
Abstract
Leaves extract of Ginkgo biloba, known in China since the most ancient times, has been widely used in the area of senile dementia thanks to its improving effects on cognitive function. A promising formulation of this botanical ingredient consists in a Ginkgo biloba-soy-lecithin-phosphatidylserine association obtained by the Phytosome® process. The precise assessment of the mixing degree between Ginkgo biloba and soy-lecithin-phosphatidylserine in this formulation is an important piece of information for understanding the reasons of its final performances. To this aim in the present study we carried out for the first time a Solid State Nuclear Magnetic Resonance investigation on Ginkgo biloba-soy-lecithin-phosphatidylserine association, on its constituents and on a mechanical mixture. The analysis of different observables highlighted a very intimate mixing (domains of single components not larger than 60 nm) of Ginkgo biloba and soy-lecithin-phosphatidylserine in their association obtained by Phytosome® process, together with a slight modification of their molecular dynamics, not observed in the case of the mechanical mixture.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ginkgo biloba L. has been used in Chinese medicine since the most ancient times, while its pharmacological properties have been investigated in Europe only in the last decades [1]. The Ginkgo biloba tree is one of the oldest plants in the world and it is often defined as a “living fossil”, being the only surviving species of the Ginkgoaceae family, originated 150 millions years ago. As reported in a review by DeFeudis [2], Ginkgo biloba, thanks to its composition including several active compounds, has a “polyvalent” character. Currently Ginkgo biloba leaves extract (GB) is especially indicated for treating cerebral insufficiency, that is a series of symptoms as lack of short term memory, confusion, apathy, affective and somatic problems, which can be related to altered cerebral circulation and ageing and are considered as early signs of senile dementia [3]. Indeed several specific terpenoids and flavonoids of GB are considered responsible for cognitive improvements, which can be particularly important in treating senile dementia [4,5,6,7].
In order to potentiate the cognitive effects associated with GB, the Phytosome® process [8,9,10,11] has been used to produce an association (GS) between GB and a soy lecithin enriched in phosphatidylserine (SLPS), a major acidic phospholipid in the brain, extensively studied in regard to its actions on brain functions. Indications that in GS a good synergy between its two nootropic agents (GB and SLPS) is realized have been obtained by Kennedy et al. [12], who found a significant improvement of memory task performance following administration of GS to healthy young humans, whereas no such benefits were evident for the same dose of GB. Products of Phytosome® process are lipid dispersions which proved to strongly increase the biovailability of natural ingredients usually characterized by low solubility in biological fluids due to large size, scarce hydrophilicity or lipophilicity [8,9,10,11]. Especially due to the natural origin of their components (both lipids and bothanicals) their characterization is quite challenging, but crucial for understanding the reasons for their performances and guide the optimization of their preparation. In particular a precise characterization of the mixing degree between GB and SLPS in GS appears very important, since the effective mixing accomplished in the formulation can be a key factor for the observed positive results. Indeed, in the pharmaceutical field, and in particular in the area of solid dispersions, a good dispersion of drug and excipients is generally a requisite for obtaining a desired biovailability [13, 14]. Several techniques, as calorimetry, diffraction methods, electronic microscopies, can provide indications on the drug-excipient mixing degree, and have been applied also to the characterization of lipid dispersions [4,5,6,7]. To this aim, among the techniques available at present, Solid State Nuclear Magnetic Resonance spectroscopy (SSNMR) is particularly attractive, since, by exploiting the 1H spin-diffusion process, it allows the obtainment of a quantitative estimate of the mixing degree between two different components in a solid system on a 10–100 Å spatial scale, which is often accessible with difficulty by microscopic techniques [13,14,15]. SSNMR is an extremely versatile technique, successfully and extensively used not only in the pharmaceutical field, but also, just citing one example, in that of advanced materials. Indeed, by exploiting a variety of observable nuclei and many nuclear properties, ranging from spectral features to various types of relaxation times, from internuclear interactions, detectable by multidimensional experiments, to the proton spin diffusion process, it is possible to unravel structural and dynamic features and interactions of complex solid systems, on very wide spatial (0.1–100 nm) and time scales (10−11–102 s). This is particularly important in the field of pharmaceutical solid dispersions, which can have very complex architectures and properties [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30].
In this paper we present a SSNMR investigation of the degree of mixing and of the dynamic behaviour of the different components of GS. In particular, we mainly used 1H on-resonance Free Induction Decay (FID) analysis and the measurement of 1H spin–lattice relaxation times, and the results were compared with those obtained for the mechanical mixture of GB and SLPS. To the best of our knowledge, this is the first study in which SSNMR is applied to the investigation of a product obtained by the Phytosome® process.
2 Materials and Methods
2.1 Materials
The following samples have been supplied by Indena SpA, Italy:
-
Ginkgo biloba dry extract (GB) was prepared according to the procedure reported in patent EP0360556 [31]. Briefly, Ginkgo biloba ground leaves were extracted with 60% w/w acetone, at 60 °C, until exhaustion. The leachates were combined and vacuum concentrated, obtaining a suspension that was left aside overnight. The supernatant was separated by decantation and extracted repetitively with butanol. The butanol layers were combined and vacuum concentrated to obtain a soft mass. Water and ethanol were added to have a solution with 40% alcohol concentration and 20% dry residue. The solution was extracted repeatedly with hexane. The hydroalcoholic solution was concentrated to dryness and the residue was dried overnight to obtain the Ginkgo biloba dry extract.
-
Soy-lecithin-phosphatidylserine (SLPS) is a soy lecithin enriched in phosphatidylserine (about 20%, W/W). Average composition of SLPS includes phospatidylserine (about 20%), phospatidylcholine (about 20%), phosphatidylinositol (about 15%), phosphatidylethanolamine (about 10%), and other minor phospholipids for a total of about 80% glycerophospholipids.
-
Ginkgo Biloba—Soy-lecithin-phosphatidylserine association (GS) VIRTIVA®. GS is a GB and SLPS mixture obtained through the Indena technology process according to the procedure reported in patent WO 2005/074,956 [32]. Briefly, GB and SLPS (1:3; W/W) were solubilized in ethyl acetate under reflux. The solution was filtered hot. The mother liquors were concentrated to soft mass and the residue was dried in oven. The dry product was passed through a 20 mesh grid.
-
Ginkgo Biloba—Soy-lecithin-phosphatidylserine mechanical mixture (MM). MM was obtained through a mechanical mixing of GB and SLPS in same proportions of GS. GB and SLPS powders were passed through a 50 mesh grid and mixed with the aid of an orbital shaker at 250 rpm for 1 h.
2.2 Solid State NMR
A two-channel Varian InfinityPlus 400 NMR spectrometer, working at 399.89 MHz for proton, was used for the high-resolution measurements here reported. The spectrometer was equipped with a Varian 7.5 mm Cross-Polarisation Magic Angle Spinning (CPMAS) probe.
The 1H 90° pulse duration was 4 μs. In the Cross-Polarization (CP) experiments contact times of 3, 1 and 2 ms were used for SLPS, GB and GS, respectively. All the CP spectra were acquired with a TPPM decoupling scheme using a decoupling field of 45 kHz.
Indirectly-detected Inversion Recovery was here used for the high-resolution measurement of 1H spin–lattice relaxation times in the laboratory frame (T1). This pulse sequence [33] allows 1H T1′s to be measured by means of 13C observation through 1H-13C transfer of magnetisation via CP; in this way, it was possible to exploit 13C spectral resolution to measure individual relaxation times for GB and SLPS components. The measurements were performed under MAS conditions, with a spinning frequency of 5 kHz. For GS and MM samples a minimum of 600 transients were accumulated with a recycle delay of 10 s. T1 values were obtained by fitting the peak integrals vs the Inversion Recovery variable delay to the corresponding exponential recovery function:
where I(t) is the peak integral, t is the Inversion Recovery variable delay, M0 is the equilibrium value of the magnetization and α is a correction factor that accounts for an imperfect inversion (α ≤ 1, with α = 1 for a perfect inversion).
Low-resolution measurements were performed on a Varian XL-100 spectrometer, interfaced with a Stelar DS-NMR acquisition system, equipped with a 5-mm probe and working at 25.00 MHz for proton.
The 1H Free Induction Decays (FIDs) were recorded under on-resonance conditions after application of a solid echo pulse sequence. The data were analyzed directly in the time domain, without any processing of the FID, by means of a package of Mathematica [34] suitably written by some of us. 1H T1′s were also measured, using the inversion-recovery pulse sequence followed by solid echo; T1 values were obtained by fitting the trends of the intensity of the first point of the on-resonance FID vs the Inversion Recovery variable delay to the suitable exponential recovery function, corresponding to Eqs. 1 and 2 in case of mono- and bi-exponential trends, respectively.
where I(t), t, M0 and α have the same meaning as in Eq. 1, T1a and T1b are the two T1 components, and wa and wb their corresponding fractional weights.
All measurements were performed at a temperature of 25 °C. 13C chemical shifts were referred to hexamethylbenzene as secondary reference and TMS as the primary one.
3 Results and Discussion
Both high- and low-resolution SSNMR techniques were applied in order to investigate the way GB and SLPS interact in GS. In particular, the 1H FID, acquired on-resonance under low-resolution conditions, contains purely dynamic information, since it is not affected by the spin diffusion process. The on-resonance 1H FID can be reproduced by a sum of analytical decaying functions, corresponding to motionally distinguishable domains [35]. Each function is characterized by a T2 and a weight, which is proportional to the number of protons belonging to that domain. T2, or an equivalent decay parameter, is about 10–20 μs in the “rigid lattice regime” (occurring when the molecular motions have characteristic frequencies lower than the static line width, usually of the order of tens of kHz), and it monotonically increases with increasing motional characteristic frequencies above this limit. On the other hand, proton spin–lattice relaxation times in the laboratory frame (T1), which can be measured with both high- and low-resolution experiments, are determined by molecular motions (with characteristic frequencies in the MHz regime) and spin diffusion. In particular, spin diffusion can partially or completely average different intrinsic proton spin–lattice relaxation times throughout the sample: the more intimate is the mixing among different components the more effective is the averaging process. A single T1 is measured, \(\frac{1}{{T_{1}^{av} }} = \sum\limits_{i} {\frac{{w_{i} }}{{T_{1i} }}}\) (where T1i and wi are the different intrinsic T1 values and their corresponding fractional weights), when the average domain dimensions are smaller than about 100 Å [36, 37].
3.1 Low-Resolution SSNMR Measurements
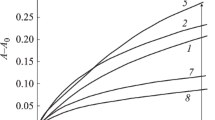
Proton T1′s were first measured at a Larmor frequency of 25.00 MHz using a low-resolution technique, acquiring a single relaxation recovery curve for all the protons in the sample. Both the pure components, GB and SLPS, showed a mono-exponential relaxation recovery curve (see Fig. 1), with a T1 of 404 and 90.1 ms, respectively. This difference allowed us to investigate the degree of mixing between GB and SLPS in both MM and GS samples, by looking at the degree of averaging between these two values performed by the proton spin diffusion process in the composite systems.

T1 recovery curves for the samples: a SLPS, b GB, c MM, e GS, reproduced by a single exponential recovery function; d for sample MM reproduced by a two-exponential function as described in the text. The parameter α defined in Eq. 1, taking into account incomplete inversion of the magnetization after the 180° pulse, was found to be equal to 0.92–0.93 in all cases
For the sample MM a single exponential recovery curve was clearly unsuitable to reproduce the experimental trend (see Fig. 1), indicating an incomplete mixing of GB and SLPS domains on a 100-Å spatial scale. In order to reproduce the experimental trend, a bi-exponential function was necessary, but the fitting parameters appeared quite correlated. Under the assumption that the MHz dynamics of both components does not change from pure components to MM, in the fitting the T1 values were fixed to those previously found for the “pure” components (404 ms for GB and 90.1 ms for SLPS). A very satisfactory description of the data was obtained (see Fig. 1), indicating that the degree of mixing between the domains of GB and SLPS is very scarce. The weights found by the fitting of the relaxation curve (88.8 and 11.2% for the faster and slower-relaxing component, respectively). provide an estimate of the percentages of protons belonging to the two components, which cannot be a priori determined due to the unknown exact chemical composition of Gingko biloba extract and SLPS [38].
Contrary to what observed for MM, the relaxation curve of sample GS was satisfactorily reproduced by a single exponential function with a T1 of 108.4 ms (see Fig. 1), which is indeed close to the T1av value (98.7 ms) calculated using the T1 values of the single components (90.1 and 404 ms) and the weights estimated from the relaxation curve fitting of MM. Even though this result comes from a low-resolution measurement, and cannot be considered as a proof of a truly complete spin diffusion averaging process (the presence of T1′s differing by a factor of less than two–three would be undetectable by this method), this result clearly indicates that the domains of GB and SLPS are much better mixed in GS than in MM. A quantitative estimate of the degree of mixing between GB and SLPS in GS could be obtained using the equation that relates the maximum diffusing path length (L) to the spin–lattice relaxation time T1 (Eq. 3) [14 and references therein].
Taking a value of 0.8 nm2ms−1 for the spin-diffusion coefficient in organic solids and the measured T1 value, L ≈ 23 nm is obtained, which can be considered as the maximum size of GB and SLPS domains. It must be noticed that the measured value slightly differs from that (98.7 ms) expected by fully averaging the T1 values measured for the pure components. This suggests that the phytosome process slightly altered the MHz dynamic behaviour of one or both components of the composite material. The spin–lattice relaxation times measured through low-resolution techniques are summarized in Table 1.
3.2 High-Resolution SSNMR Measurements
In order to obtain additional information on the degree of mixing between GB and SLPS domains in samples MM and GS, the measurement of 1H T1 was also carried out through 1H-13C CP, exploiting the 13C spectral resolution. However, since almost all the signals of the two components resulted heavily superimposed in the 13C CPMAS spectra (see Fig. 2), the measurement of individual relaxation times for GB and SLPS components in the composite samples resulted quite difficult, also considering the much smaller intensity of GB signals respect to SLPS. These problems were overcome by recording a very large number of transients for the measurement of T1 (up to 5 days acquisition time for each sample) and, afterwards, by applying peak deconvolution techniques to the spectral regions exhibiting the minor superposition between GB and SLPS signals.

13C CP MAS spectra recorded with spinning frequency of 5 kHz (a) and expansion of the 100–200 ppm region (b) of, from bottom to top, GB, SLPS, MM, GS
For both the pure components (GB and SLPS) a single T1, equal within the experimental error for all the signals, was measured: 2.12 s and 573 ms for GB and SLPS, respectively. This allowed us to select some representative signals of each component in the spectra of samples MM and GS. For GB the three signals resonating at about 162, 157 and 146 ppm were chosen, since they are the only ones in a spectral region relatively free from SLPS signals. In spite of that, given the very low intensity of GB signals, the application of a spectral deconvolution was necessary to remove the contribution of the tail of the SLPS signal resonating at 173 ppm. As far as SLPS signals are concerned, none of them is present in a spectral region free from GB resonances, but that at about 30 ppm is very intense and superimposed to a very small GB signal, so it can be safely considered completely arising from SLPS. The T1 values so determined for sample MM were about 2.9 s and 710 ms for GB and SLPS, while the corresponding values for sample GS were about 800 and 650 ms. This result strongly confirms what previously observed from low-resolution measurements: there is no intimate mixing on a 100 Å spatial scale between GB and SLPS components in sample MM, while an almost complete mixing at the same scale is achieved in sample GS, for which the relaxation times of the two components are almost completely averaged to a single value. It is worth to notice that the actual measurement of two slightly different T1 values must also be related to the intrinsic larger capability of the high-resolution experiment, with respect to the low-resolution one, to reveal slightly differing T1 components. By applying Eq. 3 it is possible to estimate a maximum size for GB and SLPS domains in GS of about 60 nm. The spin–lattice relaxation times measured through high-resolution techniques are summarized in Table 2.
In order to obtain information about the possible change of dynamic properties occurring in passing from pure components to their composites, we performed low-resolution FID analyses. The 1H FID of GB was well reproduced by a combination of a Pake [39] and an exponential function. Two distinct dynamic regimes can be therefore identified: that described by the Pake function, involving 93% of protons of the sample, is typical of very rigid phases, while that described by the exponential function concerns protons in a slightly more mobile environment, even though the very short T2 value (35.6 μs) indicates the absence of a remarkable molecular mobility above the kHz regime. The 1H FID of SLPS was more complex and needed at least three different functions (one Gaussian and two exponentials) to be satisfactorily reproduced. The Gaussian function (T2 = 22.4 μs and weight = 12.4%) is relative to a quite rigid phase, while the two exponential functions (T2 = 49.8 and 175 μs with weights of 35.9 and 51.7%, respectively) arise from more mobile environments. It is therefore evident that SLPS globally experiences a much faster molecular dynamics than GB. In principle, the simultaneous presence of GB and SLPS in the two samples MM and GS should give rise to a 1H FID, which should be similar to the simple sum of the FIDs of the pristine components if weak interactions occur between them. On the other hand, we would expect that an intimate mixing between the two components should involve stronger interactions between them, and, consequently, a modification of the molecular dynamics of the single components, thus originating deviations from the sum of the FIDs of the pure components. However, before discussing the 1H FIDs of MM and GS in terms of their similarity with those of the pure components, we needed to unravel some problems arising from the complexity of these systems. Indeed, the numerical analysis of the FIDs of MM and GS could not be performed by preserving the same fitting functions used in the FIDs of the pure components, since the parameters of functions characterized by similar decay rates are strongly correlated, giving rise to completely unreliable numerical results of the fitting procedure. It was therefore necessary to find a balance between the high number of functions that could better describe our systems, and the short number of functions that could keep low the correlation among fitting parameters, as well as permit a physical interpretation of the results. The best compromise was represented, in our case, by the use of a set of three functions, similar to that used for fitting the 1H FID of SLPS, one Gaussian and two exponentials. The corresponding T2 values, which resulted to be very similar in the fittings of MM and GS, and which therefore were kept equal for the two samples in order to facilitate a comparative analysis of the results, were 18.2, 44.1 and 166 μs for the Gaussian (“G”) and the two exponential functions (“E1” and “E2”), respectively. The results, summarized in Table 3 for all the samples, showed that a non negligible difference is present for the weight percentages of the three FID functions between MM and GS: in particular, while E2 has the same weight in both samples, the very rigid fraction, described by the Gaussian function, has a 3.1% higher weight in GS with respect to MM.
It might be useful to attempt a comparison with the pristine components of the two mixtures: to this purpose, fortunately, the three dynamic regimes highlighted in MM and GS, in the following for simplicity indicated as “Rigid”, “Intermediate” and “Mobile”, can be directly compared to those previously found in the analysis of the pure components: the Gaussian function (“Rigid” regime) can be direcly compared with both the Pake function of GB and the Gaussian function of SLPS; E1 (“Intermediate regime”) should include both the exponential function of GB and the exponential function of SLPS with T2 = 49.8 μs; finally, E2 (“Mobile regime”) corresponds to the sole exponential function of SLPS with the longest T2. Even though this is an approximation, a comparison with the amount of protons present in the three distinct dynamic regimes in the pure components, as well as in MM and GS, can be attempted, the reliability of the results being satisfactorily guaranteed by the good separation of the decaying parameters among these three dynamic regimes. As already said, due to the complexity of GB and SLPS, their exact compositions are unknown and therefore the proton weight percentage of GB and SLPS in MM and GS cannot be known a priori. However, by assuming that the percentage of SLPS protons is 88.8%, i.e., the percentage previously found from low-resolution 1H T1 measurements, we obtained the results reported in Table 4. It must be noticed that, while in MM the percentages of protons present in the three dynamic regimes are in good agreement with those expected from the FID analyses of the pure components (differences are less than 2%), in the case of GS the deviations from the “calculated” values are bigger (by about + 3%, − 5% and + 2% for “Rigid”, “Intermediate” and “Mobile” regimes, respectively). This suggests the occurrence in GS of a slight, but detectable modification of the dynamic behaviour of the two components induced by the interactions between their domains.
4 Conclusions
The proton relaxation study carried out in this work allowed a clear difference to be highlighted between samples MM (mechanical mixture) and GS (obtained by the Phytosome® process), concerning both the degree of mixing between the two components GB (Ginkgo biloba) and SLPS (soy-lecithin-phosphatidylserine) and their dynamic properties. In particular, while MM is clearly heterogeneous on a 100 Å spatial scale, on the same scale GS is almost completely homogeneous and the mixing between GB and SLPS is intimate, being the maximum size of GB and SLPS domains not larger than 60 nm as observed from high-resolution spin–lattice relaxation measurements. In the assumption that also the spin–lattice relaxation time measured for GS with low-resolution techniques can be considered reliable, i.e., if the intrinsic relaxation times of GB and SLPS do not differ by a factor of less than two–three, it is possible to improve the estimate of the maximum domains size down to about 20 nm. Moreover, the FID analysis carried out on the whole set of samples indicated a different molecular dynamic behaviour of GS with respect to MM: while in the latter the dynamic behaviour is very similar to that individually experienced by GB and SLPS in the pristine components, for GS some modifications, reasonably ascribable to physical interactions at the GB-SLPS domain interfaces, could be detected.
From the results obtained it clearly emerges a relation between the success of GS and the intimate mixing of its active ingredient and phospholipids. Encouraged by the good results obtained for the first time on such a complex system, it would be interesting to extend this kind of investigation to other successful or newly designed phytosomes. Moreover, even if the complexity of the components is remarkable, in the perspective of better characterizing the molecular dynamic properties, as well as of trying to understand possible molecular interactions between the active ingredient and the lipids, it would be interesting to further exploit the power of SSNMR, extending the experiments to high-resolution mono- and bidimensional techniques for the observation of 31P, 13C and 1H nuclei.
References
Van Beek TA, Bombardelli E, Morazzoni P, Peterlongo F (1998) Ginkgo Biloba. Fitoterapia 69:195–238
DeFeudies FV (1991) Ginkgo biloba extract: pharmacological activities and clinical applications. Elsevier, Paris
Birks J, Evans JG (2009) Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst Rev 1:CD003120
Liu H, Ye M, Guo H (2020) An updated review of randomized clinical trials testing the improvement of cognitive function of Ginkgo biloba extract in healthy people and Alzheimer’s patients. Front Pharmacol 10:1688–1718
Arrigo FG, Cicero AFG, Fogaccia F, Banach M (2018) Botanicals and phytochemicals active on cognitive decline: the clinical evidence. Pharmacol Res 130:204–212
Zuo W, Yan F, Zhang B, Li J, Mei D (2017) Advances in the studies of Ginkgo biloba leaves extract on aging-related diseases. Aging Dis 8:812–827
Pothineni NVK, Karathanasis SK, Ding Z, Arulandu A, Varughese KI, Mehta JL (2017) LOX-1 in atherosclerosis and myocardial ischemia. JACC 69:2759–2768
Santos AC, Rodrigues D, Sequeira JAD, Pereira I, Simões A, Costa D, Peixoto D, Costa G, Veiga F (2019) Nanotechnological breakthroughs in the development of topical phytocompounds-based formulations. Int J Pharm 572:118787-1–118816
Bresciani L, Favari C, Calani L, Francinelli V, Riva A, Petrangolini G, Allegrini P, Mena P, Del Rio D (2020) The effect of formulation of curcuminoids on their metabolism by human colonic microbiota. Molecules 25:940–1 10
Lu M, Qiu Q, Luo X, Liu X, Sun J, Wang C, Lin X, Deng Y, Song Y (2019) Phyto-phospholipid complexes (phytosomes): a novel strategy to improve the bioavailability of active constituents. Asian J Pharm Sci 14:265–274
Matias D, Rijo P, Pinto Reis C (2017) Phytosomes as biocompatible carriers of natural drugs. Curr Med Chem 24:568–589
Kennedy DO, Haskell CF, Mauri PL, Scholey AB (2007) Acute cognitive effects of standardised Ginkgo biloba extract complexed with phosphatidylserine. Hum Psychopharmacol 22:199–210
Paudel A, Geppi M, Van Den Mooter G (2014) Structural and dynamic properties of amorphous solid dispersions: the role of solid-state nuclear magnetic resonance spectroscopy and relaxometry. J Pharm Sci 103:2635–2662
Urbanova M, Gajdosova M, Steinhart M, Vetchy D, Brus J (2016) Molecular-level control of ciclopirox olamine release from poly(ethylene oxide)-based mucoadhesive buccal films: exploration of structure-property relationships with solid-state NMR. Mol Pharm 13:1551–1563
Policianova O, Brus J, Hruby M, Urbanova M, Zhigunov A, Kredatusova J, Kobera L (2014) Structural diversity of solid dispersions of acetylsalicylic acid as seen by solid-state NMR. Mol Pharm 11:516–530
Geppi M, Mollica G, Borsacchi S, Veracini CA (2008) Solid-state NMR studies of pharmaceutical systems. Appl Spectrosc Rev 43:202–302
Vogt FG (2015) Characterization of pharmaceutical compounds by solid-state NMR. eMagRes 4:255–268
Ashbrook SE, Hodgkinson P (2018) Perspective: current advances in solid-state NMR spectroscopy. J Chem Phys 149:04901–4914
Lu X, Huang C, Lowinger MB, Yang F, Xu W, Brown CD, Hesk D, Koynov A, Schenck L, Su Y (2019) Molecular interactions in posaconazole amorphous solid dispersions from two-dimensional solid-state NMR spectroscopy. Mol Pharm 16:2579–2589
Carignani E, Borsacchi S, Blasi P, Schoubben A, Geppi M (2019) Dynamics of clay-intercalated ibuprofen studied by solid state nuclear magnetic resonance. Mol Pharm 16:2569–2578
Hughes DJ, Badolato Bönisch G, Zwick T, Schäfer C, Tedeschi C, Leuenberger B, Martini F, Mencarini G, Geppi M, Alam MA, Ubbink J (2018) Phase separation in amorphous hydrophobically modified starch-sucrose blends: glass transition, matrix dynamics and phase behavior. Carbohyd Polym 199:1–10
Deligey F, Bouguet-Bonnet S, Doudouh A, Marande P-L, Schaniel D, Gansmüller A (2018) Bridging structural and dynamical models of a confined sodium nitroprusside complex. J Phys Chem C 122:21883–21890
Chakravarty P, Lubach JW, Hau J, Nagapudi K (2017) A rational approach towards development of amorphous solid dispersions: experimental and computational techniques. Int J Pharm 519:44–57
Geppi M, Borsacchi S, Carignani E (2016) In: Descamps M (ed) Disordered pharmaceutical materials, 1st edn. Wiley-VCH Verlag GmbH & Co. KgaA, Weinheim
Folliet N, Roiland C, Bégu S, Aubert A, Mineva T, Goursot A et al (2011) Investigation of the interface in silica-encapsulated liposomes by combining solid state NMR and first principles calculations. J Am Chem Soc 133:16815–16827
Aso Y, Yoshioka S (2006) Molecular mobility of nifedipine-PVP and phenobarbital-PVP solid dispersions as measured by 13C-NMR spin-lattice relaxation time. J Pharm Sci 95:318–325
Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS, Strohmeier M et al (2010) Analysis of amorphous solid dispersions using 2D solid-state NMR and H-1 T-1 relaxation measurements. Mol Pharm 7:1667–1691
Tatton AS, Pham TN, Vogt FG, Iuga D, Edwards AJ, Brown SP (2013) Probing hydrogen bonding in cocrystals and amorphous dispersions using 14N–1H HMQC solid-state NMR. Mol Pharm 10:999–1007
Abraham A, Crull G (2014) Understanding API—polymer proximities in amorphous stabilized composite drug products using fluorine—carbon 2D HETCOR solid-state NMR. Mol Pharm 11:3754–3759
Yuan X, Sperger D, Munson EJ (2014) Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol Pharm 11:329–337
Bombardelli E, Mustich G, Bertani M (2006) New extracts of ginkgo biloba and their methods of preparation. EP0360556
Morazzoni P, Petrini O, Scholey A, Kennedy DO (2005) Use of a Ginkgo complexes for the enhancement of cognitive functions and the alleviation of mental fatigue. WO/2005/074956
Kitamaru R, Horii F, Murayama K (1986) Phase structure of lamellar crystalline polyethylene by solid-state high-resolution carbon-13 NMR detection of the crystalline-amorphous interphase. Macromolecules 19:636–643
Wolfram Research Inc. Mathematica v.12 (2020)
Hansen EW, Kristiansen PE, Pedersen B (1998) Crystallinity of polyethylene derived from solid-state proton NMR free induction decay. J Phys Chem B 102:5444–5450
Kenwright AM, Say BJ (1996) Analysis of spin-diffusion measurements by iterative optimisation of numerical models. Solid State Nucl Magn Reson 7:85–93
Geppi M, Harris RK, Kenwright AM, Say BJ (1998) A method for analysing proton NMR relaxation data from motionally heterogenous polymer systems. Solid State Nucl Magn Reson 12:15–20
Van Beek TA, Montoro P (2009) Chemical analysis and quality control of Ginkgo biloba leaves, extracts, and phytopharmaceuticals. J Chromatogr A 1216:2002–2032
Look DC, Lowe IJ, Northby JA (1966) Nuclear magnetic resonance study of molecular motions in solid hydrogen sulfide. J Chem Phys 44:3441–3452
Acknowledgements
Open access funding provided by Università di Pisa within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carignani, E., Geppi, M., Lovati, M. et al. Solid State NMR Study of the Mixing Degree Between Ginkgo Biloba Extract and a Soy-Lecithin-Phosphatidylserine in a Composite Prepared by the Phytosome® Method. Chemistry Africa 3, 717–725 (2020). https://doi.org/10.1007/s42250-020-00165-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-020-00165-0