopodi, D., Tshepho, R. et al. Optimization of the QuEChERS Extraction of Mycotoxins in Maize Using Response Surface Methodology (RSM). Chemistry Africa (2024). https://doi.org/10.1007/s42250-024-00965-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42250-024-00965-8