
Abstract
Introduction
Langerhans cell histiocytosis (LCH) localised in the hypothalamic-pituitary region (HPR) is very rare, especially in adults. Diabetes insipidus (DI) is considered to be a hallmark of HPR LCH, while anterior pituitary abnormalities are usually seen as consequences of surgery, radiotherapy or chemotherapy.
Case description
We present a patient with localised HPR LCH with dominant anterior pituitary dysfunction and tumour mass effects but without DI. Seven years after surgery and local radiotherapy, she is stable. Control MRI shows no residual tumour growth and thorough physical examination is still without any signs of disease spread.
Conclusions
Anterior pituitary deficiency can appear without DI and not only as a consequence of LCH treatment. All patients with LCH should be screened for this endocrine abnormality so that appropriate substitution therapy may be provided.
Similar content being viewed by others

Introduction
Langerhans cell histiocytosis (LCH) is a rare disease which is characterised by nonspecific granulomatous deposits in many different tissues. The disorder may involve the lungs, bones, hypothalamus and pituitary gland, skin and mucous membranes, lymph nodes, liver and other soft tissues, including the testes [1]. It is usually diagnosed in childhood [2, 3]. Less than 30% of all reported cases are adults, with an incidence of 1.8 per 106 [4, 5]. According to the number of affected tissues, LCH is classified as localised or multifocal. Localised LCH is defined as the involvement of one soft tissue with or without the skeletal system being affected, while the multifocal form denotes the involvement of two or more soft tissues again with or without the skeletal system being affected [6]. The hypothalamic-pituitary region (HPR) is infiltrated in 5–50% of all patients with LCH, but most commonly in those with the multifocal disease form [2, 7,8,9]. Isolated presence of LCH in the HPR is very rare [1]. Diabetes insipidus (DI), the most frequent hormonal abnormality, occurs in 15–50% patients with HPR LCH [4, 7, 9, 10]. Pituitary dysfunction occurs in only 5–20% of patients, almost always accompanied by DI [9, 11, 12]. Herein, we present a patient with localised hypothalamic-pituitary LCH who had dominant tumour mass effects and pituitary dysfunction without DI and her 7-year follow-up. Procedures, data collection, and presentation were approved by the hospital ethics committee (decision no.: 37232/1). Written informed consent was obtained from the patient to participate and for publication of this case report and any accompanying data.
Case report
A 31-year-old woman was admitted to the Endocrinology Clinic for secondary amenorrhea, weight gain, and hands, legs, and face oedema. After pregnancy and delivery 11 years previously, her menstrual cycles became irregular and completely disappeared a year ago. She gained 25 kg during the past year. In the last 3 months, she has been experiencing frequent headaches and blurred vision. Besides obesity (BMI 34.5 kg/m2), physical examination at admission was unrevealing. Blood pressure and heart rate were 130/90 mmHg and 68/min and respiratory rate was 19/min. Initial laboratory investigation results (blood cell count, blood sugar, electrolytes, urea, creatinine, hepatogram, amylase) were normal.
Endocrinological examination revealed elevated prolactin (PRL), secondary hypothyroidism, and hypogonadotropic hypogonadism, while the rest of the pituitary hormones remained normal (Table 1).
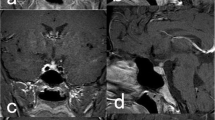
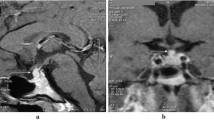
Magnetic resonance imaging (MRI) of the HPR was performed. MRI revealed a suprasellar mass close to the optic chiasm, measuring 15 × 15 × 13 mm (KKxLLxAP), expanding to the sella turcica and causing pituitary stalk encasement. It also extended to the optic nerves, mainly to the left nerve, but without obvious invasion of their prechiasmatic parts. Tumour mass signal intensity was T1w, T2w evidently stronger after the application of contrast. Radiological diagnosis was optic chiasm-hypothalamic glioma (Fig. 1). A visual field testing mapped temporal hemianopsia in the right eye and nasal hemianopsia in the left (Fig. 2).

MRI of hypothalamic-pituitary region

Perimetry findings before and after surgery
Since there were no clinical signs of systemic disease, while radiological characteristics of the tumour were suspicious for glioma, standard preoperative examination was performed comprising chest X-rays, cardiological examination (ECG, echocardiography), and laboratory tests including coagulation status. All results were normal. Left pterional craniotomy and subsequent tumour reduction starting from the optic chiasm were performed. The tumour was moderately vascularized, dark red in colour and distinct from the surrounding tissues. Maximal reduction of the tumour was carried out, except near the hypothalamus where the demarcation between the brain and the tumour was not clear.
During the postoperative course, the patient developed transitory DI, which completely vanished after 2 weeks, thus the desmopressin therapy was gradually reduced and stopped.
Histopathological examination revealed medium-sized to large-sized Langerhans cells mixed with non-neoplastic inflammatory cells (HE × 200) (Fig. 3a). A further immunohistochemical study verified Langerhans cells with abundant cytoplasm, typically positive for S100 protein, and admixed with infiltrating T cells (LSAB × 200) (Fig. 3b).

a Histopathological examination. Langerhans cell histiocytosis of the suprasellar region. b Immunohistochemical results. Langerhans cells cytoplasm positive for S 100 protein
To assess the stage of the disease, the following diagnostic tests were performed: a thorough physical examination, including a complete evaluation of the skin and oral mucosa, which were normal; abdominal echosonography and liver function tests, performed to evaluate possible liver involvement, were in the referent range; a control blood cell count ruled out cytopenia; a skeletal system investigation that included bone X-rays. Since somatostatin has been identified in Langerhans cells, we performed a 99mTc-Tektrotid scan, which was also normal. Control chest X-rays and echocardiography remained normal, as they were before the operation. Since all additional examinations did not reveal the involvement of any other tissues or organs, the diagnosis remained localised LCH of the HPR.
Follow-up hormone measurements after surgery revealed secondary hypocortisolism and confirmed secondary hypothyroidism, both requiring substitution with hydrocortisone and l-thyroxin. Even though insulin-like growth factor 1 (IGF1) concentration was normal, the glucagon-stimulating test was performed to exclude the possibility of GH deficiency. The test was performed after adequate replacement of hydrocortisone and l-thyroxin, and a peak GH level higher than 3 mU/l excluded GH deficiency. Previously, elevated PRL levels were normalised. The levels of the rest of the pituitary hormones were lower compared to values before surgery (Table 1).
Control MRI showed an irregularly shaped formation, with unclear borders on the same site and with the same characteristics of the previously diagnosed tumour. Its diameters were 8 × 6 mm (KK, LL). After contrast application, the formation was non-homogeneous with strong signal intensity. Radiological findings were consistent with the diagnosis of recurrent tumour (Fig. 1).
Repeated perimetry showed significant visual field improvement in the right eye and partial withdrawal of nasal hemianopsia in the left after surgery (Fig. 2).
According to the histopathology, radiation therapy was indicated and performed (7 fractions of 12.6 Gy), but without significant tumour reduction. We continued with once a year follow-ups, along with MRI and hormonal status controls and ongoing substitution therapy. Seven years after the surgery, she is stable. Follow-up MRI shows no remnant tumour growth and on thorough physical examination, there are still no signs of the disease spreading.
Discussion
Langerhans cell histiocytosis is a rare disease mainly occurring in childhood, with very low incidence in adults. In the multifocal disease form, the CNS and HPR could be affected, but rarely as the primary sites of disease. MRI showing a mass in the HPR in patients with known multifocal LCH is usually considered conclusive for the diagnosis [13]. However, localised HPR LCH is practically indistinguishable from suprasellar germinoma [13, 14]. In Howarth’s retrospective study of 314 patients with LCH, recruited during a 50-year period, 44 patients had pituitary-thalamic axis LCH along with other affected tissues and only two patients had isolated thalamic axis LCH, while all patients had DI [1]. Anterior pituitary dysfunction (APD) is, however, neglected in the majority of studies. Because it is very rare, usually associated with DI and often recognised after LCH treatment, it is considered to be a consequence of surgery, irradiation and chemotherapy. Kaltsas et al. evaluated 12 adult patients with HPR LCH; all of them had DI and 8 of 12 developed some anterior pituitary hormonal deficiencies during 20 years of follow-up [15]. There are several patients presented in the literature who had APD and DI when LCH was diagnosed [16,17,18,19]. Tabarin et al. reported a patient with hypothalamic LCH who had APD without DI; however, it was not isolated since cutaneous involvement was present [20].
Each pathological substrate in the HPR presents with symptoms and signs that are divided into the following groups: (1) symptoms/signs suggestive of anterior and posterior pituitary hormone excess or deficiencies, (2) non-endocrine hypothalamic dysfunction (NHD) and (3) tumour mass effects (TMF). The patient herein presented had polymorph symptoms and signs from all three groups.
Symptoms and signs of pituitary hormone excess or deficiencies
PRL levels
History and physical and laboratory examination excluded the possibility of physiological and medicamentous hyperprolactinemia in our patient. MRI did not reveal any pathological substrate in the anterior pituitary gland which could be identified as prolactinoma. Moderately increased prolactin levels pointed to possible stalk disconnection. At the same time, radiological findings verified pituitary stalk encasement by a suprasellar mass: this may account for the patient’s hyperprolactinemia as being part of the “stalk syndrome” in which physical pressure on the hypothalamic-pituitary stalk stops PRL-inhibiting hormone, dopamine, leading to the increased PRL synthesis and secretion. Normalisation of PRL levels in our patient after surgery confirmed that hyperprolactinemia occurred due to pituitary stalk encasement.
FSH-LH deficiency
The gonadotropin deficiency revealed in our patient could have been a consequence of a slow progression of the tumour mass, damaging the hypothalamic area producing gonadotropin-releasing hormone (GnRH). On the other hand, hyperprolactinemia per se could disrupt pulsatile FSH/LH secretion, this leading to hypogonadotropic hypogonadism. Gonadotropin deficiency was the second most common pituitary hormone deficiency in a 20-year follow-up of adult patients with HPR LCH [15]. Hypogonadotropic hypogonadism in our patient persisted after surgery either as a consequence of irreversible hypothalamic damage due to the tumour mass or as a consequence of the operation.
TSH deficiency
The hypothyroidism in our patient with low levels of thyroid hormones and TSH was diagnosed as secondary hypothyroidism. Thyroid LCH, which is very rare, usually presents as isolated or multiple hypoechoic nodules localised unilaterally or bilaterally in the thyroid gland, typically with normal thyroid status [21]. The normal ultrasonography features in our patient and the absence of thyroid antibodies confirmed the laboratory interpretation of secondary hypothyroidism. TSH secretion did not recover after surgery. Hence, substitution therapy with l-thyroxin was continued indefinitely.
ACTH deficiency
ACTH deficiency is usually presented in the context of panhypopituitarism and rarely as an isolated deficiency [15]. The incidence of ACTH deficiency in patients with HPR LCH ranges from the 30% reported by Kaltsas et al. to the 50% documented in the series of seven cases presented by Modan-Moses et al. [10, 15]. Even though ACTH deficiency was not observed in our patient before surgery, it occurred afterwards, requiring hydrocortisone substitution.
GH deficiency
Patients with GH deficiency (GHD) may have abdominal obesity and poor exercise capacity as well as impaired quality of life and psychosocial function, but there is no pathognomonic clinical presentation. GHD can precede the development of all other endocrine deficiencies [15], or it can be observed as the first endocrine defect in addition to DI [2], or it may occur after radiotherapy treatment [22]. We have continued with follow-up, but it has so far not been recorded in our patient.
DI
Diabetes insipidus is virtually a hallmark of the presence of a pituitary disorder in LCH. In all 12 patients, followed up for 20 years, DI was the earliest hormonal deficiency. In four patients, it remained the only endocrine abnormality, while the majority of patients developed at least one anterior pituitary hormone deficiency [15]. Similar results are reported in the series of seven cases where anterior pituitary dysfunction occurred almost exclusively with DI [10]. Both patients reported by Isoo et al. had DI as the first noted abnormality [13]. It was the only hypothalamic-pituitary abnormality in the patient reported by Asano T et al. [23]. One of the unique features in our patient with isolated HPR LCH is the absence of clinical, laboratory, and radiological signs of DI. After surgery, the patient developed transitory DI, which completely vanished in 2 weeks.
Symptoms and signs of non-endocrine hypothalamic dysfunction
NHD can present with a wide spectrum of signs and symptoms, such as hyperphagia, obesity, thermoregulation disorder, adipsia, and a variety of memory impairments. Our patient had abnormal eating patterns, followed by weight gain and obesity with a BMI of 34.5 kg/m2. Although secondary hypothyroidism could contribute to weight gain and oedema, NHD was more likely the main cause of obesity in our patient.
Symptoms and signs of tumour mass effect
Enlargement of any intracranial tumour mass, leading to increased intracranial pressure, could present with different signs and symptoms. Frequent headaches, which our patient experienced, often followed by nausea and refractory to symptomatic analgesic therapy could be one of them.
Blurred vision and a variety of visual field impairments are more specific signs of tumour localisation in the HPR. A similar loss of the visual field was reported by Ghafoori et al. with almost the same tumour dimensions and localisation in the HPR very similar to that in our patient [18]. Cessation of headaches and significant vision improvement after surgery confirmed that these major symptoms in our patient were consequences of the “tumour mass effect”.
Response to the treatment
The very low incidence of localised HPR LCH is the main reason that there is no consensus as to optimal treatment. Complete surgical removal is the “best scenario”, but unfortunately, a very rare one. D’Avella et al. presented what appears to be a unique case of complete surgical excision of hypothalamic LCH granuloma followed by no residual tumour in a 13-month follow-up period [16]. However, LCH is usually in close proximity to the hypothalamus, pituitary stalk, optic chiasm, and/or optical nerves, thus such an aggressive approach is rarely possible to perform. Limited literature data show that low-dose irradiation (≤ 22 Gy) is usually the first-line therapy and adequate for most cases of isolated HPR LCH with sporadic recurrence of illness [1, 13, 15]. Even though it has been suggested that the course of LCH cannot be changed by the available therapy [24], many literature data show that radiotherapy applied in the early stages may be effective, with partial or even complete remission [15, 25]. Remnant tumour in our patient was not significantly reduced after radiotherapy; however, during the 7-year follow-up, we registered no tumour growth along with absence of the posterior pituitary bright spot, nor was there progression of the hypopituitarism. Single-agent chemotherapy, most commonly vinblastine [1], or multiagent chemotherapy is usually the first choice in patients with multifocal LCH and/or a progressive disease course [26]. In some patients with chemoresistant disease, cyclosporin A has been reported to be effective [27]. In the case of disease relapse, choice of therapeutic approach will depend on the coexisting LCH sites (bone, skin, lung or lymph nodes). Management of HPR LCH and multisystem LCH can be via chemotherapy alone or with radiation therapy and surgical excision. Our patient with 7-year relapse-free survival is an example suggesting that surgical excision, followed by low-dose radiation therapy, should be considered as a safe and effective therapeutic approach for isolated HPR LCH. Reviewed cases of Langerhans cell histiocytosis of the sellar region are presented in Table 2.
Conclusions
Diagnosis of localised HPR LCH in adults, especially without DI, is a considerable challenge. Clinical presentation can be a mixture of symptoms and signs caused by pituitary hormone excess or deficiencies, non-endocrine hypothalamic dysfunction, or tumour mass effect. There are no specific MRI features that are helpful in distinguishing LCH from germinoma, which means that a histological examination is essential for the final diagnosis. Anterior pituitary deficiency can appear without DI and not only as a consequence of LCH treatment. This endocrine abnormality should be considered in all patients with LCH, which subsequently should be closely followed up and regularly treated by means of substitution therapy.
References
Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ (1999) Langerhans cell histiocytosis. Diagnosis, natural history, management and outcome. Cancer 85(10):2278–2290
Broadbent V, Dunger D, Yeomans E, Kendall B (1993) Anterior pituitary function and computed tomography/magnetic resonance imaging in patients with Langerhans cell histiocytosis and diabetes insipidus. Med Pediatr Oncol 21(9):649–654
Writing Group of the Histiocyte Society (1987) Histiocytosis syndromes in children. Lancet 1(8526):208–209
Malaps SJ (1998) Langerhans cell histiocytosis in adults. Hematol Oncol Clin North Am 12(2):259–268
Malaps SJ, Norton AJ (1996) Langerhans cell histiocytosis in the adult. Med Pediatr Oncol 27(6):540–546
Gramatovici R, D’ Angio JG (1988) Radiation therapy in soft-tissue lesions in histiocytosis X (Langerhans cell histiocytosis). Med Pediatr Oncol 16(4):259–262
Vawter G, GreenbergerJS CAC (1986) A retrospective clinicopathologic analysis of diabetes insipidus in dendritic cell diseases. Med Pediatr Oncol 14(4):112
Grios N, Favara B, Mostbeck H, Prayer D (1998) Central nervous disease in Langerhans cell histiocytosis. Haematol Oncol Clin North Am 12(2):287–297
Dunger DB, Broadbent V, Yeoman E et al (1989) The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 321(17):1157–1162
Modan-Moses D, Weintraub M, Meyerovitch J, Segal-Lieberman G, Bielora B (2001) Hypopituitarism in Langerhans cell histiocytosis: seven cases and literature review. J Endocrinol Investig 24(8):612–617
French Langerhans Cell Histiocytosis Group (1996) A multicentric retrospective survey of Langerhans cell histiocytosis: 348 cases observed between 1983 and 1993. Arch Dis Child 75(1):17–24
Willis B, Ablin A, Weinberg V, Zoger S, Wara WM, Matthay KK (1996) Disease course and late sequelae of Langerhans cell histiocytosis: 25-year experience at the University of California, San Francisko. J Clin Oncol 14(7):2073–2082
Isoo A, Ueki K, Ishida T et al (2000) Langerhans cell histiocytosis limited to the pituitary-hypothalamic axis—two case reports. Neurol Med Chir (Tokyo) 40(10):532–535
Crespo-Rodriguez AM, Franco C, Lidon MC, Izquierdo B, Angulo E, Mazas-Artasona LV (2004) Hypothalamic-infundibular histiocytosis: magnetic resonance findings. Rev Neurol 39(2):125–129
Kaltsas GA, Powles TB, Evanson J et al (2000) Hypothalamo-pituitary abnormalities in adult patients with Langerhans cell histiocytosis: clinical, endocrinological and radiological features and response to treatment. J Clin Endocrinol Metab 85(4):1370–1376
d’Avella D, Giusa M, Blandino A, Angileri FF, La Rosa G, Tomasello F (1997) Microsurgical excision of a primary isolated hypothalamic eosinophilic granuloma. J Neurosurg 87(5):768–772
Horn EM, Coons SW, Spetzler RF, Rekate HL (2006) Isolated Langerhans cell histiocytosis of the infundibulum presenting with fulminant diabetes insipidus. Childs Nerv Syst 22(5):542–544
Ghafoori SH, Mohseni SH, Larijani B, Mohajeri-Tehrani MR (2015) Pituitary stalk thickening in a case of Langerhans cell histiocytosis. Arch Iran Med 18(3):193–195
Lian C, Lu Y, Shen S (2016) Langerhans cell histiocytosis in adults: a case report and review of the literature. Oncotarget 7(14):18678–18683
Tabarin A, Corcuff JB, Dautheribes M et al (1991) Hystiocytosis X of the hypothalamus. J Endocrinol Investig 14(2):139–145
Chen ED, Cheng P, Cai YF et al (2014) Ultrasonographic features of Langerhans cell histiocytosis of the thyroid. Int J Clin Exp Pathol 7(3):1229–1235
Dean HJ, Bishop A, Winter JSD (1986) Growth hormone deficiency in patients with histiocytosis X. J Pediatr 109(4):615–618
Asano T, Goto Y, Kida S, Ohno K, Hirakawa K (1999) Isolated histiocytosis X of the pituitary stalk. J Neuroradiol 26(4):277–280
Kilpatrick SE, Wenger DE, Gilchrist GS, Shives TC, Wollan PC, Unni KK (1995) Langerhans’ cell histiocytosis (histiocytosis X) of bone. Cancer 76(12):2471–2484
Rosenzweig KE, Arceci RJ, Tarbell NJ (1997) Diabetes insipidus secondary to Langerhans’ cell histiocytosis: is radiation therapy indicated? Med Pediatr Oncol 29(1):36–40
Gadner H, Heitger A, Grois N, Gatterer-Menz I, Ladisch S (1994) Treatment strategy for disseminated Langerhans cell histiocytosis. Med Pediatr Oncol 23(2):72–80
Mahmoud H, Wang W, Murphy S (1991) Cyclosporin therapy for advanced Langerhans cell histiocytosis. Blood 77(4):721–725
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Procedures, data collection and presentation were approved by the hospital ethics committee (decision no.: 37232/1). Written informed consent was obtained from the patient to participate and for publication of this case report and any accompanying data.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Radojkovic, D., Pesic, M., Dimic, D. et al. Localised Langerhans cell histiocytosis of the hypothalamic-pituitary region: case report and literature review. Hormones 17, 119–125 (2018). https://doi.org/10.1007/s42000-018-0024-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42000-018-0024-6