
Abstract
The active pharmaceutical ingredient sacubitril acts as a neprilysin inhibitor in the body and is administered to patients suffering from high blood pressure and chronic heart failure. In this paper, we report the development of a three-step setup for the synthesis of an advanced sacubitril precursor in continuous flow. The key transformation of our cascade is a Suzuki-Miyaura coupling facilitated by a heterogeneous palladium catalyst. Its implementation in a packed-bed reactor and the application of continuous flow methodologies allow intensification of the cross-coupling reaction compared to batch processing. The subsequent steps for the synthesis of the target molecule involve Boc-deprotection as well as N-succinylation, which have been optimized using the statistical “Design of Experiments” (DoE) approach. In this way, the individual as well as interactive effects of selected parameters on the output of the reactions could be investigated very efficiently. The consecutive performance of the three reaction steps using an integrated setup enabled the synthesis of a late-stage sacubitril precursor in continuous flow with 81% overall yield.

.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among the different medication options for the treatment of hypertension and related cardiovascular complaints [1], drugs affecting the renin-angiotensin-aldosterone system have proved to be highly effective [1]. One prominent representative of this class of agents is LCZ696 (Novartis International AG, Basel, Switzerland), which was approved by the FDA in 2015 for the treatment of heart failure in patients with reduced ejection fraction [2]. The fixed-dose combination drug marketed under the brand name Entresto® comprises the active pharmaceutical ingredient (API) valsartan, an angiotensin II-receptor antagonist, and the prodrug sacubitril 1 [3]. The latter compound is metabolized in the body to an active neprilysin inhibitor upon cleavage of its ethyl ester moiety [2]. The synthesis of sacubitril 1 was first reported by Ksander et al. [4] in 1995, starting from an unnatural d-amino acid derivative 3. Formation of the main chiral intermediate 2 was achieved in 8 linear steps and involved a Wittig reaction as well as stereoselective hydrogenation (Scheme 1, a). An alternative route, also running via formation of intermediate 2, was developed by Hook et al. [5], who used l-pyroglutamic acid methyl ester 4 as chiral pool for the synthesis of 2 in batch (Scheme 1, b). Recently, Ley et al. [6] published a novel approach applying the concept of continuous flow technology and integrating machine-assisted methods for the synthesis of hydrochloride salt 2*HCl. In contrast to the other groups, their strategy employs exclusively achiral starting materials 5–7 and the two chiral centers are installed by both Rh-catalyzed stereoselective hydrogenation and diastereoselective Reformatsky-type carbethoxyallylation. By a combination of two batch steps and five transformations in continuous flow, they obtained the target molecule in 54% overall yield (Scheme 1, c).

Apart from the mentioned approaches involving the formation of chiral intermediate 2, two alternative strategies for the synthesis of sacubitril 1 have been reported in literature. Firstly, Xu et al. [7] achieved formation of 1 starting from Bettibase and (S)-2-methyloxobutanoic acid, which yielded oxazolidine 8. Upon stereoselective addition of a Grignard reagent to the obtained chiral intermediate 8, the biphenyl motif is installed and 3 more steps finally give the active pharmaceutical ingredient 1 (Scheme 1, d). Secondly, the group of Wang [8] developed a chiron-based approach relying on the protected triol 9. The key transformations of their process are epoxide formation as well as an one-flask Staudinger reduction/succinic amide formation, which facilitate formation of sacubitril 1 in a total of 7 steps (Scheme 1, e).
Regarding the synthesis of sacubitril 1 on an industrial scale, Novartis International AG (Basel, Switzerland) holds various patents covering different synthetic routes. Their earlier patents comprise chiral pool approaches, similar to the cascades established by Ksander [9] and Hook [10], for formation of the two chiral centers present in the API 1. However, researchers at the Swiss pharmaceutical company have recently developed a novel organocatalytic strategy for installation of the stereocenters [11]. The first option described in the patent involves the reaction of Michael acceptor 10 with propionaldehyde employing a proline-derived organocatalyst, which is high-yielding but unsatisfactory in terms of diastereoselectivity (Scheme 2, a). Formation of the desired diastereomer 12 could be improved by the use of nucleophile 11 in combination with methacrolein and a thiourea-derived catalyst (Scheme 2, b). However, in view of the potential application of organocatalysis for the synthesis of sacubitril 1 on a large scale, diastereoselectivity still needs to be improved to meet the requirements of the pharmaceutical industry.

Recent organocatalytic approaches for the synthesis of sacubitril 1 via chiral intermediate 12 patented by Novartis [11]
Although apparently a number of different strategies for the synthesis of sacubitril 1 have been developed in the past years, it is conspicuous that almost every approach relies on traditional batch chemistry. As mentioned above, the sole exception is the combined batch and flow process published by Ley and his group [6], involving 5 transformations in continuous flow. Continuous flow methodology offers a plethora of opportunities for the pharmaceutical industry in terms of both process optimization and economics. On the one hand, flow processes benefit from a rapid heat and mass transfer as well as the opportunity for automation and process intensification [exemplarily 12, 13], in this way opening novel process windows [14]. Furthermore, improved energy efficiency, high safety and reduced waste generation associated with continuous processing [13, 15, 16] align with the principles of green and sustainable chemistry [17]. On the other hand, flow chemistry allows to reduce development times and to monitor the product quality in-line, thus saving up to 30% costs in comparison with an equivalent batch process [18,19,20]. In view of the numerous arguments speaking in favor for the implementation of continuous flow operations, it is not surprising that continuous manufacturing becomes an increasingly important topic for the pharmaceutical industry [21,22,23,24] and is also supported by the Federal Drug Administration (FDA) [25, 26].
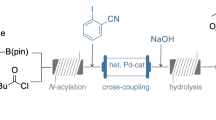
Accordingly, as part of the ONE-FLOW research project [27], we are aiming for the development of catalyzed cascade reactions for the formation of active pharmaceutical ingredients. Recently, our group has demonstrated the applicability of flow methodology for the multistep synthesis of an advanced valsartan precursor in a continuous fashion [28]. Hence, our next goal was to prove our concept also for the integrated synthesis of a late-stage sacubitril intermediate 16 in continuous flow, which we report in this paper. The key step of our envisaged reaction scheme, inspired by the paper of Ksander et al. [4], is Suzuki-Miyaura cross-coupling of functionalized sacubitril intermediate 13 (obtained via a three-step batch synthesis procedure, see Supporting Information for experimental details) with phenylboronic acid facilitated by the heterogeneous palladium catalyst Ce0.99-xSnxPd0.01O2-δ [28,29,30,31]. Subsequently, deprotection of the obtained biphenyl compound 14 and N-amidation of the free amine 15 with succinic anhydride yielded the targeted sacubitril precursor 16 (Scheme 3).

Targeted three-step approach for the integrated synthesis of sacubitril precursor 16 in continuous flow
Results and discussion
Optimization of the individual steps in continuous flow
Palladium-catalyzed C-C cross-coupling reactions have become indispensable in modern organic chemistry [32,33,34]. Due to its remarkable versatility, functional-group tolerance as well as selectivity, palladium catalysis is long since a key component in the toolbox of the pharmaceutical industry [35,36,37]. In particular, the Suzuki-Miyaura reaction between aryl halides and boronic acid derivatives plays an important role for the formation of biphenyl motifs [38,39,40]. In the past couple of years, our group has gained experience with Suzuki-Miyaura reactions in continuous flow facilitated by heterogeneous Pd-Ce-Sn-oxides [28,29,30,31]. Reviewing published literature procedures for the synthesis of sacubitril 1, only the route developed by Ksander et al. [4] comprises a Suzuki coupling step and served as our point of reference. First, we aimed for the identification of a successful cross-coupling reaction facilitated by the class of heterogeneous palladium catalysts with the molecular formula Ce0.99-xSnxPd0.01O2-δ (x = 0, 0.20, 0.495, 0.79, 0.99) [29] developed and available in our lab. Their successful utilization for the Suzuki coupling of aryl halides with boronic acid derivatives in batch as well as in continuous flow has already been reported by our group [28,29,30,31]. In previous studies and preliminary experiments, the catalyst Ce0.20Sn0.79Pd0.01O2-δ proved to be the best choice for the Suzuki cross-coupling of advanced chemical intermediates [28]. Therefore, we tested the applicability of the respective palladium catalyst (standard reaction conditions: 1 mol eq. aryl halide, 1.5 mol eq. phenylboronic acid, 1.5 mol eq. K2CO3, iPrOH:H2O = 7:3, 75 °C) for Suzuki couplings employing different cross-coupling partners. In doing so, the C-C bond formation between functionalized aryl iodide 13 and phenylboronic acid (Scheme 3, step 1) emerged to be superior and was chosen to be the key step of our reaction cascade for formation of a sacubitril precursor. Our reaction scheme was completed by Boc-deprotection of the resulting biphenyl derivative 14 as well as reaction of amine 15 with succinic anhydride, giving the sacubitril precursor 16 (Scheme 3).
Next, we had to select a suitable solvent for our targeted reaction cascade, meeting different requirements for the individual reaction steps. Concerning the Suzuki coupling step using the chosen type of catalyst, the presence of water is crucial for the reaction to occur [28, 41]. In particular, we experienced that our catalyst favors an aqueous single-phase reaction environment [28]. Regarding the solubility of substrates and reaction intermediates as well as stability of the succinic anhydride reagent in the third step, an aprotic organic cosolvent was needed. For that purpose, acetonitrile (MeCN), tetrahydrofuran as well as dioxane came into question. For toxicity as well as miscibility reasons [42, 43], we decided to use acetonitrile:water as the reaction medium for the synthesis of sacubitril precursor 16. For the first step of our cascade, Suzuki cross-coupling, a ratio of acetonitrile:water = 65:35 (v/v) turned out to provide sufficient solubility of substrate 13 (25 mM) while keeping the present potassium carbonate dissolved.
After having identified the preferred reaction medium for our targeted synthesis of sacubitril precursor 16, we aimed to optimize the individual steps of our reaction cascade in continuous flow. Initially, we focused on the first step of the reaction cascade, the Suzuki cross-coupling reaction. Previous studies [28,29,30,31] showed that Suzuki couplings employing catalyst Ce0.20Sn0.79Pd0.01O2-δ work best using the respective aryl halide in combination with both 1.5 mol eq. of boronic acid species and potassium carbonate. Hence, we adopted that approach for the targeted reaction of sacubitril intermediate 13 with phenylboronic acid in a continuous fashion. More specifically, we wanted to study the effect of the reaction temperature on measured conversion of substrate 13. For the experiment, a solution containing the cross-coupling partners and the inorganic base potassium carbonate in a mixture of acetonitrile:water = 65:35 (v/v) was pumped through an HPLC column (L x I.D. 50 × 4.6 mm) filled with particles of palladium catalyst Ce0.20Sn0.79Pd0.01O2-δ (Scheme 4, see Experimental section for details). Using a flow rate of 0.05 mL/min, the mean residence time inside the packed-bed reactor was determined to be 18.4 min regarding the measurement of a tracer-determined residence time distribution curve. According to literature [44], the employed reaction solvent starts boiling at a temperature of approximately 77 °C. However, one of the major benefits of flow chemistry compared to batch processing is the ability to heat the reaction mixture above its boiling point in a pressurized system and thus to accelerate the reaction rate [12, 13]. Hence, we evaluated the performance of our setup at 75, 100 and 125 °C, maintaining a stable pressure of 75 psi with a back pressure regulator. In doing so, a significant increase in measured conversion could be observed with higher temperature. Whereas at a temperature of 75 °C conversion of 13 was determined to be only 59% (Entry 1, Table 1), an increase to 100 °C led to an improved conversion of 84% (Entry 2, Table 1). Best results were obtained at a reaction temperature of 125 °C, giving almost quantitative conversion of 13 using the abovementioned setup (96%, Entry 3, Table 1). Isolation of biphenyl 14 synthesized in continuous flow at a temperature of 125 °C proved that only minor racemization of the chiral center occurs under the employed reaction conditions (95% enantiomeric excess, see Supporting Information for determination of enantiomeric excess by chiral HPLC).

Suzuki-Miyaura cross-coupling for formation of sacubitril intermediate 14 in continuous flow facilitated by Pd-catalyst Ce0.20Sn0.79Pd0.01O2-δ
After having demonstrated the applicability of our packed-bed reactor setup for the key Suzuki coupling reaction of our targeted cascade for the synthesis of 16, we turned our attention to the subsequent steps Boc-deprotection and N-amidation. Since we did not have a substantial amount of biphenyl intermediate 14 in hand, we wanted to keep its consumption to a minimum. Therefore, we decided to use readily available surrogate substrates for the optimization of steps 2 and 3 in continuous flow, comprising similar core structures as sacubitril precursor 14. Thus, we chose to study the Boc-deprotection of Boc-l-phenylalanine methyl ester 17 (Fig. 1) as well as N-amidation of l-phenylalanine methyl ester 18 (Fig. 2) using a coil reactor setup. Considering the fact that the outcome (e.g. conversion, yield) of a chemical transformation is the result of a number of interacting reaction parameters, the traditional one-variable-at-a-time optimization is not the most effective method to investigate the experimental space [45, 46]. For this reason, we decided to use an alternative optimization approach employing “Design of Experiments” (DoE), which is a branch of applied statistics. DoE allows the determination of individual as well as combined effects of different influencing factors on the output of an experiment. Furthermore, using DoE the required information for optimization of a given process can be obtained with the minimum amount of experimentation [46]. Being developed already in the 1920s based on the work of R. A. Fisher [47], DoE has been used extensively in various sectors including car manufacturing, biotech and steel industry [46]. In recent years, also the pharmaceutical industry has increasingly taken advantage of DoE for the efficient identification of optimal process parameters for their manufacturing processes [45, 48,49,50].

Reaction scheme and contour plot obtained by DoE analysis of Boc-deprotection of Boc-l-phenylalanine methyl ester 17 in continuous flow with MODDE Pro

Reaction scheme and contour plot obtained by DoE analysis of N-amidation of l-phenylalanine methyl ester 18 in continuous flow with MODDE Pro
Regarding the Boc-deprotection of compound 17, we first had to define how we wanted to achieve the removal of the protecting group. In view of the performance of the deprotection step in targeted cascade for the continuous synthesis of sacubitril precursor 16 after the Suzuki coupling step, the transformation had to be performed in an acetonitrile:water solution. Preliminary batch experiments showed that the deprotection of 17 utilizing commonly employed trifluoroacetic acid [51, 52] is relatively slow in aqueous media. Likewise, published deprotection on a strongly acidic ion exchange resin [53, 54] did not prove to be satisfyingly successful in water-containing solvent mixtures. In our case, best results were obtained using aqueous hydrochloric acid for removal of the protecting group. The same strategy has also been reported for achieving Boc-removal on a large scale for the synthesis of APIs in batch [55] as well as in continuous flow [24].
Considering the optimization of our Boc-deprotection step, we decided to examine the influence of the factors temperature as well as mol eq. hydrochloride (HCl) on the conversion of compound 17. Applying the Design of Experiments approach, we performed the Boc-deprotection of 17 in continuous flow at three different temperatures (70, 75, 80 °C) and varied the amount of hydrochloric acid (5, 10, 15 mol eq.) in the experiments. As we decided to utilize a full factorial DoE, we determined the conversion of 17 resulting of every possible combination of the two selected reaction parameters. Analysis of the collected experimental data with the software MODDE Pro revealed a statistically significant impact of the two interacting variables on the conversion of 17, which is visualized in Fig. 1. As expected, the contour plot illustrates an increase in conversion both at higher reaction temperature and using a larger excess of hydrochloric acid. However, the reaction temperature seems to influence the outcome of the reaction more strongly than the excess of hydrochloric acid. Exemplarily, the model suggests that using 5 mol eq. HCl the conversion can be increased from 50% to 80% by raising the temperature only by about 8 °C. To achieve the same boost in conversion at a given temperature of 70 °C, more than double the HCl concentration would be necessary. Regarding our multistep cascade for the synthesis of 16, we wanted to achieve quantitative conversion utilizing 10 mol eq. of acid. The obtained DoE model led us to expect that quantitative conversion of 17 using 10 mol eq. of HCl could be realized at a temperature of 85 °C. Indeed, the performance of an additional experiment (T = 85 °C, 10 mol eq. HCl) confirmed this assumption (see Supporting Information for a detailed description of conducted DoE study, model parameters as well as experimental data).
Concerning the reaction of nucleophiles with succinic anhydride for the synthesis of active pharmaceutical ingredients in continuous flow, O-succinylation has already been achieved using the anhydride reagent in combination with a tertiary amine base employing a tube reactor [56]. Apart from that, N-succinylation in batch has also been performed in aqueous media at room temperature [57, 58]. However, the presence of water induces the hydrolysis of succinic anhydride, which has a half-life of roughly 5 min (pH 7, RT) [59, 60]. Consequently, the targeted amidation reaction has to be faster than the hydrolysis of the anhydride reagent in order to be successful.
In terms of the continuous N-amidation of 18 in an acetonitrile:water medium at room temperature, we chose to determine the impact of the variables mol eq. succinic anhydride as well as mol eq. diisopropylethylamine (DIPEA) on the reaction outcome. In compliance with full-factorial DoE, we measured the conversion of 18 resulting from every combination of the two influencing factors (1, 1.5, 2 mol eq. succinic anhydride; 0.5, 1, 1.5 mol eq. DIPEA). Evaluation of collected data with MODDE Pro suggested a statistical relevant influence of both parameters on the conversion and revealed also interactive effects. The visual illustration of the calculated mathematical model is depicted in Fig. 2. Within the covered experimental range, maximum conversion is obtained using 2 mol eq. of succinic anhydride along with 1 mol eq. of DIPEA (see Supporting Information for a detailed description of conducted DoE study, model parameters as well as experimental data).
Multistep continuous setup for the synthesis of 16
Following the intensification of the Suzuki coupling of sacubitril precursor 13 as well as proving the feasibility of Boc-deprotection and N-amidation in a continuous fashion, we targeted the integrated synthesis of sacubitril precursor 16 in a fully continuous setup (Scheme 5). As outlined above, Suzuki-Miyaura cross-coupling of 13 was performed at a temperature of 125 °C employing a packed-bed reactor (L x I.D. 50 × 4.6 mm) containing the heterogeneous palladium catalyst Ce0.20Sn0.79Pd0.01O2-δ. Subsequently, Boc-deprotection of obtained biphenyl intermediate 14 was achieved by merging the reaction stream with a solution of hydrochloric acid and introducing it into a coil reactor (L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in.). According to the optimization of Boc-removal by DoE, the free amine 15 was generated using 10 mol eq. HCl and a reaction temperature of 85 °C. To take the partial neutralization of hydrochloric acid by present potassium carbonate (1.5 mol eq.) into account, a stock solution containing 11.5 mol eq. HCl was utilized for this purpose. Since the large excess of hydrochloric acid in the reaction solution is detrimental for the following third step of the reaction cascade, the acid was removed deploying anion exchange resin Amberlyst A21 implemented in an HPLC column (L x I.D. 120 × 8 mm). The same approach has already been reported in literature for the removal of trifluoroacetic acid from organic solvents in batch [61]. Finally, as suggested by DoE analysis, N-amidation of 15 was accomplished in a coil reactor (L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in.) after addition of 2 mol eq. succinic anhydride as well as 1 mol eq. DIPEA. Using this multistep setup (Scheme 5) exhibiting a total residence of about 90 min (including tubing), the synthesis of the late-stage sacubitril precursor 16 was successfully performed for over 5 h yielding the targeted compound with up to 98% yield. After a steady increase in the beginning, the yield of sacubitril precursor 16 was observed to slowly decrease over the course of the experiment and stabilized at approximately 81% (corresponding to a space-time-yield of 4 g/L*h). after 370 min (see Supporting Information for graphic representation). This can probably be contributed to an initial loss of palladium due to leaching from the employed heterogeneous catalyst, which has already been reported in literature [28, 30]. Determination of the residual levels of palladium, cerium and tin in the reaction solution by ICP-MS strongly support this hypothesis (Table 2). Whereas the reaction outlet collected at t = 160–180 min exhibits a palladium content of 563 ± 13 μg/kg (Entry 1, Table 2), the concentration of the metal was found to significantly decrease over the course of the continuous experiment. After a run time of 400 min, the amount of palladium was determined to be reduced to 105 ± 3 μg/kg (Entry 4, Table 2), which renders our developed multistep process highly promising to meet the strict regulatory guidelines for the production of pharmaceutical products [62]. Concerning the leaching behavior of tin from the employed heterogeneous catalyst, the residual levels in the outlet flow were measured to be ≤30 μg/kg (Entries 1–4, Table 2) in all samples. Likewise, the concentration of cerium was very low in analyzed samples (≤ 9.7 μg/kg, Entries 1–4, Table 2).

Multistep setup for the synthesis of sacubitril precursor 16 in continuous flow (SP = high-pressure syringe pump, S = syringe pump, T = T-mixer, v = 0.05 mL/min)
Apart from that, also the constant reduction of acid scavenging capacity of the deployed ion exchange resin might account for observed course of yield. As far as starting material 13 is concerned, HPLC measurements revealed full conversion throughout the whole experiment. However, it has to be noted that 13 can as well undergo Boc-deprotection and in further consequence N-amidation. Regarding the formation of by-products in the three-step continuous cascade, oxidation and homocoupling of phenylboronic acid are reported to occur upon depletion of the aryl halide coupling partner in the Suzuki coupling step [29]. In fact, the HPLC chromatogram of the outlet flow showed a small peak corresponding to biphenyl resulting from the coupling of two molecules of phenylboronic acid. Apart from that, the reaction steps seemed to be rather clean and only small peaks attributable to unidentified side-products could be detected by HPLC (λ = 237 nm) at first. However, after an experiment run time of 260 min HPLC analysis showed the appearance of a peak at 0.7 min, which seemed to increase with decreasing yield. The molecular structure of the corresponding compound could not have been elucidated so far.
Concerning the absence of other significant by-product peaks in HPLC chromatogram, the use of weakly basic anion exchange resin Amberlyst A21 for the adsorption of excess hydrochloric acid can also be regarded as an in-line purification step. It removes various kinds of acidic materials from the process stream including potentially formed hydroiodic acid [63] resulting from the use of an aryl iodide coupling partner in the Suzuki reaction as well as phenol originating from the oxidation of phenylboronic acid [64]. In addition, we observed that in our multistep continuous cascade Amberlyst A21 acts as a scavenger of unreacted phenylboronic acid too. Moreover, the respective anion exchange resin was reported to adsorb Pd(II) from aqueous solutions [65, 66] and might therefore contribute to the observed low levels of palladium in the outlet flow of the continuous setup.
Besides the reaction yield, another important quality attribute of pharmaceutical processes is the enantiopurity of the obtained target molecule. Enantiomeric excess of compound 16 synthesized in continuous flow utilizing the described integrated setup was determined to be moderate 43% (see Supporting Information for determination of enantiomeric excess by chiral HPLC). As biphenyl intermediate 14 obtained in continuous Suzuki cross-coupling was shown to be almost enantiopure, the partial racemization of the chiral center most likely occurred during the removal of the protecting group. Boc-deprotection was performed in an aqueous and strongly acidic environment at elevated temperature, conditions which presumably caused racemization of the sacubitril precursor to some extent [67, 68]. Therefore, in order to provide a process suitable for pharmaceutical industry, future work will focus on improving the enantiomeric excess of sacubitril precursor 16 obtained in developed continuous cascade by applying milder conditions for the Boc-deprotection step.
Conclusion
In summary, we have successfully applied continuous flow techniques for the synthesis of a late-stage sacubitril precursor in three steps. The optimization of interacting reaction parameters was demonstrated to be efficiently performed using statistical “Design of Experiments”. Furthermore, the use of a continuous setup comprising a packed-bed reactor as well as coil reactor modules allowed intensification of the multistep process compared to batch. After an initial equilibration phase, the target molecule was obtained with a steady 81% overall yield and moderate enantiomeric excess using an integrated continuous setup. Moreover, levels of residual palladium in the outlet flow were determined to be in the ppb range, thus proving our developed process to be attractive for pharmaceutical applications. However, in view of a conceivable implementation of developed reaction cascade into an industrial process for API production, its optimization in terms of enantiomeric excess of the final product is indispensable. Nevertheless, we could prove once more that with relatively simple equipment it is possible to synthesize advanced chemical intermediates in continuous flow. With our work, we hope to contribute to the establishment of continuous flow methodologies for process development and operation in pharmaceutical industry.
Experimental section
General information
Chemicals and solvents were purchased from commercial suppliers and used as received unless stated otherwise [Sigma Aldrich: potassium carbonate (99%), anisole (99%); LiAlH4 (1 M in diethyl ether), trifluoroacetic acid (99%), Boc-l-phenylalanine methyl ester (98%), l-phenylalanine methyl ester hydrochloride (98%), Amberlyst A21 (4.6 eq./kg dry weight); BLDpharm: Boc-4-iodo-d-phenylalanine (98%); Fluorochem:O,N-dimethylhydroxylamine hydrochloride (98%), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (99%), ethyl(hydroxyimino)cyanoacetate (98%), (carbethoxyethylidene)triphenylphosphorane (94%), succinic anhydride (99%); Carl Roth: triethylamine (99.5%), hydrochloric acid (37%); Oxchem: phenylboronic acid (95%); ACROS organics: diisopropylethylamine (98%); ChemLab: acetonitrile (HPLC grade)]. According to a reported literature procedure [69], l-phenylalanine methyl ester hydrochloride was neutralized by addition of sodium carbonate in water and extraction of the aqueous phase with ethyl acetate yielded free l-phenylalanine methyl ester. Analytical thin layer chromatography was performed on pre-coated aluminium plates (Merck, silica gel 60, F254) and spots were visualized with UV light (254 nm) or potassium permanganate stain. Column chromatography purifications were carried out using MN silica gel 60 (70–230 mesh). An Agilent 1100 series HPLC system was utilized for monitoring of the reaction progress as well as determination of enantiomeric excess (see Supporting Information for HPLC parameters). HPLC-MS measurements were performed on a Waters Acquity H-Class system equipped with a Waters Acquity SQD detector. For determination of Pd, Ce and Sn in the outlet flow of the multistep reaction cascade in continuous flow, an Agilent 7700x ICP-MS system was employed after microwave assisted acidic digestion of the samples. NMR-data were recorded on a Bruker Avance III 300 MHz spectrometer (1H: 300 MHz, 13C: 75 MHz, see Supporting Information for NMR-data). For the DoE analysis of Boc-deprotection and N-amidation in continuous flow, the software MODDE Pro (Version 12.1.0.5491, Copyright © Sartorius Stedim Data Analytics AB) was utilized.
Optimization of Suzuki cross-coupling in continuous flow
For the Suzuki coupling of intermediate 13 with phenylboronic acid in continuous flow, a packed-bed reactor setup was utilized. Therefore, an HPLC column (L x I.D. 50 × 4.6 mm) was filled with dry Pd-catalyst Ce0.20Sn0.79Pd0.01O2-δ (602 mg, containing 0.04 mmol Pd) and connected to a back pressure regulator (IDEX BPR Cartridge 75 psi Gold Coat). After equilibration of the reactor setup with the reaction solvent (MeCN:H2O = 65:35), the packed-bed reactor was heated to elevated temperature (75–125 °C) in an oil bath. Then, a solution containing intermediate 13 (25 mM), phenylboronic acid (37.5 mM, 1.5 mol eq.), potassium carbonate (37.5 mM, 1.5 mol eq.) and anisole (62.5 mM) as internal standard was prepared in MeCN:H2O = 65:35, degassed in an ultrasonic bath and filtered through a 0.2 μm membrane filter. Obtained stock solution was pumped through the reactor with a flow rate of 0.05 mL/min (τ = 18.4 min) utilizing a high-pressure syringe pump (VIT-FIT HP, Lambda Instruments). For monitoring of the reaction progress, every 10 min an aliquot (40 μL) was quenched with MeOH:H3PO4 = 75:25 (400 μL) and analyzed by HPLC (Method B).
Optimization of Boc-deprotection in continuous flow
The influence of temperature and equivalents of acid on Boc-deprotection was studied using Boc-l-phenylalanine methyl ester 17 as surrogate substrate utilizing a coil reactor setup. Solution A [Boc-l-phenylalanine methyl ester (25 mM), anisole (62.5 mM) as internal standard] and solution B [HCl (125–375 mM, 5–15 mol eq.)] were prepared in the reaction solvent MeCN:H2O = 65:35 and degassed by ultrasonic irradiation. The setup comprising a PEEK coil (L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in., V = 1.37 mL) connected to a back pressure regulator (IDEX BPR Cartridge 75 psi Gold Coat) was equilibrated with solvent and heated to elevated temperature in a water bath (70–85 °C). The two reagent solutions were merged using a T-mixing element and pumped through the reactor by means of high-pressure syringe pumps (VIT-FIT HP, Lambda Instruments) at a flow rate of 0.05 mL/min each, giving a total flow rate of 0.10 mL/min (τ = 13.7 min). After certain time points (3τ / 3.5 τ / 4 τ), aliquots (80 μL) of the outlet flow were quenched with MeOH:H3PO4 = 55:45 (400 μL) and analyzed by HPLC (Method A).
Optimization of N-amidation in continuous flow
The influence of different equivalents of succinic anhydride as well as DIPEA on N-amidation of l-phenylalanine methyl ester 18 was examined in a coil reactor setup. Solution A [l-phenylalanine methyl ester (12.5 mM), anisole (31.3 mM) as internal standard] was prepared in MeCN:H2O = 65:35, whereas solution B [succinic anhydride (25–50 mM, 1–2 mol eq.), DIPEA (12.5–37.5 mM, 0.5–1.5 mol eq.)] was prepared in anhydrous MeCN to avoid hydrolysis of the anhydride reagent. The stock solutions were degassed in an ultrasonic bath and the reactor setup composed of a PEEK coil (L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in., V = 1.37 mL) was equilibrated with the reaction solvent at ambient pressure and temperature. Stock A (flow rate 0.10 mL/min) and stock B (flow rate of 0.05 mL/min) were merged using a T-mixing element and the reaction solution was pumped through the reactor coil at a total flow rate of 0.15 mL/min (τ = 9.1 min) using high-pressure syringe pumps (VIT-FIT HP, Lambda Instruments). After certain time points (3τ / 3.5 τ / 4 τ), aliquots (120 μL) of the outlet flow were quenched with MeOH:H3PO4 = 55:45 (400 μL) and analyzed by HPLC (Method A).
Integrated synthesis of 16 in continuous flow
For the multistep synthesis of sacubitril precursor 16 in continuous flow, a modular setup comprising a packed-bed reactor and reactor coils was utilized. Stock A [(intermediate 13 (25 mM), phenylboronic acid (37.5 mM, 1.5 mol eq.), potassium carbonate (37.5 mM, 1.5 mol eq.) and anisole (62.5 mM) as internal standard] was prepared in MeCN:H2O = 65:35, dissolved by ultrasonic irradiation and filtered through a 0.2 μm membrane filter. Stock B [HCl (287.5 mM, 11.5 mol eq.)] and stock C [succinic anhydride (50 mM, 2 mol eq.), DIPEA (25 mM, 1 mol eq.)] were prepared in MeCN:H2O = 65:35 and MeCN, respectively, and degassed in an ultrasonic bath. After equilibration of the reactor system, stock A was pumped through the packed-bed reactor (L x I.D. 50 × 4.6 mm) filled with Pd-catalyst Ce0.20Sn0.79Pd0.01O2-δ (594 mg, containing 0.04 mmol Pd) at a flow rate of 0.05 mL/min (τ = 18.4 min) and a temperature of 125 °C using a high-pressure syringe pump (VIT-FIT HP, Lambda Instruments) for Suzuki-Miyaura cross-coupling. Then, the outlet flow was mixed with stock B, delivered by a second high-pressure syringe pump (VIT-FIT HP, Lambda Instruments), at a flow rate of 0.05 mL/min using a T-mixing element. The resulting reaction solution was introduced into the first coil reactor (PEEK coil, L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in., V = 1.37 mL) at a total flow rate of 0.10 mL/min and a temperature of 85 °C (τ = 13.7 min) for Boc-deprotection. After the first reaction coil, a backpressure regulator (IDEX BPR Cartridge 75 psi Gold Coat) was installed to be able to heat above the boiling point (~77 °C, [44]) of the reaction solution. For removal of excess HCl, the reagent stream was then pumped through a preparative HPLC column (L x I.D. 120 × 8 mm, τ = 46.9 min) filled with the anion exchange resin Amberlyst A21 (2 g dry weight) at ambient temperature and pressure. Subsequently, the process stream was mixed with stock C, which was pumped into the setup by means of a dual-syringe infusion pump (LA-120, Landgraf) at a flow rate of 0.05 mL/min. N-amidation was then performed in a second coil reactor (PEEK coil, L x O.D. x I.D. 3.0 m × 1/16 in. × 0.030 in., V = 1.37 mL) at a total flow rate of 0.15 mL/min (τ = 9.1 min) to obtain targeted sacubitril precursor 16 (calculated total residence time = 90.2 min). For monitoring of the reaction progress, every 10 min an aliquot of the outlet flow (120 μL) was quenched with MeOH:H3PO4 = 75:25 (400 μL) and analyzed by HPLC (Method B). To determine the residual levels of Pd, Ce and Sn in the outlet flow of the continuous setup for synthesis of 16 aliquots of the reaction solution were acidified with equal volumes of 20% HNO3/HCl (1 + 1, v/v) and analyzed by ICP-MS after microwave assisted acidic digestion.
References
Dahlöf B (2000) Valsartan and the renin-angiotensin-aldosterone system: blood pressure control and beyond. JRAAS 1:14–16
Vilela-Martin JF (2016) Spotlight on valsartan-sacubitril fixed-dose combination for heart failure: the evidence to date. Drug Des Dev Ther 10:1627–1639
Andersen MB, Simonsen U, Wehland M, Pietsch J, Grimm D (2016) LCZ696 (Valsartan/Sacubitril) - A possible new treatment for hypertension and heart failure. Basic Clin Pharmacol Toxicol 118:14–22
Ksander GM, Ghai RD, de Jesus R, Diefenbacher CG, Yuan A, Berry C, Sakane Y, Trapani A (1995) Dicarboxylic acid dipeptide neutral endopeptidase inhibitors. J Med Chem 38:1689–1700
Hook D, Riss B, Kaufmann D, Napp M, Bappert E, Polleux P, Medlock J, Zangotti-Gerosa A (2009) New processes for γ-amino-δ-biphenyl-α-methylalkanoic acid or acid ester. PCT Int. Appl. WO 2009/090251. Chem Abstr 151:198239
Lau SH, Bourne SL, Martin B, Schenkel B, Penn G, Ley SV (2015) Synthesis of a precursor to sacubitril using enabling technologies. Org Lett 17:5436–5439
Xu X-N (2015) A preparation method of sacubitril. CN Patent CN 104557600 A. Chem Abstr 162:603569
Wang Y, Chen F-E, Shi Y, Tian W-S (2016) Multigram scale, chiron-based synthesis of sacubitril. Tetrahedron Lett 57:5928–5930
Ksander G (1993) Biaryl substituted 4-amino-butyric acid amides. US Patent 5(217):996
Hook D, Ruch T, Riss B, Wietfeld B, Sedelmaier G, Napp M, Bänzinger M, Hawker S, Ciszewski L, Waykole L (2008) New process for preparation of 5-(4-biphenylylmethyl) pyrrolidin-2-one derivatives. PCT Int. Appl. WO 2008/083967 A2. Chem Abstr 149:176170
Martin B, Mandrelli F, Venturoni S-L (2018) Process and intermediates for the preparation of NEP inhibitors. U.S. Pat. Appl. US 2018/0022690 A1. Chem Abstr 165:298456
Plutschack MB, Pieber B, Gilmore K, Seeberger PH (2017) The hitchhiker's guide to flow chemistry. Chem Rev 117:11796–11893
Newman SG, Jensen KF (2013) The role of flow in green chemistry and engineering. Green Chem 15:1456–1472
Hessel V (2009) Process windows - Gate to maximizing process intensification via flow chemistry. Chem Eng Technol 32:1655–1681
Ley SV (2012) On being green: can flow chemistry help? Chem Rec 12:378–390
Wiles C, Watts P (2014) Continuous process technology: a tool for sustainable production. Green Chem 16:55–62
Anastas P, Eghbali N (2010) Green chemistry: principles and practice. Chem Soc Rev 39:301–312
Roberge DM, Zimmermann B, Rainone F, Gottsponer M, Eyholzer M, Kockmann N (2008) Microreactor technology and continuous processes in the fine chemical and pharmaceutical industry: is the revolution underway? Org Process Res Dev 12:905–910
Benaskar F, Ben-Abdelmoumen A, Patil NG, Rebrov EV, Meuldijk J, Hulshof LA, Hessel V, Krtschil U, Schouten JC (2011) Cost analysis for a continuously operated fine chemicals production plant at 10 kg/day using a combination of microprocessing and microwave heating. J Flow Chem 1:74–89
Schaber SD, Gerogiorgis DI, Ramachandran R, Evans JMB, Barton PI, Trout BL (2011) Economic analysis of integrated continuous and batch pharmaceutical manufacturing: a case study. Ind Eng Chem Res 50:10083–10092
Gutmann B, Cantillo D, Kappe CO (2015) Continuous-flow technology - a tool for the safe manufacturing of active pharmaceutical ingredients. Angew Chem Int Ed Engl 54:6688–6728
Porta R, Benaglia M, Puglisi A (2016) Flow chemistry: recent developments in the synthesis of pharmaceutical products. Org Process Res Dev 20:2–25
Baumann M, Baxendale IR (2015) The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry. J Org Chem 11:1194–1219
Heider PL, Born SC, Basak S, Benyahia B, Lakerveld R, Zhang H, Hogan R, Buchbinder L, Wolfe A, Mascia S, Evans JMB, Jamison TF, Jensen KF (2014) Development of a multi-step synthesis and workup sequence for an integrated, continuous manufacturing process of a pharmaceutical. Org Process Res Dev 18:402–409
FDA Perspective on Continuous Manufacturing (2012) US Food and Drug Administation. https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM341197.pdf. Accessed 21 Jan 2019
Statement from FDA Commissioner Scott Gottlieb, M.D., and Janet Woodcock, M.D., director of the Center for Drug Evaluation and Research on FDA’s ongoing investigation into valsartan impurities and recalls and an update on FDA’s current findings (2018) US Food and Drug Administration. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm619024.htm. Accessed 30 Oct 2018
ONE-FLOW Research Project Home Page. https://one-flow.org
Hiebler K, Soritz S, Gavric K, Birrer S, Maier MC, Grabner B, Gruber-Woelfler H (2019) Multistep synthesis of a valsartan precursor in continuous flow. J Flow Chem. https://doi.org/10.1007/s41981-019-00044-x
Lichtenegger GJ, Maier M, Hackl M, Khinast JG, Gössler W, Griesser T, Kumar VSP, Gruber-Woelfler H, Deshpande PA (2017) Suzuki-Miyaura coupling reactions using novel metal oxide supported ionic palladium catalysts. J Mol Catal A Chem 426:39–51
Lichtenegger GJ, Maier M, Khinast JG, Gruber-Wölfler H (2016) Continuous Suzuki-Miyaura reactions with novel Ce-Sn-Pd oxides and integrated crystallization as continuous downstream protocol. J Flow Chem 6:244–251
Lichtenegger GJ, Tursic V, Kitzler H, Obermaier K, Khinast JG, Gruber-Wölfler H (2016) The Plug & Play reactor: a highly flexible device for heterogeneous reactions in continuous flow. Chem Ing Tech 88:1518–1523
Bäckvall J-E (2010) Palladium-catalyzed cross couplings in organic synthesis - scientific background on the nobel prize in chemistry 2010. Sci Backgr Nobel Prize Chem 50005:1–12
Buchwald SL (2008) Cross coupling. Acc Chem Res 41:1439–1439
Magano J, Dunetz JR (2011) Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals. Chem Rev 111:2177–2250
Selander N, Szabo KJ (2011) Catalysis by palladium pincer complexes. Chem Rev 111:2048–2076
Beletskaya IP, Cheprakov AV (2009) The Heck reaction as a sharpening stone of palladium catalysis. Chem Rev 100:3009–3066
Wu XF, Anbarasan P, Neumann H, Beller M (2010) From noble metal to Nobel Prize: palladium-catalyzed coupling reactions as key methods in organic synthesis. Angew Chem Int Ed Engl 49:9047–9050
Suzuki A (2002) Cross-coupling reactions via organoboranes. J Organomet Chem 653:83–90
Miyaura N, Suzuki A (1995) Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev 95:2457–2483
Kotha S, Lahiri K, Kashinath D (2002) Recent applications of the Suzuki-Miyaura cross-coupling reaction in organic synthesis. Tetrahedron 58:9633–9695
Smith MD, Stepan AF, Ramarao C, Brennan PE, Ley SV (2003) Palladium-containing perovskites: recoverable and reuseable catalysts for Suzuki couplings. Chem Commun 3:2652–2653
Alfonsi K, Colberg J, Dunn PJ, Fevig T, Jennings S, Johnson TA, Kleine HP, Knight C, Nagy MA, Perry DA, Stefaniak M (2008) Green chemistry tools to influence a medicinal chemistry and research chemistry based organisation. Green Chem 10:31–36
Smith MD, Mostofian B, Petridis L, Cheng X, Smith JC (2016) Molecular driving forces behind the tetrahydrofuran-water miscibility gap. J Phys Chem B 120:740–747
Othmer DF, Josefowitz S (1947) Composition of vapors from boiling binary solutions. Ind Eng Chem 39:1175–1177
Gooding OW (2004) Process optimization using combinatorial design principles: parallel synthesis and design of experiment methods. Curr Opin Chem Biol 8:297–304
Eriksson L, Johansson E, Kettaneh-Wold N, Wikström C, Wold S (2008) Design of Experiments: principles and applications, 3rd edn. Umetrics AB, Umea
Mead R (1988) The design of experiments - statistical principles for practical applications. Cambridge University Press, Cambridge
(2018) Multivariate analysis in the pharmaceutical industry. 1st edn. Academic Press, Cambridge Mass
Rubin AE, Tummala S, Both DA, Wang C, Delaney EJ (2006) Emerging technologies supporting chemical process R&D and their increasing impact on productivity in the pharmaceutical industry. Chem Rev 106:2794–2810
Politis SN, Colombo P, Colombo G, Rekkas DM (2017) Design of experiments (DoE) in pharmaceutical development. Drug Dev Ind Pharm 43:889–901
López SE, Salazar J (2013) Trifluoroacetic acid: uses and recent applications in organic synthesis. J Fluor Chem 156:73–100
Qian Z, Baxendale IR, Ley SV (2010) A continuous flow process using a sequence of microreactors with in-line IR analysis for the preparation of N,N-diethyl-4-(3-fluorophenylpiperidin-4-ylidenemethyl)benzamide as a potent and highly selective delta-opioid receptor agonist. Chemistry 16:12342–12348
Liu Y-S, Zhao C, Bergbreiter DE, Romo D (1998) Simultaneous deprotection and purification of Boc-amines on ionic resin capture. J Org Chem 63:3471–3473
Zhang CX, Zheng GJ, Bi FQ, Li YL (2008) A simple and efficient synthesis of the valsartan. Chin Chem Lett 19:759–761
Coffey DS, Hawk MKN, Pedersen SW, Ghera SJ, Marler PG, Dodson PN, Lytle ML (2004) Large scale deprotection of a tert-butoxycarbonyl (Boc) group using aqueous HCl and acetone. Org Process Res Dev 8:945–947
Gilmore K, Kopetzki D, Lee JW, Horvath Z, McQuade DT, Seidel-Morgenstern A, Seeberger PH (2014) Continuous synthesis of artemisinin-derived medicines. Chem Commun (Camb) 50:12652–12655
Dulla B, Vijayavardhini S, Rambau D, Anuradha V, Rao MVB, Pal M (2014) Catalyst/surfactant free chemoselective acylation of amines in water. Curr Green Chem 1:73–79
Sumida Y, Iwai S, Nishiya Y, Kumagai S, Yamada T, Azuma M (2016) Identification and characterization of D-succinylase, and a proposed enzymatic method for D-amino acid synthesis. Adv Synth Catal 358:2041–2046
Cailla HL, Racine-Weisbuch MS, Delaage MA (1973) Adenosine 3',5' cyclic monophosphate assay at 10-15 mole level. Anal Biochem 56:394–407
Bunton CA (1963) The hydrolysis of carboxylic anhydrides. Part III. Reactions in initially neutral solution. J Chem Soc: 2918–2926
Srinivasan N, Yurek-George A, Ganesan A (2005) Rapid deprotection of N-Boc amines by TFA combined with freebase generation using basic ion-exchange resins. Mol Divers 9:291–293
ICH guideline Q3D (R1) on elemental impurities (2019) European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-32.pdf. Accessed 27 Sep 2019
Ion Exchange Resins - Removing acid from a solution (2019) DuPont Water Solutions Customer Help. https://water.custhelp.com/app/answers/detail/a_id/144/related/1. Accessed 8 May 2019
Amberlyst™ A21 Product Data Sheet (2016) The Dow Chemical Company. http://www.faran-water.ir/userfiles/files/resin/chemical/0901b803808d3096.pdf. Accessed 8 May 2019
Nagireddi S, Golder AK, Uppaluri R (2018) Role of protonation and functional groups in Pd(II) recovery and reuse characteristics of commercial anion exchange resins-synthetic electroless plating solution systems. J Water Process Eng 22:227–238
Hubicki Z, Wołowicz A (2009) Adsorption of palladium(II) from chloride solutions on Amberlyst A 29 and Amberlyst A 21 resins. Hydrometallurgy 96:159–165
Kaiser K, Benner R (2005) Hydrolysis-induced racemization of amino acids. Limnol Oceanograph: Methods 3:318–325
Yamada S, Hongo C, Yoshioka R, Chibata I (1983) Method for the racemization of optically active amino acids. J Org Chem 48:843–846
Zhang X, Cividino P, Poisson JF, Shpak-Kraievskyi P, Laurent MY, Martel A, Dujardin G, Py S (2014) Asymmetric synthesis of α,α-disubstituted amino acids by cycloaddition of (E)-ketonitrones with vinyl ethers. Org Lett 16:1936–1939
Acknowledgments
Open access funding provided by Graz University of Technology. The authors kindly acknowledge the funding by the H2020FETOPEN-2016-2017 program of the European Commission (Grant agreement number: 737266-ONE FLOW).
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Development of a multistep setup for the integrated synthesis of a late-stage sacubitril precursor in continuous flow.
• Intensification of Suzuki coupling in continuous flow employing a heterogeneous palladium catalyst implemented in a packed-bed reactor.
• Optimization of Boc-deprotection as well as N-amidation using the “Design of Experiments” approach.
Electronic supplementary material
ESM 1
(PDF 1820 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hiebler, K., Dertnig, C., Soritz, S. et al. Development of a multistep reaction cascade for the synthesis of a sacubitril precursor in continuous flow. J Flow Chem 10, 259–270 (2020). https://doi.org/10.1007/s41981-019-00058-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41981-019-00058-5