
Abstract
Wisteria vein mosaic virus (WVMV) is a potyvirus infecting Wisteria spp. worldwide, making these largely used ornamental plants unattractive and even unsalable. In 2021, nine Wisteria sinensis plants in Sarzana (Liguria, Italy) showing WVMV symptoms like vein mosaic with irregular patterns, mottling, deformation and twisting margin on leaves were reported. This work describes research on symptomatic and asymptomatic leaves to confirm the identity of the virus infection. All plants tested resulted positive to WVMV. Sequencing of NIb/CP genomic region and comparison on GenBank revealed the presence of eight new genetic variants named Sar 5–12. The eight nucleotide sequences alignments revealed identity ranging between 87.13 and 99.85%. Negative selection (dN/dS < 1) was detected suggesting well adaptation in the area here examined and stability in population. Through a phylogenetic tree, WVMV isolates were grouped in four clades with high bootstrap values, two of which included the eight Italian variants here identified. Since the late 1950 and 1960s, the present study represents the first additional report of WVMV in Italy, as well as its first molecular characterization ever. Further research is required to strengthen our understanding of the movement and transmission of WVMV. Examining how virus infected plants can disseminate this pathogen could help forestall potential risks that neighbouring species may face in their environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The genus Wisteria Nutt. (Fabaceae), characterized by deciduous woody perennial lianas with pinnately compound leaves and pendulous racemes with typical purple, violet or white papilionaceous corollas, includes species among the most beautiful ornamental flowering plants. For this reason, these plants are grown in many gardens and landscape settings worldwide (Wei and Pedley 2010). Native to China, Japan, Korea and Eastern United States (Li et al. 2013), Wisteria spp. were later introduced to United Kingdom and several other European countries (Compton 2015). Actually, non-native wisterias are now considered invasive species in some areas of the United States, especially the Southeast (Trusty et al. 2007). Other than ornamentals, wisterias are also appreciated as edible and medicinal plants, and the fiber of their stems is also used for the production of paper (Mohamed et al. 2011).
Wisteria spp. can be affected by several viruses including wisteria vein mosaic virus (WVMV; family Potyviridae; genus Potyvirus) (Brierley and Lorentz 1957), subterranean clover stunt virus (SCSV; family Nanoviridae; genus Nanovirus) (Grylls and Butler 1959), cucumber mosaic virus (CMV; family Bromoviridae, genus Cucumovirus) (Milojević et al. 2016) and wisteria badnavirus 1 (WBV1; family Caulimoviridae; genus Badnavirus) (Li et al. 2017). While these diseases are notorious for causing damage to flora, it is important that research into their impacts continue in order to protect our local ecosystems from widespread destruction. WVMV is the subject matter of this work. The main WVMV symptoms consist of leaf chlorotic mottling and mosaic, distortions and twisting, which are commonly more severe on the leaflets closest to the petioles. Visible symptoms of WVMV can significantly mar the ornamental quality and commercial viability of infected plants, presenting a difficult challenge to growers. WVMV infection has been reported in the more popular Wisteria sinensis (Chinese wisteria; De Beni 1964), Wisteria floribunda (Japanese wisteria; Brierley and Lorentz 1957), Wisteria brachybotrys (Silky wisteria; Clover et al. 2003), and its natural transmission is ascribed to aphids (Aphis craccivora, Aphis fabae and Myzus persicae; Conti and Lovisolo 1969; Valouzi et al. 2020).
This wisteria disease was likely first reported in the United States in the late 1950s (Brierley and Lorentz 1957), and later in Italy (Ciferri 1959; De Beni 1964; Conti and Lovisolo 1969) and the Netherlands (Bos 1970), but only with the serological studies by Conti and Lovisolo (1969) and Bos (1970) it was demonstrated the viral etiology and proposed the name WVMV. Later, an isolate of WVMV was obtained from Wisteria sinensis in Czech Republic (Brčák 1980), and ultrastructural analysis conducted to investigate its interaction with plant cell showed large complex inclusions previously reported for other potyviruses (Brčák and Králík 1981). First genetic characterization of WVMV was achieved on an Australian isolate by Clover et al. (2003), which confirmed the previously advised taxonomy. A few years later, Liang et al. (2006) performed a complete genome analysis on a Chinese isolate, showing that WVMV is a single strand, positive-sense RNA potyvirus of about 9,695 nucleotides excluding the poly (A) tail. Genome was characterized by one main ORF encoding for a polyprotein which cleavage generates ten mature proteins: P1, HCPro, P3, 6K1, CI, 6K2, VPg, NIapro, NIb, CP. Currently another protein is also known, namely P3N-PIPO, deriving from a short ORF positioned in the P3 coding region (Chung et al. 2008; Revers and García 2015). WVMV has had a far-reaching impact and it have been observed in seven countries across the globe in the last 18 years. Reports have emerged from China (Beijing municipality, Jiangxi and Jiangsu provinces; Liang et al. 2004; Ji et al. 2019; Zhu et al. 2019), Poland (Kaminska et al. 2006), United States (Naidu and Karthikeyan 2008), New Zealand (Ward et al. 2008), United Kingdom (Clover et al. 2015), and Iran (Al Jaberi et al. 2018), further demonstrating the virus’ spread through diverse geographical locations over time.
In Italy, the known records of wisteria viral infections by Ciferri (1959), De Beni in Wisteria sinensis (1964), and Conti and Lovisolo in Wisteria floribunda (1969), occurred in Northern regions only. No other reports came out in other Italian areas so far. In 2021, we actually reported some Wisteria sinensis plants in Sarzana district (Liguria, on the border with Central Italy) showing typical (and severe) WVMV symptoms on many leaves. This study confirms the WVMV infections and provide information on the biological properties, genetic diversity and evolutionary relationships of WVMV.
Materials and methods
Field surveys and sampling
In 2021, one Wisteria sinensis plant with some leaves showing WVMV-like symptoms (Supplementary Figure S1a) was reported in Sarzana district, Liguria, Italy (44°06’49’’N, 9°57’36’’E). Therefore, all Wisteria sinensis plants within a circular area of 0.5 km radius around the first symptomatic plant were surveyed, and further symptomatic ones were detected (i.e., showing WVMV-like symptoms). This surveying approach was repeated until no new symptomatic plants were reported in the monitored areas. In the end, nine plants were recovered in both public and private areas (e.g., streets, urban parks, gardens), all within a 1 km2 region. Plants were around ten- to fifty-year-old [according to the indications by owners, confirmed by a plant aging using a Resistograph®, Rinntech, Heidelber, Germany (Lukaszkiewicz et al. 2005)], and showed fully foliated canopies and no other diseases and infections.
In May (i.e., when symptoms were most evident on all nine plants) the foliar disease severity was classified and sorted according to a four-class pathometric scale based on the percentage of symptomatic area of leaflets: Class 0 = 0%; Class 1 = < 5%; Class 2 = 5–40%; Class 3 = > 40% (Supplementary Figure S1b). Symptomatic and asymptomatic leaves were collected from each of nine plants (i.e., two leaf samples for each plant; 18 in total) and kept refrigerated until quickly reaching the Plant Pathology Lab of the Department of Agriculture, Food and Environment, University of Pisa, where leaflets and petioles were detached and stored at -80 °C until RNA extraction.
RNA isolation and cDNA synthesis
RNA was extracted from 500 mg of leaf tissue with cetyltrimethylammonium (CTAB) buffer, according to Li et al. (2008) with some modifications (Pedrelli et al. 2021, 2023a, b). Briefly, leaves were powdered in liquid nitrogen with 5 ml CTAB 2% buffer. After incubation at 65 °C for 15 min, one volume of chloroform:iso-amyl alcohol (24:1) was added and RNA precipitated with one volume of isopropanol. Pellet was then washed with 70% ethanol, air-dried and dissolved in 80 µL of RNase/DNase free water. cDNA synthesis was finally performed using M-MMLV reverse transcriptase (GeneSpin s.r.l., Milan, Italy), according to the manufacturers’ instructions.
Virus detection
An end point PCR was performed with WVMVF1/WVMVR1 specific primers (Clover et al. 2003) targeting genomic NIb/CP region (703 bp). PCR amplification was performed on a C1000 Touch® thermal cycler (Bio-Rad, Hercules, CA, USA) using PCR conditions as follows: 30 cycles of 95 °C/30 s, 55 °C/30 s, and 72 °C/45 s and final extension at 72 °C for 10 min. Amplified products were observed on 1.2% agarose gel electrophoresis. Infected and healthy control were used as references. All samples were analysed in a qPCR to determine the presence and quality of cDNA using 18 S rRNA as internal control (Osman and Rowhani 2008).
Virus sequencing and in silico assays
The PCR products were cleaned up and directly sequenced by Sanger DNA method (Eurofins genomics, Ebersberg, Germany). The nucleotide and deduced amino acid sequences were in silico analysed using Bioedit (Hall 1999) and nucleotide sequences were also compared in BLASTn (www.ncbi.nlm.ni.gov). The role of natural selection on population using Codon Z-test by the Nei-Gojobori method and the presence of non-synonymous (dN) and synonymous (dS) single nucleotide polymorphisms (SNPs) ratio by Felsenstein (1981) model was estimated using MEGA X (Kumar et al. 2018). The recombinant events in the nucleotide sequences were evaluated by RDP4 program (v.4.101) using 3Seq, Bootscan, Chimaera, GENECONV, MaxChi, RDP, and SiScan algorithms (Martin et al. 2015) and they were accepted only if identified by at least 4 methods (p value of > 10− 5). The nucleotide sequence alignment was used to construct phylogenetic tree by the Maximum Likelihood (ML) method (Jukes-Cantor model) with 1,000 bootstrap replicates on MEGA X (Kumar et al. 2018). WVMV sequences obtained from NCBI were used as ad hoc dataset (Supplementary Table S1). An isolate of bean common mosaic virus (NC_003397.1) was used as the outgroup.
Results and discussion
Wisteria spp. are ornamental plants largely used in public and private gardens in Italy (as well as in many other countries worldwide), and WVMV represents an important and spread viral agent of a detrimental disease that makes plants unattractive and even unsaleable (Clover et al. 2003). However, information about WVMV diffusion in Italy was lacking and restricted to the few and dated publications by Ciferri (1959), De Beni (1964) and Conti and Lovisolo (1969), all of which referred to WVMV observations in Northern regions (Lombardy, Emilia-Romagna and Piedmont, respectively). To the best of our knowledge, after more than 50 years from these observations, the present work represents the first additional report of WVMV in Italy (Sarzana district, Liguria, on the border with Central Italy), as well as its first molecular characterization ever.
All the nine Wisteria sinensis plants identified in May 2021 showed typical leaf WVMV symptoms (Supplementary Figure S1a) scattered in the canopy (i.e., asymptomatic leaves were also present), which were especially noticeable in spring, whereas they became fade and masked in summer. Symptom severity assessment showed the presence of leaf samples in each class of the pathometric scale. Among symptomatic leaf samples, the harsher class 3 was mostly observed (44% of samples), followed by class 2 (34%) and 1 (22%; Table 1).
All nine tested plants resulted positive to WVMV (quality of cDNA samples was proved by 18 S rRNA gene amplification, and the Ct values of the internal control ranged from 10 to 13; data not shown). The electrophoretic analysis of WVMV amplicons identified the virus in 16 out of the 18 leaf samples (around 90%; Table 1). Specifically, nine symptomatic and seven asymptomatic leaf samples were positive, so only two asymptomatic leaves were negative (Supplementary figure S2). The presence of asymptomatic positive leaf samples was in accordance to the previous WVMV report in Wisteria floribunda by Kaminska et al. (2006), while the occurrence of the two negative leaf samples could be due to a very low infection rate in those tissues, resulting below the detection limit of the diagnostic technique (Rubio et al. 2020).
PCR amplicon sequencing identified 16 distinct sequences, eight of which were considered novel variants and deposited in GenBank as Sar 5–12 (Table 1). The most frequent variant was Sar 5 being identified in four leaf samples (25.0%), followed by Sar 6, 7, 8, 9 and 10 characterized in two leaf samples (12.5% each), and finally Sar 11 and 12 identified in only one leaf sample (6.3% each). All leaf samples infected by Sar 5 were symptomatic, whereas Sar 6, 7, 8 and 9 were found in both symptomatic and asymptomatic leaf samples of the same plant, Sar 10 only in asymptomatic leaf samples, and Sar 11 and 12 in the symptomatic and asymptomatic leaf samples of the only plant infected by these variants. Mixed strains infections included Sar 10, 11 and 12, while Sar 6, 7, 8 and 9 were present only as single infections, Sar 5 was detected in both conditions. Referring to the symptom severity, Sar 5 was always associated to the highest class (3); Sar 6, 7, 9 to the middle class (2), and Sar 8, 11 to the lowest class (1). Sar 10 and 12 were identified exclusively in class 0 (Table 1).
The nucleotide sequence alignments revealed similarity among variants ranging from 87.13 to 99.85%. In particular, Sar 5 showed the lowest similarity (87.13–87.60%) with all other variants, while Sar 10 displayed the highest one (95.69–99.85%), especially with Sar 8 and 12. The genetic variability (π) was estimated in 0.045 (± 0.005). Interesting, a viral population with higher genetic diversity is normally considered more ancient in an area (Wei et al. 2009). The RDP4 program using the 3Seq, Chimaera, MaxChi, and RDP algorithms detected a concordant result in the Sar 5 with the 242 nt beginning breakpoint and the 360 nt ending breakpoint and a mean p-value of 2.073 × 10− 2, 1.697 × 10− 2, 1.516 × 10− 2, 3.848 × 10− 2, respectively. As major parent was identified the isolate Beijing (AY656816.1) with 90.3% similarity whereas minor parent was unknown. Although four algorithms found the same putative event, the result was rejected due to the probability not meeting the accepted criterion (p value > 10− 5) for identifying a true recombinant variant. It is possible to hypothesize that a greater number of available sequences could confirm or deny the presence of this recombinant event, as reported in previous studies focused on other potyviruses (Santillan et al. 2018; Fuentes et al. 2021). The number of SNPs among the eight variants was 120. Maximum changes were recognized in Sar 5 (79 nt) with 16 dN, followed by Sar 7 (27 nt) with 4 dN, and Sar 11 (6 nt) with 2 dN (Table 1). The overall ratio between nonsynonymous and synonymous mutations (dN/dS ratio) was 0.05 (p < 0.000) within population under study. These outcomes suggest a well-adaptation of WVMV in the small infected area here reported (i.e., around one km2), a phenomenon that likely promotes a high stability of the virus genetic structure (Escriu 2017).
Comparing the novel identified sequences with those already available in GenBank (Table 1), Sar 10 revealed the maximum similarity of 99.85% with the isolate JW_2014 (KP161267.1) identified on Wisteria floribunda in England (Clover et al. 2015), followed by Sar 6, 7, 8, 9, 11 and 12 with similar rates ranging from 96.11 to 99.85%. Sar 5 showed the higher similarity percentage (89.86%) with isolate Beijing (AY656816.1) previously found on Wisteria sinensis in China (Liang et al. 2006) (Table 1).
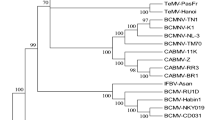
In the phylogenetic tree, WVMV isolates were grouped in four clades with high bootstrap values (i.e., > 70%), two of which included the eight Italian variants here identified (Fig. 1). Clade 1 indeed included only Chinese isolates (AY656816.1, AY519365.1, MK119779.1, MK119780.1), whereas clades 2, and 3 clustered only the Italian Sar 5 (OM417219.1) and a North American (EU677749.1), respectively. Differently, clade 4 included sequences with worldwide origin: Australia (AF484549.1), China (MK290861.1), the Netherlands (EU308593.1), New Zealand (EU308592.1), Poland (DQ009883.2), United Kingdom (KP161266.1, KP161267.1) and Iran (MN514947.1, MH558668.1), as well as the here recognized Italian Sar 6 (OM417220.1), 7 (OM417221.1), 8 (OM417222.1), 9 (OM417223.1), 10 (OM417219.1), 11 (OM417225.1) and 12 (OM417226.1). The deduced amino-acid sequences of our population revealed identity ranging from 94.59 to 100.00% with higher distance values identified between Sar 5 (94.59–95.05%) and other sequences. The alignment showed conserved amino acids in the population as A56 in NIb and V140, E191, I192 in CP, which were replaced by specific changes in the Sar 5, namely V56 in NIb, L140, S191 and V192 in CP, respectively. Moreover, the 83SKKNA87 motif was changed in 83QRKTT87 motif in Sar 5. No variation in the 78DAG80 motif associated with the transmissibility of potyviruses by aphid vectors was instead observed (Kaminska et al. 2006; Fig. 2). A comparison of the partial CP amino acid sequences of our and ad hoc WVMV database isolates showed that the difference between isolates within Clade 1 is due to a V110, Clade 2 is characterized by the distinctive feature of 13QRKTT19 motif and Clade 3 displayed specific amino acids substitutions, namely D13, A27, E36, G123, and 13SKKST19 motif. Interestingly, Clade 4 showed high similarities between the isolates and V72, E123, I124 as conserved amino acid sites. These results were in accordance with our previous analyses and with the observations on WVMV CP amino acids by Valouzi et al. (2020; Fig. 3).

Phylogenetic tree of wisteria vein mosaic virus (WVMV) reconstructed from partial CP genomic region. The trees were generated by Maximum Likelihood (ML) using the Jukes-Cantor model of evolution for nucleotide. The significance of each branch was evaluated by constructing 1,000 trees in bootstrap analysis. Only bootstrap values > 70% are shown. The scale represents a distance of 0.05 substitutions per site. The isolates sequenced in this study are in bold. Bean common mosaic virus (NC_003397.1) was used as the outgroup

Alignment of the deduced amino acid sequences of wisteria vein mosaic virus isolates recovered in this study. The 78DAG80 motif are shown in dashed rectangles. Conserved amino acids and motif change are shown in black rectangles. Polyprotein processing site, i.e. NIb/ CP junction, is indicated with a black line

Comparison of the partial coat protein amino acid sequences identified in this study and reported in the ad hoc WVMV database. Amino acid substitution in the Clade 1 is shown in dashed rectangles. Grey area underlines the distinctive motif of the Clade 2. Specific amino acids substitutions in the Clade 3 are highlighted by dotted rectangles. Conserved amino acids in Clade 4 are shown in black rectangles. The sequence of the Beijing isolate was used as the reference sequence and others were sorted as in the result of the phylogenetic analysis of the nucleotide sequences
The phylogenetic analysis and the observation of deduced amino acid sequences supported the division in four clades, each characterized by specific amino acids in the variable region of N-terminus of CP. Interesting, partial clustering based on country of origin was observed, while no one by host was identified. The Italian variants showed a different clustering with Sar 5 alone between the “Chinese” and the “American” clusters and all the other Italian variants with worldwide isolates in the same cluster. Overall, the analysis allowed to hypothesize a spread of the virus from the original area in other worldwide areas, probably due to exchange of propagating material, as previously observed for other ornamental plants (Valverde et al. 2012). Moreover, the phylogenetic outcomes associated with the π and the selective pressure seems to suggest a long-term introduction of virus and a co-evolution between W. sinensis and WVMV in the studied area (LaTourrette and Garcia-Ruiz 2022). This interpretation is supported by the presence in Italy of several plants (e.g., Leonardo’s W. sinensis in Milan and Goethe’s W. sinensis in Tivoli, near Rome) which seem to have been attested before wisteria fortune as ornamental species starting from the early 1800s.
Conclusions
Since the late 1950 and 1960 s, the present study represents the first additional report of WVMV in Italy, as well as its first molecular characterization ever. Here, eight new Italian variants with different performances were identified, with Sar 5 showing the highest distances from other variants. According to LaTourrette and Garcia-Ruiz (2022), data obtained could suppose a long-term introduction of virus and a co-evolution between W. sinensis and WVMV in the studied area. Moreover, the presence of asymptomatic-positive leaf samples enforces the essential role of monitoring plants and regular phytosanitary surveys to detect viral infections potentially dangerous for different species and cultivars. As Wisteria sinensis (as well as other wisterias) is a major woody perennial plant widely grown as ornamental in public and private gardens, more research should be carried out to improve our knowledge on WVMV diffusion (the collection and analysis of further samplings is highly encouraged), especially in Italian districts where the production and marketing of highly priced ornamental plants is crucial, as well as to elucidate the risk of growing virus infected plants that could represent a source for virus transmission to other potential neighbouring susceptible species.
References
Al Jaberi M, Zakiaghl M, Mehrvar M (2018) First report of wisteria vein mosaic virus on Wisteria sinensis in Iran. New Disease Reports 38:18. https://doi.org/10.5197/j.2044-0588.2018.038.018
Bos L (1970) The identification of three new viruses isolated from wisteria and pisum in the Netherlands, and the problem of variation within the potato virus Y group. Neth J Plant Pathol 76:8–46. https://doi.org/10.1007/BF01976763
Brčák J (1980) A Prague isolate of wisteria vein mosaic virus. Biol Plant 22:465–469. https://doi.org/10.1007/BF02880488
Brčák J, Králík O (1981) Additional data on complex inclusion evoked by wisteria vein mosaic virus in pea leaf cells. Biol Plant 23:237–239. https://doi.org/10.1007/BF02894896
Brierley P, Lorentz P (1957) Wisteria mosaic and peony leaf curl, two diseases of ornamental plants caused by viruses transmissible by grafting but not by sap inoculation. Plant Disease Reporter 41:691–693
Chung BY-W, Allen Miller W, Atkins JF, Firth AE (2008) An overlapping essential gene in the Potyviridae. PNAS 105:5897–5902. https://doi.org/10.1073/pnas.0800468105
Ciferri R (1959) Recenti risultati nelle ricerche sulle virosi delle piante arboree da frutto. La Ricerca Scientifica 29:1880–1884
Clover GRG, Tang Z, Smales TE, Pearson MN (2003) Taxonomy of wisteria vein mosaic virus and extentions to its host range and geographical distribution. Plant Pathol 52:92–96. https://doi.org/10.1046/j.1365-3059.2003.00798.x
Clover GRG, Denton JO, Denton GJ (2015) First report of wisteria vein mosaic virus on Wisteria spp. in the United Kingdom. New Disease Reports 31:1. https://doi.org/10.5197/j.2044-0588.2015.031.001
Compton JA (2015) Wisteria sinensis on the slow boat from China: the journey of wisteria to England. Curtis’s Bot Magazine 32:248–293. https://doi.org/10.1111/curt.12112
Conti M, Lovisolo O (1969) Observations on a virus isolated from Wisteria floribunda DC in Italy. Rivista di Patologia Vegetale ser 5(4):115–132
De Beni PV (1964) Ricerche sui virus delle piante. Virus, scheda: VI. Maculatura clorotica della glicine (Wisteria sinensis Sweet). La Ricerca Scientifica 4:21–24
Escriu F (2017) Diversity of plant virus population: a valuable tool for epidemiological studies. In: Bitz L (ed) Genetic diversity. Intechopen, London. pp150
Fuentes S, Gibbs AJ, Adams IR, Wilson CR, Botermans M, Fox AM, Kreuze J, Boonham N, Kehoe MA, Jones R (2021) Potato virus A isolates from three continents: their biological properties, phylogenetics, and prehistory. Phytopathology 111:217–226. https://doi.org/10.1094/phyto-08-20-0354-fi
Grylls NE, Butler FC (1959) Subterranean clover stunt, a virus disease of pasture legumes. Aust J Agric Res 10:145–159
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposyum Series 41:95–98
Ji ZL, Zhu PX, Ji YH, Xu F, Zhu F (2019) First report of wisteria vein mosaic virus in chinese wisteria in Jiangxi Province in China. J Plant Pathol 101:1259–1260. https://doi.org/10.1007/s42161-019-00318-2
Kaminska M, Malinowski T, Rudzinska-Langwald A, Diaz LC (2006) The occurrence of wisteria vein mosaic virus in Wisteria floribunda DC plants in Poland. J Phytopathol 154:414–417. https://doi.org/10.1111/j.1439-0434.2006.01118.x
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
LaTourrette K, Garcia-Ruiz H (2022) Determinants of virus variation, evolution, and host adaptation. Pathogens 11:1039. https://doi.org/10.3390/pathogens11091039
Li R, Mock R, Huang Q, Abad J, Hartung J, Kinard G (2008) A reliable and inexpensive method of nucleic acid extraction for the PCR-based detection of diverse plant pathogens. J Virol Methods 154:48–55. https://doi.org/10.1016/j.jviromet.2008.09.008
Li J, Jiang JH, Fu CX, Tang DQ (2013) Molecular systematics and biogeography of wisteria inferred from nucleotide sequences of nuclear and plastid genes. J Syst Evol 52:40–50. https://doi.org/10.1111/jse.12061
Li Y, Deng C, Qiao Y, Zhao X, Zhou Q (2017) Characterization of a new badnavirus from. Wisteria sinensis Archives of Virology 162:2125–2129. https://doi.org/10.1007/s00705-017-3322-4
Liang WX, Song LM, Li Y, Tian GZ, Li HF, Fan ZF (2004) First report of wisteria vein mosaic virus in China. Plant Pathol 53:516. https://doi.org/10.1007/s42161-019-00318-2
Liang WX, Song LM, Tian GZ, Li HF, Fan ZF (2006) The genomic sequence of wisteria vein mosaic virus and its similarities with other potyviruses. Arch Virol 151:2311–2319. https://doi.org/10.1007/s00705-006-0780-5
Lukaszkiewicz J, Kosmala M, Chrapka M, Borowski J (2005) Determining the age of streetside Tilia cordata trees with a DBH-based model. J Arboric 31:280–284. https://doi.org/10.48044/jauf.2005.036
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. https://doi.org/10.1093/ve/vev003
Milojević K, Radović N, Stanković I, Vučurović A, Nikolić D, Bulajić A, Krstić B (2016) First report of cucumber mosaic virus infecting Wisteria sinensis in Serbia. Plant Dis 100:1799. https://doi.org/10.1094/PDIS-01-16-0096-PDN
Mohamed MA, Hamed MM, Abdou MA, Ahmed WA, Saad AM (2011) Antioxidant and cytotoxic constituents from Wisteria sinensis. Molecules 16:4020–4030. https://doi.org/10.3390/molecules16054020
Naidu RA, Karthikeyan G (2008) First report of wisteria vein mosaic virus in Wisteria sinensis in the United States of America. Plant Health Progress 9:1–3. https://doi.org/10.1094/PHP-2008-0818-01-BR
Osman F, Rowhani A (2008) Real-time RT-PCR (TaqMan®) assays for the detection of viruses associated with rugose wood complex of grapevine. J Virol Methods 154:69–75. https://doi.org/10.1016/j.jviromet.2008.09.005
Pedrelli A, Panattoni A, Materazzi A (2021) Occurrence of grapevine Pinot gris virus in Chianti vineyards. Agrochimica Special Issue 65:81–87. https://doi.org/10.12871/00021857202201
Pedrelli A, Panattoni A, Cotrozzi L (2023a) First molecular characterization of plum pox virus strains in stone fruits of Tuscany (Central Italy). J Plant Pathol 105:1045–1053. https://doi.org/10.1007/s42161-023-01430-0
Pedrelli A, Panattoni A, Nali C, Cotrozzi L (2023b) Occurrence of fig mosaic disease in Tuscany, Central Italy: Characterization of new fig mosaic virus isolates, and elucidation of physiochemical responses of infected common fig cv. Dottato. Sci Hortic 322:112440. https://doi.org/10.1016/j.scienta.2023.112440
Revers F, García JA (2015) Molecular biology of potyviruses. Advance in Virus Research 92:101–518 99. https://doi.org/10.1016/bs.aivir.2014.11.006
Rubio L, Galipienso L, Ferriol I (2020) Detection of plant viruses and disease management: relevance of genetic diversity and evolution. Front Plant Sci 11:1092. https://doi.org/10.3389/fpls.2020.01092
Santillan F, Fribourg CE, Adams IR, Gibbs A, Boonham N, Kehoe MA, Maina S, Jones R (2018) The biology and phylogenetics of potato virus S isolates from the Andean Region of South America. Plant Dis 102:869–885. https://doi.org/10.1094/pdis-09-17-1414-re
Trusty JL, Lockaby BG, Zipperer WC, Goertzen LR (2007) Identity of naturalized exotic Wisteria (Fabaceae) in the south-eastern United States. Weed Res 47:479–487. https://doi.org/10.1111/j.1365-3180.2007.00587.x
Valouzi H, Hashemi SS, Wylie SJ, Ahadiyat A, Golnaraghi A (2020) Wisteria vein mosaic virus detected for the first time in Iran from an unknown host by analysis of aphid vectors. Plant Pathol J 36:87–97. https://doi.org/10.5423/PPJ.OA.10.2019.0268
Valverde RA (2012) Viruses that enhance the aesthetics of some ornamental plants: beauty or beast? Plant Dis 96:600–601. https://doi.org/10.1094/PDIS-11-11-0928-FE. Sabanadzovic S, Hammond J
Ward LI, Tang JZ, Clover GRG (2008) First report of wisteria vein mosaic virus on Wisteria sinensis in New Zealand. Plant Dis 92:1134. https://doi.org/10.1094/PDIS-92-7-1134B
Wei Z, Pedley L (2010) Wisteria. In: Wu ZY, Raren PH, Hong DY (eds) Flora of China. Science Press & St. Louis: Missouri Botanical Garden Press, Beijing, pp 188–189
Wei T, Yang J, Liao F, Gao F, Lianming L, Zhang X, Li F, Wu Z, Lin Q, Xie L, Lin H (2009) Genetic diversity and population structure of rice stripe virus in China. J Gen Virol 90:1025–1034. https://doi.org/10.1099/vir.0.006858-0
Zhu F, Zhu PX, Xu F, Ji ZL (2019) First report of wisteria vein mosaic virus infecting Chinese wisteria in Jiangsu Province in China. J Plant Dis Prot 126:373–377. https://doi.org/10.1007/s41348-019-00217-9
Acknowledgements
This paper is dedicated to Dr. Alberto Materazzi, a long-time head of the phytovirological lab, on the occasion of his retirement from the Department of Agriculture, Food and Environment at Pisa University.
Funding
Open access funding provided by Università di Pisa within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pedrelli, A., Panattoni, A. & Cotrozzi, L. First molecular analysis of wisteria vein mosaic virus in Italy: eight new variants reported in Wisteria sinensis. J Plant Pathol 106, 117–125 (2024). https://doi.org/10.1007/s42161-023-01526-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-023-01526-7