
Abstract
Bitter gourd wilt caused by Fusarium oxysporum f. sp. momordicae (FOM) is a devastating crop disease in China. A total of 173 isolates characteristic of typical Fusarium oxysporum with abundant microconidia and macroconidia on white or ruby colonies were obtained from diseased plant tissues. BLASTn analysis of the rDNA-ITS of the isolates showed 99% identity with F. oxysporum species. Among the tested isolates, three were infectious toward tower gourd and five were pathogenic to bottle gourd. However, all of the isolates were pathogenic to bitter gourd. For genetic differences analysis, 40 ISSR primers were screened and 11 primers were used for ISSR-PCR amplification. In total, 127 loci were detected, of which 76 were polymorphic at a rate of 59.84%. POPGENE analysis showed that Nei’s gene diversity index (H) and Shannon’s information index (I) were 0.09 and 0.15, respectively, which indicated that the genetic diversity of the 173 isolates was low. The coefficient of gene differentiation (Gst = 0.33 > 0.15) indicated that genetic differentiation was mainly among populations. The strength of gene flow (Nm = 1.01 > 1.0) was weak, indicating that the population differentiation caused by gene drift was blocked to some degree. The dendrogram based on ISSR markers showed that the nine geographical populations were clustered into two groups at the threshold of genetic similarity coefficient of 0.96. The Shandong and Henan populations were clustered into Group I, while the Guangdong, Hainan, Guangxi, Fujian, Jiangxi, and Hubei populations constituted Group II. Results of the genetic variation analysis showed that the Hunan and Guangxi populations had the highest degree of genetic differentiation, while the Hubei population had the lowest genetic differentiation. Our findings enrich the knowledge of the genetic variation characteristics of FOM populations with the goal of developing effective disease-management programs and resistance breeding programs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bitter gourd (Momordica charantia L.) wilt caused by Fusarium oxysporum f. sp. momordicae Sun & Huang (FOM) is one of the most damaging crop diseases in China (Chen et al. 2014), Japan (Imamura et al. 2007), India (Rakholiya et al. 2003), and the Philippines (Cumagun et al. 2008, 2010). Sun and Huang first reported the occurrence of bitter gourd wilt disease in Taiwan (Sun and Huang 1983), where it has become prevalent since 1996 (Lin et al. 1996). In the past 20 years, bitter gourd wilt has been identified in Zhejiang, Fujian, Guangxi, Guangdong, Hunan, and Jiangxi provinces (Rao and Li 1999; Yang et al. 2010; Zhu and Qi 1998). In recent years, the consumption of bitter gourd, a very popular vegetable in China, has increased rapidly and led to an increase in planted areas. However, effective disease control has not been easily achieved even with effective chemical use (Chen et al. 2015; Cumagun et al. 2008). Bitter gourd wilt affects the quality and yield of bitter gourd and has become a major factor limiting the production of this crop (Chen et al. 2015).
Soil-borne pathogens always invade the plant root hair epidermis via wounds and then extend into the vascular tissue. FOM colonizes the xylem vessels and produces masses of mycelia and conidia to block water transportation. Therefore, typical bitter gourd wilt symptoms are characterized by the initial wilting of leaves, followed by leaf yellowing and withering, and the darkening and thinning of stems closest to the ground. Eventually, the entire plant wilts and dies (Guo and Mo 2007). No commercially available bitter gourd cultivars are immune to FOM ( Zhu and Qi 1998). To control this disease, bitter gourds are grafted onto rootstocks of Cucurbita ficifolia and C. moschata, which are resistant to FOM (Li et al. 2008). To some degree, the disease is controlled by this grafting technique; however, this method is time-consuming and labor-intensive. Breeding disease-resistant cultivars are still the most economical and effective strategy to control this disease (Chen et al. 2015). An understanding of the genetic diversity and population structure of FOM is a crucial prerequisite for developing resistant host genotypes as well as effectively deploying available resistant cultivars. However, details of the genetic diversity of FOM remain largely unclear.
To date, several techniques have been developed to characterize the genetic variability of different Fusarium oxysporum forma specialis isolates, such as vegetative compatibility groups (VCG), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), and simple sequence repeats (SSRs) (Altinok and Can 2010; Assigbetes et al. 1994; Balmas et al. 2005; Duan et al. 2008). Cumagun et al. (2010) clustered 21 FOM isolates into four VCGs, while the VCG diversity ratio was much lower. It was even presumed that most F. oxysporum isolates infecting bitter gourd should belong to a single VCG. Chen et al. (2015) revealed that 48 FOM isolates were classified into twelve AFLP groups with distinct genetic variation. Owing to the contradiction in results from previous studies, the genetic variation in FOM populations is still not clearly understood.
The inter simple sequence repeat (ISSR) amplifies intermicrosatellite sequences at multiple loci throughout the genome with a single 16–18 bp primer used for PCR reactions (Zietkiewicz et al. 1994). The amplified DNA fragments are often polymorphic between different individuals owing to strict annealing temperatures (Mccall et al. 2004). Additionally, the cost of the ISSR analysis is relatively lower compared to that of AFLP, and ISSR produces good reproducibility (Esselman et al. 1999). This technique has therefore been extensively used in population genetics studies of different fungal species, such as Fusarium oxysporum (Mohannadi et al. 2011), Verticillium dahliae Kleb (Liu et al. 2010), Botryosphaeria dothidea (Moug.) Ces & De not (Feng et al. 2017), Valsa mali Miyabe et Yamada (Zang et al. 2012), and Exserohilum turcicum (Pass.) Leonard et Suggs (Gu et al. 2008).
Therefore, the objectives of this study were to (1) detect FOM pathogenicity in different bitter gourd varieties and other cucurbit hosts and (2) to calculate and analyze the genetic variation and population construction within and among different populations using ISSR analysis.
Materials and methods
Sample collection and pathogen isolation
Diseased bitter gourd samples were collected from the Fujian, Guangdong, Guangxi, Hainan Hubei, Henan, Hunan, Jiangxi, and Shangdong provinces from 2016 to 2017. The diseased stem tissue was washed with tap water and cut into small pieces. The tissue was immersed in 75% ethanol for 1.5 min, followed by two washes with sterile water. Lastly, tissue pieces were placed on the surface of potato dextrose agar (PDA) plates amended with cefotaxime sodium. The plates were incubated in the dark at 28 °C for three days. Tips of mycelia were cut from the colony margin, transferred to a new plate, and continually incubated in darkness at 28 °C until the fungal colony fully covered the plates. Small pieces (2 × 2 mm) from the colony were placed into 2-ml cryogenic vials containing 1.6 ml of 20% glycerinum and stored at -80 °C.
Pathogenicity and formae speciales determination
The pathogenicity of the isolates was tested on seedlings of three bitter gourd cultivars (cv. Changlv, Ruyu33, and Ruyu41) and seven cucurbit crops, including muskmelon, cucumber, watermelon, tower gourd, bottle gourd, pumpkin, and cantaloupe. The pathogen isolates from bitter gourd plants were cultured on PDA plates for 7 days at 28 °C. Conidia were obtained from 7-day-old cultures grown on PDA at 28 °C, filtered through two layers of cheesecloth and re-suspended in sterile distilled water at 1 × 106 conidia/ml. The roots of 7-day-old bitter gourd plants were dipped into 500 mL of the 1 × 106 conidial suspension for 30 min, transplanted to plastic pots (diameter = 10 cm) containing organic substrate, and kept in an illumination incubator (28 °C, RH = 80%, light:darkness ratio = 12:12). Control plants were treated with sterile distilled water. Disease severity was calculated based on the disease severity standard described by Chen et al. (2015).
DNA extraction and purification
DNA extraction and purification were performed as described by Raeder and Broda (1985) and Nel et al. (2006). Briefly, the mycelia of 5-day liquid culture in 50-ml flasks at 28 °C was collected using double-layer cheesecloth, followed by rinsing with distilled water and removing the extra water using filter paper. The mycelial pads were ground in an atmosphere of liquid nitrogen for DNA extraction using the CTAB protocol. RNA was removed from the DNA sample with RNase (100 μg/μL). The purified genomic DNA was separated on a 1% agarose gel (w/v) stained with ethidium bromide and photographed under UV illumination. DNA concentration and quality were measured using an ND-1000 spectrophotometer (Thermo Scientific Inc, Grand Island, NY). The DNA working concentration was adjusted to 100 ng/μL and stored at -20 °C.
rDNA-ITS region amplification
The rDNA-ITS region was amplified with the universal primer pairs ITS1 (5′- TCCGTAGGTGAACCTGCG-3′) and ITS4 (5′-TCCTCCGCTTATTGATAT-3′). The PCR reactions were carried out using a 25-μL mixture containing 1X Taq buffer (75 mM Tris–HCl [pH 8.8], 20 mM (NH4)2SO4, and 0.01% Tween 20), 10 mM each of deoxyribonucleotide triphosphates (dNTPs), 25 mM MgCl2, each primer at 0.4 μM, 1 U Taq, and approximately 10 ng DNA as template. The amplification conditions were as follows: 94 °C for 4 min; followed by 35 cycles of 94 °C for 30 s, 51 °C for 30 s, and 72 °C for 1 min; and a final extension at 72 °C for 10 min. The above reaction without the DNA template served as a negative control.
Five microliter aliquots of PCR products were separated on a 1.5% agarose gel stained with ethidium bromide. PCR products that produced bright and distinct bands were sequenced independently along both strands by Songon Biotech Co. Ltd. (Shanghai). The sequences were aligned on the NCBI website using BLASTn.
ISSR PCR primer selection and ISSR-PCR amplification
To select primers that generated more polymorphic bands, a total of 40 ISSR primers were screened using genomic DNA from six representative isolates of FOM from different locations. These primers consist of di- or trinucleotide repeats. The screened polymorphic primers were used to characterize the 173 FOM isolates. The 25-μL mixture contained 2 μL 10 × PCR buffer, 1.8 mM MgCl2, 0.1 mM dNTPs, each primer at 100 μM, 50 ng template DNA, 1.5 U Taq polymerase (TaKaRa Biomedical Technology Co. Ltd, China), and double-distilled water. Amplifications were performed using a thermal cycler (Eppendorf, Hamburg, Germany) with the following PCR program: 5 min of initial denaturation at 94 °C; 30 cycles of 94 °C for 30 s, 30 s for annealing at the primer-specific melting temperature, and 72 °C for 3 min; followed by a final extension of 10 min at 72 °C. The above reactions performed without template DNA served as a negative control. The PCR products were separated at 120 V for 1 h on a 2% agarose gel pre-stained with ethidium bromide. The gel was visualized under UV light and photographed using the InGeniusLHR gel imaging system (Syngene, Frederick, MD). All PCR amplifications were performed at least twice for each isolate. A 3000-bp DNA ladder was used as a size marker.
Data analysis
The repeatable, clearly visible bands generated when using ISSR primers were converted into a binary data set with 0 and 1, where 1 and 0 represent the presence and absence of bands, respectively. Population genetic diversity, Nei’s genetic diversity, Shannon’s diversity index, and genetic variation were analyzed using POPGENE version 1.32. The dendrogram relationship of different geographical populations was analyzed using the unweighted pair-group mean average (UPGMA) method in the SHAM module based on genetic distance data in NYSTS Version2.10. Meanwhile, the neighbor-joining (NJ) dendrogram was constructed using the NJ function in the R package of phangorn version 2.5.5. STRUCTURE (version 2.3.4) software was used to obtain the hierarchical organization of the genetic structure of the nine geographical populations.
Results
Pathogen isolation and identification
A total of 173 isolates were obtained from diseased bitter gourd plants. The shape of spores and colony characteristics were initially used to identify the isolates. The isolates showed typical Fusarium oxysporum characteristics with abundant microconidia and macroconidia on white or ruby colonies (Fig. 1). A single DNA fragment of approximately 600 bp was amplified with the universal primers ITS1 and ITS4. Aligning with the published sequences on the NCBI website by BLAST, all isolate sequences tested were similar to those of F. oxysporum formae speciales with identities over 99%. Therefore, the tested isolates were identified as F. oxysporum.

The morphology of Fusarium oxysporum f. sp. momordicae isolates on PDA plates a, b, c, g, h: the colony frontal view of GuX10, HuN16, HuN15, GuX11, GuX3; d, e, f, j, k: the colony opposite view of isolates GuX10, HuN16, HuN15, GuX11, GuX3 ; i: Microconidia of HeN15; l: Macroconidia of HeN15. The bar of i and l was 10 μm
Pathogenicity and formae speciales determination
Nine days after inoculation, typical wilt symptoms were observed on the inoculated bitter gourd seedlings. The inoculated bitter ground showed typical yellowing and wilting symptoms on the lower leaves. The veins between the main veins yellowed, while the control plants did not show any symptoms (Fig. 2). All isolates could infect the three bitter gourd cultivars. The cv. Changlv was relatively resistant to the pathogen, while the other two cultivars, Ruyu33 and Ruyu41, were susceptible. With the exception of eight isolates, FuJ10, GuX7, HeN15, HuN1, JiX12, JiX15, ShD6, and ShD9, which could infect the towel gourd and bottle gourd, the other isolates were not pathogenic to the seven cucurbit crops tested (Table 1). Thus, the isolates used in this study were identified as F. oxysporum f. sp. momordicae (FOM).

Pathogenicity of Fusarium oxysporum f. sp. momordicae isolates HeN15 on cv. Ruyu41 CK: inoculated with sterile distilled water; IN: inoculated with HeN15 conidial suspensions; CK-Leaf: The healthy leaf from the CK treatment; IN-Leaf: the diseased leaf from IN treatment
Primer screening and ISSR polymorphisms
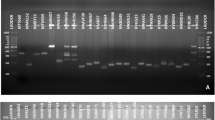
Among the 40 ISSR primers tested, only 11 ISSR primers produced PCR products that displayed distinct and bright polymorphic bands, and almost no bands were observed in the PCR products when the remaining 29 primers were used (Fig. 3). When applied to the 173 FOM isolates, the 11 primers produced 127 DNA fragments, of which 76 polymorphic fragments ranged from 250 to 3000 bp. The percentage of polymorphic bands was 59.84% (Table 2). The number of bands generated from each primer ranged from 8 to 13, with an average of 11.50 per primer. The average number of polymorphic bands was 6.9.

The ISSR-PCR amplification products of some FOM isolates with the primer of VCA025. Lanes 1-24 respectively were: ShD1, ShD14, ShD18, HeN5, HeN22, HeN24, HaN7, HaN8, HaN12, HuN8, HuN10, HuN16, GuX8, GuX28, GuX31, GuD9, GuD18, Gu22, FuJ1, FuJ13, FuJ18, JiX1, JiX8, JiX17 FOM isolates. M: 3000bp DNA marker. CK: negative control
Genetic diversity among different geographical populations
The genetic parameters of the nine populations were calculated using the PopGene software package. Results indicated that the different populations had different genetic diversity characteristics. The Jiangxi population had the greatest genetic diversity, with Nei’s gene diversity index (H) and Shannon’s information index (I) values of 1.07 and 0.06, respectively, while the Hubei population had the lowest genetic diversity, with H and I values of 0.01 and 0.02, respectively. The average value showed that the number of alleles (Na) and the effective number of alleles (Ne) were 1.13 and 1.08, respectively, while H and I were 0.05 and 0.07, respectively, in the nine geographical populations (Table 3).
At the species level, the Na, Ne, H, and I were 1.57, 1.13, 0.09, and 0.15, respectively (Tables 3 and 4). According to the evaluation criterion of population diversity richness, H and I values should be greater than 0.20 and 0.30, respectively. Therefore, the genetic diversity of the 173 tested isolates from nine geographical populations was relatively low. The values of total heterozygosity (Ht), intraspecific heterozygosity (Hs), coefficient of gene differentiation (Gst), and gene flow (Nm) were 0.09, 0.06, 0.33, and 1.01, respectively. Gst indicated that the genetic differentiation among the nine geographical populations was generally high (Gst = 0.33 > 0.15). The Nm value revealed that the population differentiation caused by gene drift was blocked to some degree (Nm = 1.01 > 1.0) (Table 4).
The genetic identity of the nine geographical populations ranged from 0.947 to 0.986. The maximum genetic identity appeared between the Hunan and Hainan populations. The genetic distances for these populations ranged from 0.017 to 0.055. The maximum genetic distances appeared between the Hubei and the Shandong populations (Table 5).
Based on the genetic distance on the topological structure of the dendrogram, the nine geographical populations were clustered into two distinct groups at 0.962 of the genetic similarity coefficient value (Fig. 4). Group I comprised the Shangdong and Henan populations, which were both located in the north of the Qinling Mountain and Huai River. Group II consisted of the Guangdong, Hainan, Fujian, Jiangxi, Hubei, Hunan, and Guangxi populations. In contrast, these populations all lay to the south of the Qinling Mountain and Huai River (Fig. 5). With a genetic similarity coefficient value of 0.968, Group II was divided into three subgroups. Subgroup 1 included the Guangdong, Hainan, Fujian, Jiangxi, and Hubei populations. The Hunan and Guangxi populations formed subgroups 2 and 3, respectively. The topological structure of neighbor-joining (NJ) was similar to the UPGMA dendrogram, which gave strong support in the UPGMA clustering (Fig. 6).

The UPGMA dendrogram of nine FOM geographical populations based on the genetic distance

The geographical distribution of nine FOM geographical populations on the Chinese map The red ellipse stand for the group I and the red circle means the group II, the dashed line in the middle of the map stands for the approximate location of the Qinling Mountain and Huai River, which was the diving line between north and south China

The neighbor-joining (NJ) dendrogram of nine FOM geographical populations based on the genetic distance
Results from the nine geographical population structure analysis showed that different populations had different levels of genetic differentiation (Fig. 7). The most obviously differentiated populations were that of the Hunan and Guangxi geographical populations. There was a large genetic difference among the isolates in the two geographical populations. The degree of population differentiation of the Hubei geographical population was the lowest. The genetic differences among the isolates within this population were minimal. The Shangdong, Henan, Guangdong, Hainan, Fujian, and Jiangxi geographical populations had obvious population genetic differentiation; however, the degree of population genetic differentiation was much weaker than that of the Hunan and Guangxi geographical populations.

The hierarchical organization of the genetic structure of nine geographical populations in China The different colors means different groups based on the method of Evanno (K=4), the length of the colored segment suggested the estimated membership proportion of each sample in the designed group
Discussion
In this study, morphological characteristics, rDNA-ITS sequencing, and formae speciales tests were combined to ensure that all the isolates studied were FOM. In general, Fusarium pathogens cannot be identified to the species level with the rDNA-ITS sequence alone; however, Fusarium oxysporum appears to be an exception and can be identified specifically using the rDNA-ITS sequence. With the supplement of formae speciales test results, all isolates were identified as FOM. The precision and reliability of the ISSR analysis were demonstrated.
In pathogenicity experiments, we found that the FOM isolates had different aggressivity on Cucurbitaceae crops. Three FOM isolates (HuN1, JiX12, and ShD9) could infect the tower gourd (Luffa cylindrica). Five FOM isolates, FuJ10, GuX7, HeN15, JiX15, and ShD6, were pathogenic to bottle gourd (Lagenaria ciceraria). Although it has been reported that there is host specificity among the different formae speciales of F. oxysporum, the cross infectivity of some formae speciales has been mentioned in several studies (Bouhot 1981; Kim et al. 1993; Mccallum et al. 2001; Snyder and Hansen 1940). Zhu and Qi (1998) showed that FOM from bitter gourd was pathogenic to bottle gourd seedlings and club bottle gourd seedlings and grown plants. Cumagun et al. (2010) found that some FOM isolates from bitter gourd could infect 7-day-old bottle gourd seedlings, but not 1-month-old plants. Chen et al. (2015) also reported that some bitter gourd FOM isolates produced mild infections in bottle gourd (Lagenaria siceraria var. clavata) seedlings. F. oxysporum f. sp. luffae was also found to infect bitter gourd cultivars (Lv et al. 2000). These findings were identical to our results. However, contradictory results have also been demonstrated in previous reports. F. oxysporum f. sp. momodicae was first found on bitter gourd in Taiwan in 1983 and at the time was not considered pathogenic to Cucurbitaceae crops (Lin et al. 1996; Snyder and Hansen 1940). Yang et al. (2010) also showed that FOM isolates could specifically infect bitter gourd, but did not infect white gourd, tower gourd, watermelon, and muskmelon. These contradictory results may be attributed to the host age and pathogen-adaptive variation. Gerlagh and Blok (1988) stated that the formae speciales specificity of F. oxysporum causing seedling wilt in Cucurbitaceae is limited. Bouhot (1981) demonstrated that mutation in one forma specialis could be induced to convert to another forma specialis with a pathogenic capacity to another host.
Information on population genetic variation and population structure of FOM is essential and vital for understanding the roles of genetic diversity in the future. In this study, 173 FOM isolates from nine geographical populations were studied using the ISSR technique. According to the values of H and I of different geographical populations, the genetic differentiation within populations was relatively small. This indicates that the genetic homogeneity within the nine populations was high. Cumagun et al. (2008) reported that four VCGs occur in F. oxysporum f. sp. momordicae isolates. However, it is speculated that FOM isolates may belong to a single VCG because most isolates belonged to VCG1, with the exception of one isolate each from VCG3 and VCG4 and three isolates from VCG2. Chen et al. (2014) also found that the polymorphic bands amplified by RAPD molecular markers from FOM isolates were less; therefore, it was assumed that a high genetic similarity existed in FOM isolates. Later, they showed that the genetic differentiation within the population was lower using the AFLP approach (Chen et al. 2015). The low genetic diversity within the population might be attributed to the strictly asexual reproduction of F. oxysporum and limited gene flow between populations.
Gst is an important parameter to measure whether genetic differentiation exists among populations. When the Gst value was greater than 0.15, genetic differentiation was considered large (Wright 1978). Nm is a vital factor affecting genetic differentiation. When the Nm value was less than 1.00, the gene flow was blocked to some degree (Wright 1931). In our study, the Gst and Nm values were 0.33 and 1.01, respectively. These data showed that genetic differentiation among geographical populations was high, while gene exchanges between these populations were relatively rare; that is, each geographical population was a relatively independent unit. The most likely reason was the high level of adaptability of pathogens to local climate, soil conditions and host variety. In China, the climate and soil conditions of different bitter gourd-producing regions are quite different. It is hot and rainy in Fujian, Guangdong, Guangxi, Hainan, Hubei, Hunan, and Jiangxi provinces, which are all located to the south of the Qinling Mountain and Huai River, whereas it is arid in Henan and Shandong, which are located in the north of the Qinling Mountain and Huai River. The main cultivated bitter gourd variety and cultivation methods are relatively stable. The same varieties could be used for many years in the same region. Thus, the selection pressures that the pathogen faces will be relatively stable and the pathogen will trend toward homogeneity.
Conclusion
In summary, ISSR is an effective tool for detecting the genetic variation among and within F. oxysporum f. sp. momordicae populations. The genetic variation within the pathogen populations was relatively low, but was slightly greater among the populations. Gene exchanges between different populations were blocked to some degree. These findings enrich our knowledge of genetic variation characteristics in pathogen populations and may be used for breeding Fusarium wilt disease-resistant bitter gourd in the future.
References
Altinok HH, Can C (2010) Characterization of Fusarium oxysporum f.sp. melongenae isolates from eggplant in Turkey by pathogenicity. VCG and RAPD analysis Phytoparasitica 38:149–157
Assigbetes KB, Fernandez D, Doubios MP, Geiger JP (1994) Differentiation of Fusarium oxysporum f.sp. vasinfectum races on cotton by random amplified polymorphic DNA (RAPD) analysis. Phytopathology 84:622–626
Balmas V, Scherm B, Primo PD, Rau D, Marcello A (2005) Molecular characterization of vegetative compatibility groups in Fusarium oxysporum f.sp. radicis-lycopersici and f.sp. lycopersici by random amplification of polymorphic DNA and microsatellite-primed PCR. Eur J Plant Pathol 111:1–8
Bouhot D (1981) Some aspects of the pathogenic potential in formae speciales and races of Fusarium oxysporum on Cucurbitaceae. In: Nelson PE, Toussoun TA, Cook RJ (eds) Fusarium: Disease, Biology, and Taxonomy. The Pennsylvania State University Press, Pennsylvania, pp 479–504
Chen ZD, Huang RK, Li QQ, Wen JL, Yuan GQ (2015) Development of pathogenicity and AFLP to characterize Fusarium oxysporum f.sp. momordicae isolates from bitter gourd in China. J Phytopathol 163:202–211
Chen ZD, Yuan GT, Li QQ, Qin J, Jiang YQ, Huang RK (2014) Identification and genetic diversity of Fusarium oxysporum isolates from bitter gourd. Acta Phytopathologica Sin 44:36–45
Cumagun CJR, Aguirre JA, Relevante CA, Balatero CH (2010) Pathogenicity and aggressiveness of Fusarium oxysporum Schl. in bottle gourd and bitter gourd. Plant Pro Sci 46:51–58
Cumagun CJR, Oribiana ZC, Tolentino MS, Relevante CA, Balatero CH (2008) Vegetative compatibility among Fusarium oxysporum isolates from bitter gourd and bottle gourd in the Philippines. J Plant Prot Res 48:283–293
Duan HJ, Zhang CY, Li XH, Guo XM, Ma ZY (2008) Assessment of genetic diversity in Fusarium oxysporum f.sp. niveum by RAPD. ISSR and AFLP analysis Mycosystema 27:267–276
Esselman EJ, Li JQ, Crawford DJ, Windus JL, Wolfe AD (1999) Clonal diversity in the rare Calamagrostis porteri spp. insperata (Poaceae): comparative results for allozymes and random amplified polymorphic DNA (RAPD) and intersimple sequence repeat (ISSR) markers. Mol Ecol 8:443–451
Feng HJ, Wen CY, Yin XM, Hong KQ, Zang R (2017) Population genetic diversity analysis of Chinese jujube branch canker pathogens (Botryosphaeria dothidea) in China using ISSR markers. J Fruit Sci 34:977–987
Gerlach M, Blok WJ (1988) Fusarium oxysoprum f.sp. cucurbitacearum n.f. embracing all formae speciales of F.oxysporum attacking cucurbitaceous crops. Neth J Plant Pathol 94:17–31
Gu SQ, Fan YS, Li P, Dong JG (2008) Optimization of ISSR reaction and genetic diversity analysis of Exserohilum turcicum. Acta Phytophylacica Sin 35(5):427–432
Guo TX, Mo JY (2007) Resistance to the blight disease (Fusarium oxysporum f.sp. momordicae) of several bitter gourd varieties. Guangxi Agr Sci 38:408–410
Imamura Y, Kawagoe Y, Kuno k, Jinkawa T, Sugita T, (2007) Occurrence of Fusarium wilt caused by Fusarium oxysporum in balsam pears (Momordica charantia) in Miyazaki, Japan and analysis of the pathogen based on the rDNA-IGS sequence. Kyushu Plant Protection Research 53:1–8
Kim DH, Martyn RD, Magill CW (1993) Mitochondrial DNA(mtDNA)-Relatedness among formae speciales of Fusarium oxysporum in the Cucurbitaceae. Phytopathology 83:91–97
Li DZ, Wen QF, Kang JB, Li YP, Xue ZZ (2008) Studies on engraft preventing fusarium wilt of Balsam pear. Southwest China J of Agri Sci 21:888–890
Lin YS, Huang CH, Kuo MS (1996) Occurrence and dissemination of Fusarium wilt of bitter gourd in Taiwan. Plant Pathol Bull 5:38–46
Liu YX, Hou LJ, Li W, Yang JR, Xiao R (2010) Optimization of ISSR reaction and analysis of genetic diversity of Verticillium dahliae in cotton. Acta Phytophylacica Sin 37:425–430
Lv PK, Liu WZ, Duan BS, Zhang BD (2000) The continuation of atlas of insect pests and diseases of vegetable crops in China. YuanFang Press, Hohhot, pp 102–103
Mccall AC, Kelly D, Chapman HM (2004) Little geographic or host plant variation in a Chionochloa (Poaceae) seed predator (Cecidomydiidae: undescribed species). N Z J Ecol 28:215–224
Mccallum BD, Tekauz A, Gilbert J (2001) Vegetative compatibility among Fusarium graminearum (Gibberella zeae) isolates from barley spikes in southern Manitoba. Can J Plant Pathol 23:83–87
Mohammadi N, Goltapeh EM, Babaie AA, Puralibaba H (2011) Pathogenic and genetic characterization of Iranian isolates of Fusarium oxysporum f.sp. lentis by ISSR analysis. J Agric Technol 7:63–72
Nel B, Steinberg C, Labuschagne N, Viljoen A (2006) Isolation and characterization of nonpathogenic Fusarium oxysporum isolates from the rhizosphere of healthy banana plants. Plant Pathol 55:207–216
Raeder U, Broda P (1985) Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol 1:17–20
Rakholiya KB, Jani SM, Chheladiya KK, Kikani BK (2003) Fusarium wilt of bitter gourd. Indian Phytopathol 4:503–504
Rao XQ, Li RK (1999) Studies on identification of resistance to bitter gourd wilt at seedling stage with the filtrate of Fusarium oxysporum Schl. f.sp. momordicae. Acta Agric Univ Jiangxiensis 21:367–369
Snyder WC, Hansen HN (1940) The species concept in Fusarium. Am J Bot 27:64–67
Sun SK, Huang JW (1983) A new Fusarium wilt of bitter gourd in Taiwan. Plant Dis 67:226–227
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1978) Evolution and the genetics of populations: a treatise in four volumes: Variability within and among natural populations. University of Chicago Press, Chicago
Yang Y, Zhou Q, Ji H, Wei F, Xu XB, Jiang DH (2010) Isolation and identification of Fusarium species from wilted bitter gourd. Acta Agric Zhejiangensis 22:354–357
Zang R, Huang LL, Li ZL, Gao XN, Kang ZS (2012) Polymorphism analysis of Valsamali isolated from Shaanxi province using ISSR markers. Acta Phytophylacica Sin 39:51–57
Zhu TS, Qi PK (1998) Study on the pathogen of bitter gourd wilt in Guangdong. J S China Agric Univ 19:14–18
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome finger-printing by simple sequence repeat(SSR)-anchored polymerase chain reaction amplification. Genomics 20:176–183
Acknowledgments
We are grateful to Dr. Zhengdong Chen in Guangxi Academy of Agricultural Science for their tremendous efforts in sample collection. We appreciate Prof. Qinsheng Gu in Zhengzhou Fruit Research Institute for his precious advances in experiment design. Our thanks also extend to Prof. Shengli Ding and Hezhong Wang in Henan Agricultural University for earlier manuscript improvement. This work is supported by the grants from the Special Fund for Agro-scientific Research in the Public Interest (201503110).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zang, R., Zhao, Y., Guo, K. et al. Population genetic variation analysis of bitter gourd wilt caused by Fusarium oxysporum f. sp. momordicae in China using inter simple sequence repeats (ISSR) molecular markers. J Plant Pathol 103, 787–797 (2021). https://doi.org/10.1007/s42161-021-00759-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-021-00759-8