
Abstract
This paper reviews the properties of methanesulfonic acid (MSA) and its potential for use in hydrometallurgy. Although MSA is much less known than sulfuric, hydrochloric or nitric acid, it has several appealing properties that makes it very attractive for the development of new circular flowsheets in hydrometallurgy. Unlike other organic acids such as acetic acid, MSA is a very strong acid (pKa = − 1.9). In addition, it is very stable against chemical oxidation and reduction, and has no tendency to hydrolyze in water. In terms of its environmental impact, MSA has low toxicity and is biodegradable. In nature, it is part of the geochemical sulfur cycle. A useful property is the high solubility of its salts in water: methanesulfonate salts have a much higher solubility in water than sulfate salts. Additionally, MSA and its salts are compatible with the electrowinning of metals because the anode reaction involves the formation of oxygen gas (unlike chlorine gas formation in chloride electrolytes) and no cathodic reduction of the anion occurs (unlike nitrate reduction in nitrate electrolytes). MSA is particularly interesting for lead hydrometallurgy, where it offers more environment-friendly alternatives to HBF4 and H2SiF6. However, MSA can also be adopted in all hydrometallurgical processes that require strong Brønsted acids. It can be used in the metallurgy of copper, zinc, cobalt, nickel, and rare earths, as well as in the recycling of metals from end-of-life products. Although MSA itself is a non-oxidizing acid, in combination with hydrogen peroxide it yields strongly oxidizing lixiviants that can leach copper from chalcopyrite or dissolve metallic silver. The global production of MSA is expected to increase rapidly in the near future thanks to both the industrialization of a new sustainable synthesis process and its many applications (cleaning fluids, electrolytes for electroplating, redox-flow batteries, catalysts in organic synthesis, and as a solvent for high-molecular-weight polymers). As a result, MSA will become more widely available and a lower price will make it an increasingly attractive option.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sulfuric acid (H2SO4) is by far the most often used acid in hydrometallurgy. It is widely applied for leaching metals from ores, concentrates, industrial process residues and other metal-containing streams, and it is the acid of choice in electrolytes for the electrowinning and electrorefining of metals [1, 2]. Sulfuric acid has many advantages that stimulate its widespread use in hydrometallurgy. First, it is widely available and produced on a very large scale, making it the cheapest acid available. In fact, it is the number one chemical in terms of quantity produced by the chemical industry. Often, metallurgical plants produce their own sulfuric acid from the sulfur dioxide that is released during the roasting of sulfuric minerals or during other pyrometallurgical unit processes. Sulfuric acid is also formed naturally by the bacterial oxidation of sulfidic minerals, and it is a lixiviant that is generated in-situ in dump and heap leaching of sulfidic ores.
As aqueous sulfuric acid solutions have a low corrosivity, there are few constraints with respect to the choice of materials used for equipment in sulfate hydrometallurgy. Sulfuric acid is not volatile, and it is odorless. It also has a low toxicity and can be considered as an environment-friendly acid. Electrowinning processes based on sulfuric acid electrolytes have the advantage that oxygen gas is formed at the anode. Sulfuric acid is also essential in copper hydrometallurgy.
Notwithstanding all these advantages, there is one major disadvantage associated with the use of sulfuric acid in hydrometallurgy: metal sulfate salts have a relatively low solubility in aqueous solutions compared to other salts, such as chlorides and nitrates. Some metal salts are very insoluble in water. Examples include PbSO4 (anglesite) and CaSO4·2H2O (gypsum). This is problematic for the extractive metallurgy of lead. Calcium sulfate scaling causes many issues in hydrometallurgical operations. Furthermore, in the electrometallurgy of copper, the relatively low solubility of CuSO4·5H2O in sulfuric acid solutions limits the maximum copper concentration in the advance electrolyte. This has a negative effect on the efficiency of copper electrowinning and electrorefining [3].
The poor solubility of metal sulfates compared to the corresponding chloride and nitrate salts is a driving force for the development of chloride hydrometallurgy (based on HCl) [4, 5] and nitrate hydrometallurgy (based on HNO3) [6]. Metal nitrates, in particular, have a high solubility in aqueous solutions. In fact, there are no poorly soluble metal nitrates. Metal chloride salts are, in general, much more soluble than sulfate salts, but there are some poorly soluble chloride salts of relevance for hydrometallurgy, e.g., PbCl2 and AgCl. The solubility of a poorly soluble metal chloride will drastically increase the formation of anionic chloride complexes in the presence of high concentrations of added chloride salts (for instance in brine solutions). Chloride solutions and especially hydrochloric acid solutions are corrosive, so that attention must be paid to the material selection for equipment and piping. In the electrowinning of metals from chloride electrolytes, chlorine gas rather than oxygen gas is formed at the anode. Although HNO3 solutions are less corrosive than HCl solutions, nitrate electrolytes are problematic because of the nitrate reduction at the cathode during electrowinning or electrorefining.
There is a third acid, besides HCl and HNO3, that forms metal salts that are much more soluble in aqueous solutions than the corresponding sulfates: methanesulfonic acid (MSA) [7]. MSA is a strong organic acid with the formula CH3SO3H. It is the smallest member of the series of the alkane sulfonic acids. MSA has many properties that make it an ideal acid for use in hydrometallurgy. Although, so far, it has been largely overlooked by hydrometallurgists. But perhaps that is not very surprising. Although MSA has been known for more than 100 years [8], and is produced on an industrial scale since the 1950s [9], prior to 2000, MSA had only some niche applications (e.g., MSA has been used by the microelectronic and electroplating industries since the 1980s [10]). This was mainly due to its rather high price and limited availability. However, this situation changed around 2000. As a result, the number of industrial applications of MSA has grown accordingly. Nowadays, MSA is being used as a catalyst or reagent for organic reactions, including the production of biodiesel [11]; in the pharmaceutical industry, as a solvent for high molecular weight polymers [12]; in cleaning fluids for rust removal [13]; for descaling of pipes in the oil industry [14, 15]; electrolytes for electroplating of metals [7, 10]; and in redox-flow batteries [16]. The yearly global production of MSA is approximately 50,000 tons, and this number is expected to increase in the future [17].
After many years of the total neglect of MSA by metallurgical engineers, the tide is turning. A literature search of the scientific and patent literature yields a significant increase in the number of papers on applications of MSA in hydrometallurgy in recent years. Still, most researchers in the field of hydrometallurgy remain unfamiliar with this “new” acid, although it has great potential for the development of circular hydrometallurgical flowsheets. Therefore, in this review we describe the properties of MSA and methanesulfonate salts, and we summarize the studies that were published on the applications of MSA for the leaching of different ores, concentrates, industrial process residues and urban waste, as well as the use of MSA in aqueous electrometallurgy. To the best of our knowledge, this is the first review of MSA in hydrometallurgy.
Industrial Production Processes
The first industrial process for the production of MSA was developed in the 1940s by Standard Oil of Indiana (USA) [7, 9]. This process is now obsolete and was based on the NOx-based air oxidation of methyl mercaptan (methyl sulfide, CH3SH), followed by a stripping procedure to remove the residual NOx from the MSA product. This was a cheap process (using air as a reagent), but it suffered from a poor product quality and explosion hazards. In 1967, the Pennwalt Corporation (USA) disclosed a process based on the direct chlorine oxidation of an aqueous emulsion of methyl mercaptan. The raw material is purified by extraction. The process is still used by Arkema SA (France) for the production of highly pure MSA (chlorine-oxidation process). Disadvantages include the use of toxic and expensive raw materials, as well as the formation of large amounts of HCl.
BASF SE (Germany) developed a chlorine-free process. BASF combines methanol, hydrogen and sulfur to make dimethyldisulfide (DMDS), followed by the catalytic air oxidation of methyl mercaptan to MSA [18]. Methanol is obtained from the syngas (CO/H2 mixture) that is derived from natural gas. Sulfur is available from oil refineries as a product of the desulfurization of crude oil. The crude MSA is then further purified by distillation. In 2003, BASF commenced the production of highly pure MSA at its site in Ludwigshafen (Germany).
In 2016, Grillo-Werke AG (Germany) developed an innovative process that makes it possible to prepare MSA by the direct activation of methane: the Grillo-Methane-Sulfonation (GMS) process [17, 19]. Methane (CH4) reacts with sulfur trioxide (SO3) in an oleum solution (fuming sulfuric acid, H2SO4 saturated with SO3), in the presence of a sulfonyl peroxide initiator. Methane can originate from natural gas or biogas. The reaction occurs at moderate temperatures (~ 50 °C) and pressures (~ 100 bar). No expensive metal-containing catalyst is required. The GMS process is the first example of the conversion of methane in a single reaction step to result in a high-value product. Hence, this is a very environment-friendly process, as was corroborated by LCA studies [20], especially when the methane originates from biogas. BASF acquired the process approach for producing MSA from Grillo-Werke AG in 2019 to implement it on an industrial scale.
At present, the two main manufacturers of MSA are Arkema and BASF, with BASF being the leading global producer. It has recently increased its production capacity to 50,000 metric tons per year. BASF is marketing MSA under the brand name Lutropur®. Lutropur® MSA is a 70% aqueous solution of MSA, whereas Lutropur® MSA 100 is anhydrous MS (99.98%).
Properties of MSA
Methanesulfonic acid (MSA) is commercially available in anhydrous form (> 99.5%) and as a concentrated aqueous solution (70 wt%) [11, 21]. At room temperature the anhydrous MSA is a colorless, odorless viscous liquid. Its melting point is + 19 °C and its kinematic viscosity is 8.3 mPa·s at 23 °C. It has a high boiling point and cannot be distilled at atmospheric pressure without decomposition. At a pressure of 1 mm Hg (133 Pa) anhydrous MSA boils at 122 °C. Upon heating in air, it shows no signs of thermal decomposition at temperatures up to 180 °C. When heating in an inert solvent, the thermal stability range can be extended to about 225 °C. Because of its high boiling point, the vapor pressure of MSA at room temperature is low. The 70 wt% aqueous solution has a melting point of − 54 °C. MSA is miscible in all proportions with water.
MSA is an organic compound with the chemical formula CH3SO3H. It has a sulfonic acid functional group. With its methyl group (CH3), it is the smallest member in the homologous series of alkane sulfonic acids, having the general formula CnH2n+1SO3H, with n being the number of carbon atoms (n = 1 for MSA). The chemical structure of MSA can be seen as that of sulfuric acid in which one of the two OH groups has been replaced by a CH3 group. MSA is a strong Brønsted acid, with pKa = − 1.9 [22]. Thus, MSA is a much stronger acid than other organic acids, such as formic acid, acetic acid or citric acid. In fact, its acid strength is comparable with that of strong mineral acids such as H2SO4, HCl or HNO3. MSA is almost completely ionized in a 0.1 M solution. The heat of formation of the methanesulfonate ion in an aqueous solution is − 163.79 ± 1.04 kcal mol−1 and the heat of formation of liquid MSA is − 152.39 ± 1.12 kcal mol−1 (at 25°) [23].
MSA is a very stable chemical compound [9, 11]. It is not attacked by strong oxidizing agents such as hydrogen peroxide, concentrated nitric acid, potassium permanganate or even chromosulfuric acid (a strongly oxidizing solution of a dichromate salt in concentrated sulfuric acid). MSA is also highly resistant to strong reducing agents such as nascent hydrogen. It is not hydrolyzed by boiling water or hot alkali solutions, not even after a prolonged time. It does not react with chlorine gas at ambient pressures either.
Anhydrous MSA has an electrochemical window of about 3.8 V, which is much wider than that of water (1.23 V). Aqueous solutions of MSA have a high electrical conductivity, which is important for electrochemical applications such as electrowinning, electrorefining and electroplating [7].
MSA will readily dissolve oxides, hydroxides and carbonates. At high concentrations, it will react with many sulfides, with the release of hydrogen sulfide gas (H2S). MSA is a non-oxidizing acid, in contrast to concentrated sulfuric acid or nitric acid. However, a mixture of MSA and hydrogen peroxide is strongly oxidizing and can dissolve metallic copper [24], nickel [24], tin [25], lead [25], and silver (but not gold or platinum-group metals) [26].
Most types of stainless steel are resistant to exposure to a 70% aqueous MSA solution. MSA does not attack glass, polyethylene, polypropylene or fluorinated polymers. Notwithstanding the low reactivity of MSA towards many construction materials, it is recommended to perform material compatibility tests before a final decision is made about the choice of material for equipment in contact for prolonged times with concentrated MSA [11]. This is especially relevant to hydrometallurgy, since it has been reported that, unlike purified MSA, MSA containing different impurities like sulfate, chloride and nitrate, may cause corrosion problems in different types of stainless steel [27].
In Table 1 the main properties of MSA are compared with those of sulfuric acid, hydrochloric acid and nitric acid. The prices are indicative, because they fluctuate over time, show large regional differences, and depend on the purity. Although the current price of MSA (70% solution) seems to be high compared to the other acids, it is expected to decrease due to higher production capacities and cheaper processes. They are of the order of magnitude of technical grade organic solvents. However, we should not forget that if sulfuric acid is used to regenerate MSA by metal sulfate precipitation (vide infra), the consumable is the sulfuric acid and not the MSA.
MSA in the Environment
MSA is part of the geochemical sulfur cycle [28]. It is formed in the atmosphere by the photochemical oxidation of dimethyl sulfide (DMS). DMS originates from the decomposition of algae, cyanobacteria and marine-plant biomass in oceans and salt marshes. Once formed in the atmosphere, MSA is stable and does not undergo photochemical decomposition. It partitions in aerosols and takes place in the formation of cloud condensation nuclei, returning to Earth in the form of rain and snow [29]. It has been discovered in the perennial ice layers of the Antarctic and Greenland. Up to 5 ppm of MSA is found in surface waters, with only 100 to 200 ppb in shallow ground water and close to 0 ppb in deep water wells [11].
The low concentration of MSA in the environment is due to its rapid biodegradation. Certain organisms use MSA as a source of sulfur, but not carbon, whereas the methyl group of MSA supports the growth of methylotropic bacteria. Methylotropic bacterial communities utilize MSA as a source of carbon and energy [29]. All MSA-utilizing bacteria seem to share a common enzyme: MSA monooxygenase.
Experiments have shown that MSA is readily biodegradable according to the OECD guidelines 301A, 306 and 311, forming carbon dioxide, sulfate, water and biomass [11]. There is a low oxygen demand for degradation because MSA is a C1 compound (1 carbon atom per molecule). Since no phosphorus is present, MSA does not contribute to eutrophication or algal growth. Because of the very low vapor pressure, MSA is also virtually free of volatile organic compounds (VOCs).
Metal Methanesulfonate Salts
Salts of MSA are called methanesulfonate or mesylate salts (symbol: OMs). One of the most remarkable and most useful properties of MSA and its compounds is the very high solubility of metal methanesulfonate salts in aqueous solutions. All methanesulfonate salts are well soluble, much higher than the solubility of the corresponding sulfate salts. Concerning the chlorides, the trends are somewhat less clear. Some metal ions form methanesulfonate salts with a much higher solubility than their chlorides (Pb2+, Ag+), whereas other metal ions have a similar solubility (Cu2+, Cd2+). Zinc(II) is an exception, because ZnCl2 is much more soluble in water than Zn(CH3SO3)2. When comparing different literature sources, we must carefully check whether the solubilities are expressed in metal concentrations in the saturated solution, or as the solubility of the metal salt. For instance, for Pb2+, an aqueous saturation solubility of 2.60 mol L−1 is provided in reference [7], which corresponds to a metal concentration of 539 g L−1, whereas in reference [11] a value of 1075 g L−1 is mentioned, which refers to the solubility of the Pb(CH3SO3)2⋅H2O salt (MW = 415.41 g mol−1).
It must be mentioned that the solubility data remain fragmentary, in the sense that only experimental data are available at room temperature. No information is available on the temperature dependence of the metal methanesulfonates, whereas it is known that highly soluble metal salts often show a strong dependence of their solubility on the temperature. It has been observed that the solubility of silver(I) methanesulfonate decreases with an increasing free MSA concentration in solution [30]. So far, this phenomenon has not been explained, although probably the common-ion effect plays an important role. Although the solubility of methanesulfonate salts in water is very high, metal methanesulfonates are poorly soluble in anhydrous MSA, especially at room temperature. This is evident when anhydrous MSA is being used to leach solid materials; precipitates are often formed upon cooling the solution [31, 32].
Not much information about the solution chemistry of methanesulfonate salts is available in the literature. The methanesulfonate anion is a weakly coordinated anion so that it can be assumed that in diluted solutions it resides only in the second coordination sphere, which means that the metal ions have only water molecules in the first coordination sphere. The formation of metal complexes between lead(II) and methanesulfonate ions has been studied by polarography [33]. The authors mention the stepwise formation of the complexes [Pb(CH3SO3)]+, [Pb(CH3SO3)2] and [Pb(CH3SO3)3]− and determined the overall stability constants βi (i = 3). These values are very small, indicating a weak tendency to form complexes. The authors also state that at MSA concentrations above 1.8 M, the [Pb(CH3SO3)3]− complex dominates. However, we must be cautious with the interpretation of these results, because there is no real evidence for the formation of inner-sphere complexes (i.e., the methanesulfonate ions in the first coordination sphere). There is obviously a need for more detailed studies on (1) the speciation of metal methanesulfonate complexes in concentrated aqueous solutions and (2) the determination of the corresponding complex formation constants.
The solid–liquid equilibria of the Zn(CH3SO3)2 − H2O and Cu(CH3SO3)2 − H2O binary systems have been investigated [34]. Parts of the phase diagrams from sub-solidus temperatures up to 70 °C have been determined by differential scanning calorimetry (DSC). Below 26 °C, the solubility of zinc(II) methanesulfonate decreases abruptly due to hydrate formation. A new hydrate has been identified: Zn(CH3SO3)2∙12H2O. The crystal structure Zn(CH3SO3)2∙12H2O could be determined by single-crystal X-ray diffraction and the structure is formed by hexaaquazinc(II) cations [Zn(H2O)6]2+, CH3SO3− anions and lattice water molecules. There is no direct chemical bond between the methanesulfonate anion and the zinc(II) cation. For the copper(II) system, a new hydrate was also discovered: Cu(CH3SO3)2∙8H2O. However, its crystal structure could not be determined. Vapor-pressure measurements have been performed for the Zn(CH3SO3)2−H2O and Cu(CH3SO3)2−H2O systems, and the data used to calculate the water activity and the Pitzer–Simonson–Clegg interaction parameters between the ions [35]. Sections of the phase diagram of the ternary system ZnCl2−Zn(CH3SO3)2−H2O were determined between − 10.8 and 25 °C [36]. The density and water activity were measured at different temperatures for Zn(CH3SO3)2 solutions. The work of the Russian group is one of the rare examples of thermodynamic studies and phase-diagram determinations of systems involving metal methanesulfonates and water. The results indicate that these systems are more complicated than expected due to hydrate formation. We can predict that more surprising results will be reported in the future if these studies are refined and extended.
The high solubility of methanesulfonate salts compared to sulfate and chloride salts is also advantageous for the regeneration of MSA in process solutions after the removal of the valuable metals. For instance, if CaO, Ca(OH)2 or CaCO3 is used for neutralization of the excess MSA after leaching and the metals have been removed from the pregnant leach solution (PLS) by precipitation as hydroxides or sulfides, the raffinate will be rich in dissolved calcium methanesulfonate. The addition of sulfuric acid to this solution will precipitate CaSO4·2H2O (gypsum), while MSA is regenerated:
Unlike the gypsum that is formed by the addition of CaO, Ca(OH)2 or CaCO3 to a sulfuric acid solution for neutralization of the PLS after sulfuric acid leaching, the gypsum formed by the addition of sulfuric acid to a calcium methanesulfonate solution is expected to be of a sufficient quality for use in the construction industry.
In a similar way, silver can be recovered from MSA solutions by the addition of hydrochloric acid. Poorly soluble silver(I) chloride will be precipitated [37]:
Rare-earth elements (REEs) can be recovered from MSA solutions by the addition of oxalic acid [38, 39]:
Lead Hydrometallurgy
The extractive metallurgy of lead is an obvious field of application for MSA. Sulfidic lead ores (galena, PbS) are typically processed by pyrometallurgy because the autogenous smelting of high-grade concentrates makes this process energy efficient. Oxidic lead ores (PbO, Pb(OH)2, PbCO3, PbSO4) are not very suitable as feed for pyrometallurgical smelting operations because of the lack of intrinsic fuel value of their concentrates [40]. Therefore, they should be processed via hydrometallurgy, by leaching, followed by electrowinning or precipitation. However, there are serious issues related to the conventional acids that are used for leaching. Sulfuric acid cannot be used as a lixiviant because PbSO4 is poorly soluble. Hydrochloric acid leads to PbCl2 precipitates. Nitric acid causes problems in the electrowinning step because of nitrate reduction at the cathode. Oxidic lead ores can be solubilized by brine leaching (using a concentrated NaCl solution), but here the formation of chlorine gas at the anode during electrowinning is problematic. Alkaline leaching with NaOH is an alternative, but electrowinning from alkaline solutions is less straightforward due to the tendency of PbO2 formation at the anode and the alkaline corrosion of lead at the cathode [41]. For all these reasons, the conventional acids are not very suitable for the electrorefining of lead.
Therefore, hydrometallurgical processes for lead are typically based on fluoroboric acid (fluoboric acid, tetrafluoroboric acid, HBF4) or fluorosilicic acid (hexafluorosilicic acid, H2SiF6), because Pb(BF4)2 and PbSiF6 are well soluble in water [42]. These acids have been commercially available since the early 1900s. Hexafluorosilicic acid is cheap and widely available because it is a byproduct of the production of phosphate fertilizers. An electrorefining process for the purification of impure lead anodes in a fluorosilicic acid electrolyte is known as the Betts process [43, 44]. The RSR process comprises leaching lead-battery sludge converted to PbCO3, PbCO3·Pb(OH)2 and PbO by HBF4 or H2SiF6, followed by the electrowinning of metallic lead [45]. Hydrometallurgical processes for primary and secondary lead resources based on the use of fluoroboric acid have been developed by the Italian company Engitec [46]. The company further developed a process for the oxidative dissolution of lead bullion in a Fe(BF4)3 solution, followed by the electrowinning of metallic lead and the regeneration of the oxidizing agent Fe(BF4)3 at the anode in a divided electrolysis cell (LEADBOR process) [47]. However, BF4− and SiF62− are susceptible to hydrolysis in acid media, leading to the formation of dangerous hydrofluoric acid (HF). The emission of HF to the surrounding atmosphere creates an unhealthy working environment. In the presence of HF, dissolved lead will precipitate as PbF2. Although HBF4 solutions can be stabilized by the addition of boric acid (H3BO3) and H2SiF6 by the addition of silicic acid (H4SiO4), it is very difficult to completely avoid hydrolysis and HF emission to the air above the process solutions.
Therefore, MSA has been proposed as a more suitable alternative to both fluoroboric and fluorosilicic acid in lead hydrometallurgy. MSA is an excellent electrolyte for the electrowinning and electrorefining of lead [40, 48]. Lead(II) methanesulfonate is highly soluble in water, just like the corresponding fluoroborate and fluorosilicate salts, but MSA is stable against hydrolysis and more environmentally friendly. There is no danger of the formation of HF fumes. Additional advantages for electrometallurgy are the high electrical conductivity of the MSA-based electrolytes and the oxygen gas formation at the anode. Although the standard reduction potential of the Pb2+/Pb couple (− 0.126 V versus SHE) is more negative than that of the H+/H2 couple (0.00 V versus SHE), lead can be electrodeposited from strongly acidic electrolytes with a cathode efficiency of close to 100% thanks to the high overpotential for hydrogen gas formation on a lead electrode.
Hydrometallurgical processes for lead, based on MSA, have been developed by the University of British Columbia (UBC, Canada), in collaboration with BASF (Germany) [49, 50]. Different modifications of the MSA process make it possible to treat oxidic and sulfidic lead ores. The process has been demonstrated for cerussite concentrate [51]. In the primary leach, cerussite was first leached with MSA and readily dissolved. The leaching kinetics were fast: most of the cerussite was dissolved within 5 min. The residue of the primary leach, which consisted largely of anglesite, was treated in a desulfurization step with Na2CO3 solution to convert the anglesite to cerussite. The desulfurized residue was leached with MSA in the secondary leach. In total, more than 98% of the lead present in the cerussite concentrate could be solubilized. The lead was recovered from the pregnant leach solution by electrowinning. The advance electrolyte had a concentration of 100–150 g L−1 of lead, as Pb(CH3SO3)2, and 0.25–0.5 M free MSA. A graphite anode and a stainless-steel cathode were used in the undivided electrolysis cell. The current density was between 215 and 250 A m−2, and the temperature of the electrolyte was between 35 and 40 °C. Different additives were tested in the electrowinning circuit, and 2.5 g L−1 of calcium ligninsulfonate was found to give the highest suppression of dendrite formation while resulting in the best morphology of the obtained lead metal. A schematic representation of the flowsheet is provided in Fig. 1.

Copyright Elsevier 2014
Schematic flowsheet for the processing of cerussite concentrate by MSA. Redrawn from reference [51]. Reproduced with permission.
The process had to be adapted for the leaching of galena concentrates because the direct leaching of PbS in MSA is inefficient and triggers the formation of toxic H2S gas. Galena can be oxidatively leached by Fe(CH3SO3)3 in MSA [52]. By controlling the experimental conditions, it is possible to convert the sulfide ions either to elemental sulfur or to oxidize them further to sulfate ions. The sulfate ions will react with the lead(II) ions in solution and precipitate as PbSO4, but the latter compound can be converted in a desulfurization step with Na2CO3 to PbCO3. The proposed flowsheet is similar to that for the processing of cerussite concentrate, with the difference being that the electrowinning is performed in a divided electrolysis cell to regenerate the Fe(CH3SO3)3 lixiviant at the anode.
The BASF-UBC process has been further studied at the bench, pilot and demonstration scales on a lead concentrate of the Paroo Station lead mine in Western Australia, owned by LeadFX [53]. LeadFX secured the process rights from BASF and UBC. The process involves a three-step leaching of a lead concentrate in MSA (“Leaching–Desulfurization–Releaching”), followed by the electrowinning of lead metal. Cerussite can be directly dissolved in the spent MSA electrolyte of the electrowinning circuit (step 1), but minor lead minerals such as pyromorphite must be processed first by sulfuric acid to convert them into PbSO4, which can be treated by Na2CO3 by desulfurization and conversion to PbCO3 (step 2). Step 3 is a re-leach with MSA of the PbCO3 formed in the desulfurization step. Galena can be leached by adding Fe(CH3SO3)3 to the MSA solution. Fe(CH3SO3)3 can be partially regenerated in the electrowinning circuit, but regeneration is aided by the addition of hydrogen peroxide to the spent electrolyte. The advance electrolyte of the electrowinning circuit contains more than 300 g L−1 of dissolved lead. The lead electrowinning circuit, comprising 45 lead cathodes and 46 graphite anodes, is designed to produce up to 80,000 tons per year of cathode lead. Cathode lead with a purity of more than 99.995% is produced at current densities between 300 and 400 A m−2. The electrowon lead cathodes are melted in an induction furnace and cast to lead ingots.
Copper Hydrometallurgy
For oxidic copper ores, MSA offers limited advantages over sulfuric acid in the leaching−solvent-extraction−electrowinning process (L-SX-EW process) taking into account the huge scale of the leaching operations and the much higher price of MSA compared to sulfuric acid. Although limited information is available about the leaching behavior of such ores in MSA, it was reported that malachite, Cu2CO3(OH)2, can be readily dissolved in MSA [54].
Several studies have been devoted to the leaching of chalcopyrite (CuFeS2) in MSA solutions. Although chalcopyrite is the most abundant copper mineral, it is very refractory towards acids under ambient conditions because of the formation of a surface passivation layer [55]. MSA itself is a poor lixiviant for chalcopyrite because of its non-oxidizing nature. However, a mixture of MSA and hydrogen peroxide can efficiently leach the copper from chalcopyrite, provided that hydrogen peroxide is occasionally added during the leaching process to keep the ORP sufficiently high [56]. Using hydrogen peroxide as an oxidant, the copper-extraction yield was up to 60% after 96 h at 75 °C. This yield could be boosted to 99% by the periodic addition of hydrogen peroxide of 0.9% every 24 h with 30 g L−1 MSA. The sulfide ions were oxidized to elemental sulfur. Further studies indicated that the chalcopyrite leaching was very dependent on the hydrogen peroxide concentration [57]. The reaction mechanism is diffusion controlled through a protective sulfur layer at lower temperatures, and controlled by the surface chemical reaction at temperatures in excess of 55 °C. Other oxidizing agents have been added to MSA to enhance the leaching kinetics of chalcopyrite in MSA. These include FeCl3 [58, 59], K2Cr2O7 [60], and NaNO3 [60]. Although such studies are of interest from a fundamental point of view, as they help to reveal the leaching mechanism, they are less interesting for industrial applications because of the added costs and the more complex mixed-anion leachates.
There has been a proposal to treat sulfidic copper ores or concentrates from the Olympic Dam mine in Australia with mixtures of MSA and hydrochloric acid, nitric acid or amidosulfonic acid to remove the natural radionuclides (Olympic Dam is a combined copper−uranium mine) [61].
Zinc Hydrometallurgy
Smithsonite (ZnCO3) readily dissolves in MSA [62]. As expected, the leaching rate is enhanced with increasing MSA concentration, reaction temperature, stirring speed, and decreasing particle size.
The dissolution process follows the kinetic law of the shrinking core model. Similar results were obtained for the leaching of hemimorphite (Zn4Si2O7(OH)2·H2O) in MSA [63]. Sphalerite (ZnS) can be leached efficiently by iron(III) methanesulfonate [64]. A solution of 0.8 M iron(III) methanesulfonate could extract 99.3% of zinc from sphalerite particles (particle size: 106−150 μm) after leaching at 70 °C for 96 h. Zinc extraction was slightly promoted by the presence of oxygen and was significantly boosted by the presence of lattice iron in the (Zn,Fe)S sphalerite solid solution. Leaching of high-Fe sphalerite (13.94 wt% Fe) was about three times faster than for low-Fe sphalerite (0.40 wt% Fe). Fe(CH3SO3)3 was found to be more efficient than Fe3(SO4)2 or FeCl3 for leaching of sphalerite.
Silver Hydrometallurgy
As part of the development of a new MSA-based process for the refining of crude silver recycled from secondary resources, the dissolution characteristics of silver metal granules in MSA were studied [30, 65]. While MSA alone did not dissolve the granules, the addition of hydrogen peroxide as an oxidizing agent resulted in dissolution. Silver granules, containing approximately 94% of silver along with other precious metals like gold and PGMs, were successfully leached with a mixture of MSA and hydrogen peroxide. Leaching yields in excess of 90% were found, with solid-to-liquid ratios of up to 550 g L−1 and a threefold stoichiometric excess of hydrogen peroxide. An optimum yield was observed between 65 and 85 °C. A high selectivity with respect to palladium was achieved: only 7% of the palladium was co-dissolved. The leaching residue mainly consisted of gold and unleached silver, as well as small amounts of palladium and platinum. A negative correlation was observed between the solubility of the silver(I) methanesulfonate and the concentration of the free MSA after leaching [30].
Industrial Process Residues and Production Scrap
Zinc Industry Residues
An MSA-based flowsheet was developed for the recovery of lead and silver from the leaching residue of the roast-leach-electrowinning process in the zinc industry (Fig. 2) [66]. Although the composition of the residue is highly dependent on the composition of the raw materials and the leaching steps, the residue is mainly composed of lead (4−20 wt%), zinc (1.5−20 wt%), iron (6−23 wt%), calcium (2−4 wt%), and silver (300−1000 ppm). Therefore, this residue is an interesting secondary source of lead, zinc, and silver. The most common way of processing this residue is via pyrometallurgy; however, such a process does not give direct access to the valuable silver. Therefore, hydrometallurgical processes are of interest, and an MSA-based process offers the advantage of the high solubility of lead(II) and silver(I) methanesulfonate. The direct leaching of the residue with MSA resulted in a low lead-leaching efficiency and high co-dissolution of gypsum. The gypsum was selectively recovered via Randall extraction by immersion in hot water, which was continuously reused. A carbonation (desulfurization) reaction with a Na2CO3 solution was used to transform the anglesite into cerussite, which is well soluble in MSA. It was found that it is important to perform the carbonation after the Randall extraction to avoid high Na2CO3 consumption by the calcium still present in the residue. The leaching efficiency of lead drastically increased after the carbonation reaction. With the prospective flowsheet, nearly 80% of the lead and silver could be recovered. It was proposed to recover the lead and silver from solution by electrowinning. The authors suggested concentrating the MSA at the end of the process by distillation to remove part of the water, but they admitted that distillation is energy intensive and that cheaper alternatives should be sought.

Conceptual flowsheet for the MSA-based recovery of lead and silver from zinc leach residue. Redrawn from reference [66]
Similarly, MSA was tested for the leaching of lead and zinc from the iron-rich jarosite residue of the zinc industry [31]. The leaching of lead, zinc and iron increased as a function of the MSA concentration in water up to 90 vol% MSA. At higher MSA concentrations, a precipitate was formed due to the limited solubility of the iron(III) and zinc(II) methanesulfonate salts in water-lean MSA. Lead(II) methanesulfonate has a significantly higher solubility in pure MSA than the corresponding iron(III) and zinc(II) compounds. As a result, the lead did not precipitate along with the iron and zinc. The pregnant leach solution obtained after carrying out the optimized leaching procedure contained 100% of the lead, 50% of the zinc and 9% of the iron from the original jarosite residue sample. A conceptual flowsheet was designed (Fig. 3), and tested in a 1 L temperature-controlled reactor. The MSA was recovered from the leachate by vacuum distillation and successfully reused up to three times.

Conceptual flowsheet for the recovery of lead and zinc from the iron-rich jarosite residue of the zinc industry. Redrawn from reference [31]
Phosphogypsum
Large volumes of phosphogypsum are produced during the recovery of phosphoric acid from phosphate rock by leaching with sulfuric acid. Phosphogypsum is an interesting secondary resource of rare-earth elements (REEs) [67]. Moderate leaching efficiencies for REEs were observed by treating phosphogypsum by a 3 M MSA solution: 78% of the total REE content could be recovered [68]. It was suggested to use the cleaned phosphogypsum for building material applications.
Coal Ash Residues
MSA showed good selectivity for the leaching of REEs from coal ash, containing a total REE concentration of 530 ppm [69]. At a concentration of 1 M the MSA leached out about 70% of the REEs after 1 h at 90 °C, with negligible quantities of other coal ash elements co-dissolved (< 10%). XRD analysis of the coal ash residue after leaching with the MSA revealed that the coal ash matrix was not modified during the process, unlike when mineral acids were used. Kinetic studies at different temperatures indicated that leaching by MSA followed the classic shrinking-core model. The rates of leaching for both the slow and fast reaction stages were found to be diffusion controlled.
Mining Waste
In a comparative study of the performance of acids for the dissolution of copper and arsenic from legacy mine tailings in Devon (UK), it was found that MSA exhibited a similar leaching ability for arsenic as H2SO4 and HCl, but higher than for citric acid [70]. For copper removal, MSA was found to perform worse than H2SO4 and HCl, but significantly better than citric acid. The authors suggest that MSA is a suitable alternative to H2SO4 and HCl for soil washing and heap leaching. The biodegradability of MSA is seen as an advantage for this application. The MSA leaching of historic sulfidic mining waste in the Plombières tailings pond (Belgium) showed high leaching yields for both lead and zinc, but also with a significant co-dissolution of iron [71].
Nuclear-Fuel Production Scrap
A more exotic MSA application is the recycling of thoria (ThO2) from thorium-based nuclear-fuel production scrap. Thoria is the most stable of the oxides and is notoriously difficult to dissolve. For instance, the dissolution of thoria in concentrated nitric acid is extremely slow. The only method that shows some promise for use in the nuclear industry is the use of the so-called THOREX solution, which comprises HNO3, Al(NO3)3, and a catalytic amount of HF [72]. The Al(NO3)3 is used as an additive to prevent HF-induced corrosion of the equipment, as a salting-out agent, and to avoid precipitation of ThF4. Despite the addition of Al(NO3)3, HF still causes corrosion, implying that special recipients have to be used for this process. Although aqueous solutions of MSA cannot dissolve thoria, the fluorinated analogue of MSA, trifluoromethylsulfonic acid (triflic acid, CF3SO3H), is able to do so [73]. An aqueous CF3SO3H solution with concentrations ranging between 30 and 98 wt% and temperatures of 160–200 °C made it possible to dissolve thoria powder and pellets. It was shown that a closed-loop process could be developed when thorium(IV) oxalate was precipitated from the solution by the addition of solid oxalic acid [72, 74]. Thorium(IV) oxalate was calcined to thoria. The triflic acid could be recycled by distillation and re-used in the next dissolution step. A conceptual flowsheet is shown in Fig. 4.

Conceptual flowsheet for the recycling of thoria (ThO2) from thorium-based nuclear-fuel scrap by triflic acid. Redrawn from reference [74]
Urban Mining and Recycling
Lead-Acid Batteries
Given the high potential of MSA for the processing of primary lead ores and concentrates, it is not surprising that MSA can be used for the recycling of lead-acid batteries. Here again, MSA offers a safer and environmentally friendly alternative to the well-established technologies based on fluoroboric acid or fluorosilicic acid. The challenge is to develop a method that can efficiently recover the lead from the lead paste in lead-acid batteries, which mainly consists of a mixture of PbSO4, PbO and PbO2 [42]. Metallic lead is easier to treat via smelting operations. The approach is to desulfurize the lead paste by treating it with a NaOH solution, followed by leaching of the solid material with MSA. The lead can be recovered from the resulting lead(II) methanesulfonate electrolyte by electrowinning (Fig. 5) [75]. Hydrogen peroxide was added as a reducing agent to convert the insoluble PbO2 to soluble PbO [76]. We should note that hydrogen peroxide reacts as an oxidizing agent towards metals, but as a reducing agent towards strongly oxidizing agents such as PbO2. The PbSO4 desulfurization and PbO2 reduction steps could be combined by treating the lead paste by a solution containing a mixture of Na2CO3 and Na2SO3 [77].

Conceptual flowsheet for the recycling of lead-acid batteries. Redrawn from reference [75]
Lithium-Ion Batteries
MSA was shown to be an efficient solvent for the dissolution of LiCoO2 cathode material of lithium-ion batteries [78]. In the presence of a small amount of hydrogen peroxide as a reducing agent (0.9 vol%), Lithium and cobalt could be leached very efficiently by a 1 M MSA solution. MSA performed much better than other organic acids that have been tested (citric acid, malonic acid, succinic acid and oxalic acid). The dissolved cobalt was precipitated as CoCO3 and calcined to Co3O4, whereas dissolved lithium was precipitated as Li2CO3 by addition of Na2CO3 solution. Li2CO3 and Co3O4 were combined via a solid-state to new LiCoO2 cathode material.
Lamp-Phosphor Waste
The use of MSA for the recovery of REEs from the lamp-phosphor waste of end-of-life fluorescent lamps has been studied in detail by the SOLVOMET group at KU Leuven (Belgium). This waste stream is a valuable secondary resource of these critical raw materials [79, 80]. Prior to the recovery of valuable REEs from the waste powder, the crushed fluorescent lamps are sieved to remove the coarse glass particles. The mercury present in the lamp powder can be removed in a separate process step. The lamp phosphor waste powder fraction is a complex mixture of inorganic compounds. The largest fraction is the white phosphor (Sr,Ca)10(PO4)6(Cl,F)2:Sb3+,Mn2+ (HALO). HALO does not contain REEs, but it is relatively rich in antimony. The other phosphors are the red phosphor Y2O3:Eu3+ (YOX), the green phosphors LaPO4:Ce3+,Tb3+ (LAP), CeMgAl11O19:Tb3+ (CAT), (Ce,Gd)MgB5O10:Tb3+ (CBT), and the blue phosphor BaMgAl10O17:Eu2+ (BAM). Additionally, the waste powder contains fine glass particles and alumina from the binder. Most research studies have focused on the recovery of REEs from YOX phosphor, because this phosphor is the most valuable one, as it mainly consists of yttrium and europium. In a typical recycling process, the lamp-phosphor waste is treated with a dilute acid solution to dissolve first the HALO phosphor, followed by the dissolution of YOX in a more concentrated acid solution. After the dissolution of YOX, the REEs can be recovered from the solution by precipitation with oxalic acid or the solution is further purified by solvent extraction. The other phosphors in the lamp waste fraction are much more refractory than the HALO or YOX. The residue after the dissolution of YOX is often landfilled, although it contains a high content of valuable terbium. Lamp-phosphor recycling processes can become economically feasible if not just yttrium and europium are recovered, but terbium as well. LAP is the most interesting terbium-containing compound in the waste fraction, because of its high terbium content and because it is easier to dissolve than CAT and CBT. Nevertheless, very long leaching times with concentrated solutions of acids are required to dissolve LAP. To dissolve LAP in a reasonably short period of time, harsh conditions must be applied, similar to those used to treat monazite ore. Typically, the LAP is treated with concentrated sulfuric acid at temperatures above 250 °C or by alkali fusion. A milder MSA-based process was developed to dissolve the terbium present in LAP phosphor waste [81]. The process was applied on a residue obtained after leaching lamp-phosphor waste with a H2SO4 solution. By leaching with anhydrous MSA at 200 °C, high leaching efficiencies (74% Tb, 78% Ce and 95% La) were obtained after 1 h. Most of the REEs present in the green phosphor LAP could be recovered, whereas MSA did not dissolve the green phosphors CAT and CBT. Further processing of the pregnant leach solution by solvent extraction, precipitation by oxalic acid, followed by calcination, made it possible to obtain the purified oxides Tb4O7, CeO2 and La2O3. A conceptual flowsheet is shown in Fig. 6. The leaching with anhydrous MSA can be considered as a form of solvometallurgy [82].

Conceptual flowsheet for the recovery of terbium from a fluorescent lamp-phosphor residue by high-temperature leaching with anhydrous MSA. Redrawn from reference [81]
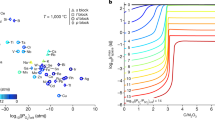
This work was further extended to the development of a closed-loop, selective process for the recovery of the three main phosphors of the lamp-phosphor waste (HALO, YOX, and LAP) using only MSA as the lixiviant [83]. The solubility of the HALO, YOX, and LAP phosphors was found to be greatly affected by the MSA concentration and the leaching temperature. Interesting observations were that (1) the HALO solubility goes through a minimum as a function of the MSA concentration, and (2) the solubility of YOX in MSA strongly decreases as a function of the MSA concentration with a very low solubility in 100% MSA (Fig. 7). The process variables, i.e., the MSA concentration and temperature, were modified to selectively leach the three types of lamp phosphors. Pure MSA (10%) could selectively leach the HALO phosphor at 25 °C. The calcium-rich PLS was purified by vacuum distillation, and both calcium and the phosphoric acid that was formed were recovered. The calcium-free residue was leached with diluted MSA to selectively leach the YOX phosphor. The remaining residue could be leached with pure MSA at elevated temperatures to recover the LAP phosphor. The Y-rich PLS and the La-rich PLS were purified via solvent extraction with D2EHPA and stripping precipitation with oxalic acid to produce a highly pure Y2O3 + Eu2O3 final product after calcination.

Reproduced from reference [83]
Effect of MSA concentration on the leaching efficiency of the lamp phosphors YOX, HALO, and LAP. The leaching conditions were: a liquid-to-solid ratio (L/S) = 15 L/kg, t = 2 h, T = 25 °C; b L/S = 15 L/kg, t = 2 h, T = 80 °C.
Note that all these processes for the recycling of REEs from lamp phosphors were developed at a time when fluorescent lamps were an expanding market. At present, the demand for lamp phosphors for fluorescent lamps is much lower due to the popularity of light-emitting diodes (LEDs) for lighting applications. This has a significant effect on the demand for highly pure europium and yttrium oxides, with much lower prices for these compounds as a consequence. The situation for terbium is somewhat different, since the terbium recycled from end-of-life fluorescent lamps could be reused in REE permanent magnets.
Cathode-Ray-Tube (CRT) Phosphor Waste
Closely related to the work on the recovery of REEs from fluorescent-lamp-phosphor waste is the recovery of europium and yttrium from cathode-ray-tube (CRT) phosphor waste from old television and computer screens [84]. The phosphor composition is different from that of fluorescent lamps. In CRTs, the blue and green phosphors are based on zinc sulfide (ZnS:Ag for the blue phosphor and ZnS:Cu for the green phosphor), while the red phosphor is Y2O2S:Eu3+. The latter phosphor is rich in yttrium and europium. The developed flow sheet was based on the sequential steps of (1) roasting, (2) leaching with organic acids (acetic acid and MSA) and (3) precipitation (Fig. 8). Zinc was efficiently removed from the roasted CRT phosphors by leaching with acetic acid, giving access to the REE content. Yttrium and europium were quantitatively leached from the residue by a 1 M MSA solution. Precipitation with oxalic acid gave a mixed Y/Eu oxalate that could be calcined to a mixed Y/Eu oxide of high purity (> 99 wt%). This mixed Y/Eu oxide can be reused as a recycled YOX phosphor, or further treated for the separation of the two elements. It must be realized that the available volumes of CRT-phosphor waste are much smaller than that of lamp-phosphor waste, and this trend will continue in the future since CRT technology become obsolete several years ago.

Conceptual flowsheet for the recovery of europium and yttrium from cathode-ray-tube (CRT) phosphor waste. Redrawn from reference [84]
Photovoltaic Modules
Palitzsch at Loser Chemie (Germany) developed an innovative process for recycling end-of-life thin-film photovoltaic (PV) modules, in which MSA plays an essential role [85, 86]. In contrast to conventional recycling technologies, the photovoltaic modules are not shredded so that the glass is not broken. The sandwich structure of the modules is opened, and the glass panels are taken out in one piece. These glass panels are coated with a thin layer of a semiconductor material, such as cadmium telluride (CdTe), gallium arsenide (GaAs), copper indium selenide (CIS) or copper indium gallium selenide (CIGS). The molybdenum, indium tin oxide (ITO) and silver conducting layers and contacts can be dissolved in a mixture of MSA and hydrogen peroxide (50%). Silver can be recovered from the polymetallic solution by the precipitation of silver(I) chloride (AgCl) upon the addition of hydrochloric acid [85, 87]. Recovery of the other elements is not disclosed. The cleaned glass panels can be reused or used as a raw material in the production of floated glass. The dissolution step with MSA can also be applied to the silver contacts of silicon solar cells [88].
Korean researchers have developed a process to recover silver from crystalline silicon wafers in end-of-life photovoltaic modules by oxidative dissolution of silver metal by a mixture of MSA and hydrogen peroxide [89]. Experiments were carried out to optimize the MSA:H2O2 ratio. The MSA was regenerated by addition of hydrochloric to precipitate AgCl. The precipitated AgCl was converted to silver powder by reaction with NaOH and H2O2. Since the recovered silver metal has a low purity (99.8%,) due to lead and chlorine impurities, it was further purified to high-purity silver (99.995%) by electrorefining via a AgNO3 electrolyte. In a later modification of the process, electrorefining was done via a Ag-MSA electrolyte [90]. The concentration of MSA in the regenerated solution was increased by removing water by fractional distillation [91].
Waste Printed-Circuit Boards (PCBs)
Mixtures of MSA and hydrogen peroxide have also been used to selectively dissolve the solder (lead–tin alloy) for the dismantling of electronic components from waste printed-circuit boards (PCBs) [25]. The MSA and H2O2 concentrations and the reaction time needed to be optimized to selectively dissolve the lead and tin, with a minimum co-dissolution of the copper present underneath the solder. A lixiviant containing 3.5 M MSA and 0.5 M H2O2, and a reaction time of 45 min, yielded the optimum conditions for selective desoldering. After the dissolution of the solder, the electronic components could be easily separated from the PCBs. It must be mentioned that metallic tin can also be dissolved in MSA if oxygen is present. In the electroplating processes of tin from MSA electrolyte, the loss of tin in the electrolyte is replenished externally, by pumping the tin(II) methanesulfonate electrolyte from the plating bath to a column reactor filled with tin metal particles and fed with oxygen gas [92]. Thereafter, the more concentrated tin(II) methanesulfonate solution is pumped back to the electroplating bath. Hydrogen evolution can be neglected during the dissolution of the tin in the presence of oxygen.
Research Opportunities and Needs
In the previous sections, an overview has been given of the literature on the properties of MSA and the methanesulfonate salts, as well as of studies of the use of MSA in hydrometallurgy and closely related fields (e.g., the electroplating industry). This review highlights that this work is still rather fragmentary, compared to the huge body of literature on the applications of other acids in hydrometallurgy. Therefore, in this section, we identify possible knowledge gaps that may inhibit the widespread use of MSA in the metallurgical industry.
Before describing these knowledge gaps, it is important to stress that the likelihood that metallurgical engineers will adopt MSA in genuine, cost-effective industrial flowsheets is far higher than is the case for neoteric solvents such as ionic liquids or deep-eutectic solvents (DESs). The properties of MSA are simply too good to be ignored. The higher price of MSA compared to other acids used in hydrometallurgy might still be a hurdle, but the prices are expected to decrease over time due to increasing production capacity for MSA in the chemical industry (corrected for higher prices of natural gas and energy). It is also an asset that the applications of MSA are not restricted to hydrometallurgy, as this acid finds applications in many other industries: e.g., oil, metal-finishing (electroplating), the chemical and pharmaceutical industries. Furthermore, MSA has great potential for future generations of redox-flow batteries (i.e., a large volume application). This widespread range of applications, in combination with the low toxicity and environmental friendliness of MSA, makes it unlikely that MSA will disappear from the market in the near future (unlike what happened to many extractants for solvent extraction in hydrometallurgy, such as Cyanex 301). Of paramount importance is that MSA is fully compliant with the REACH regulations of the European Union, and with similar regulations in other parts of the world [93]. This gives it a huge competitive advantage compared to neoteric solvents, as design engineers prefer well-known, commercially available chemicals over novel compounds because of the easily available data and the mitigation of safety risks and process-performance risks. The same holds for health, safety and environment (HSE) advisors and the authorities who deliver the required safety and environmental permits to authorize the building and startup of a new plant.
So, what are the remaining (fundamental) knowledge gaps for MSA, which need to be tackled to allow design engineers to fully adopt MSA in novel hydrometallurgical processes? Which fundamental and applied research topics are of interest to facilitate the widespread use of MSA in the metallurgical industry?
Fundamental Research Needs
First of all, there is only a limited amount of experimental data about the physicochemical properties of MSA electrolytes. Likewise, the available data on the liquid–solid and vapor–liquid equilibria is rather fragmentary. Similarly, only few experimental phase diagrams of binary systems of methanesulfonate salts with water exist [34, 35]. This is even less the case for ternary systems, comprising either MSA—methanesulfonate-salt-water, or phase diagrams of mixtures of methanesulfonate salts with salts of other anion (mixed-anion systems) [36].
As mentioned above, the solubilities of methanesulfonate have only been determined at room temperature and not at higher temperatures, so that the temperature dependence of the solubility is not known. Even at the standard temperature of 25 °C, the available data are not accurate. Likewise, there are no systematic studies on the effect of the concentration of free MSA on the solubility of the methanesulfonate salts. That being said, the accurate determination of solubilities for highly soluble salts such a methanesulfonates is not straightforward [94]. Incorrect solubility data can arise from the failure to achieve true equilibrium between properly defined solid and solution phases. Highly soluble metal salts often form hydrates, and we have to characterize the solid that is in equilibrium with the saturated solution. The hydrates might undergo solid-state conversions, and the attainment of the true equilibrium is often a very slow process. Because highly soluble metal salts often have very large temperature coefficients of solubility, the saturated solution might crystallize out in the syringe needle that is used for sampling. Highly concentrated solutions of metal salts are often very viscous so that they are difficult to sample or to filter.
Likewise, densities of concentrated solutions have not been measured systematically yet. With a few exceptions, the vapor pressure of MSA electrolytes has not been studied as a function of salt concentration, and the water activity of these solutions is not known [35, 36]. Measurements should not be restricted to single-metal systems; binary systems must be considered as well. Such data are required for a determination of the parameters for activity-coefficient models, such as the Pitzer interaction parameters [95]. Hardly any standard thermodynamic properties are available for the methanesulfonate salts (Gibbs free energy, enthalpy, entropy, heat capacity). Measurements of thermodynamic data must be complemented by speciation studies in (concentrated) solutions. It is not known yet whether methanesulfonate forms inner-sphere complexes with metal ions. Further application of computational chemistry, such as density functional theory (DFT) or molecular dynamics (MM) calculations, to MSA and its salts is encouraged as well [96, 97]. Such fundamental studies might seem at first sight not relevant to the application of MSA in hydrometallurgy, but such data are essential as input for the software for predictive thermodynamic modeling (e.g., with OLI Systems or HSC Chemistry), and in an extension for flowsheet modelling by metallurgical engineers.
Applied Research Needs
From a more applied research point of view, we suggest undertaking a systematic comparison between the reactivity of minerals with MSA and the reactivity with other acids (sulfuric acid in particular), because these studies give valuable information about the leachability of minerals in different types of ores. These studies must go beyond the conventional tests in which only a few process variables, such as temperature, time, particle size, acid concentration and stirring speed, are varied. Such studies merely provide trivial conclusions, for instance that the leaching rate increases with higher temperature, decreasing particle size, or enhanced stirring speed [98]. The kinetics should be studied in detail and should be modelled to the appropriate kinetic models.
Not only leaching studies should be performed, but also studies about the recovery of excess MSA after leaching (rather than neutralization by the addition of a base), purification of the pregnant leach solution, and recovery of metals. The removal of impurity anions (sulfate, chloride, phosphate, arsenate) is particularly challenging. Essential for the design of circular hydrometallurgical flowsheets is the regeneration of the acid [99]. Unlike hydrochloric acid, MSA cannot be recovered by processes such as pyrohydrolysis or reactive distillation [4], because MSA has a very low vapor pressure and cannot be volatilized. If the solution contains high concentrations of calcium methanesulfonate, MSA can be regenerated by the addition of sulfuric acid, so that calcium sulfate precipitates (vide supra). However, new approaches to the regeneration of MSA from the raffinate after metal recovery must be developed. Careful washing of leaching residues with water must be carried out after solid–liquid separation of the residues from the PLS in order to minimize the losses of MSA. Adsorption of MSA on the surface of solid particles could lead to prohibitively high acid losses. It will be a challenge to recover MSA from these dilute washing solutions. Losses of MSA by inefficient washing has a negative impact on the process economics, especially because of the higher price of MSA compared to that of other acids used in hydrometallurgy.
The low volatility and high thermal stability make MSA a very suitable reagent for dry digestion, in which the ore is mixed with a small amount of acid to a moist mixture that is heated for some time at a temperature between 100 and 150 °C, followed by water leaching [100]. This procedure is not only very efficient in terms of water usage, but it also makes it possible to hydrometallurgically process ores that contain easily soluble silica that might form silica gel in the pregnant leach solution. Although MSA shares this property with sulfuric acid, the resulting methanesulfonate salts are more soluble than the corresponding sulfates. On the other hand, unlike sulfuric acid, MSA cannot be used for a process similar to sulfation roasting because MSA is not stable at the high temperatures required.
Systematic studies on solvent extraction (SX) and ion exchange (IX) from MSA solutions could be useful, not only for the purification of the pregnant leach solution and for the metal recovery, but also for rejuvenating the electrolyte solutions that are used for electroplating. Very little is known about the solvent extraction of MSA or concerning the extraction of metal ions from MSA media. However, because of the very poor tendency of methanesulfonate to form anionic metal complexes, it is expected that only acidic extractants (such as D2EHPA, Cyanex 272 or Ionquest 290) are efficient for the extraction of metal ions from MSA solutions. Solvating and basic extractants will not give high extraction yields. For the same reason, it is anticipated that only cation-exchange resins or chelating resins will be efficient for the uptake of metal ions from MSA solutions.
Finally, more research on MSA in the field of copper electrorefining would be useful, given the ability to obtain more concentrated electrolytes with MSA. Currently, copper refineries are making use of electrolytes of copper(II) sulfate dissolved in sulfuric acid. Although CuSO4 is known to be very soluble in water, its solubility rapidly decreases in the presence of sulfuric acid. For instance, a saturated CuSO4 solution contains 395 g of dissolved salt per liter at 15 °C, but upon the addition of 100 g per liter of H2SO4, the solubility of CuSO4 is reduced to 215 g per liter [101]. Although the solubility of copper(II) methanesulfonate is undoubtedly lower in the presence of MSA than in aqueous solutions without free MSA (no experimental data available yet), the approach of using this salt for copper refining is worth investigating, since the solubility of copper(II) methanesulfonate in water is about 65% higher than that of copper(II) sulfate [102].
Conclusions
In this review we presented MSA as a “new” acid in hydrometallurgy. Compared to the more conventional acids presently used in hydrometallurgy (H2SO4, HCl, HNO3), MSA is blessed with a multitude of (potential) advantages. One major benefit is the very high solubility of methanesulfonate salts in water, implying that very concentrated solutions can be used without the risk of unwanted precipitation or the crystallization of solids. The high chemical stability of MSA, the large solubility of its metal salts, the high electrical conductivity of its solutions, as well as the oxygen evolution as the anode reaction indicate that MSA electrolytes are very interesting for the electrowinning and electrorefining of metals. Its low corrosivity puts fewer constraints on the material selection with respect to chloride hydrometallurgy. Although MSA itself is a non-oxidizing acid, its mixtures with hydrogen peroxide are strongly oxidizing and can be used for the oxidative leaching of difficult-to-dissolve ore minerals such as chalcopyrite. The low vapor pressure, the low toxicity and its biodegradability contribute to the “green” character of MSA. One of the few disadvantages of MSA is its relatively high price compared to other acids, and especially sulfuric acid. That being said, MSA is a bulk chemical, commercially produced by reliable companies at a reasonable cost, and REACH-compliant, giving it a competitive advantage over neoteric solvents such as ionic liquids. Furthermore, it is very likely that the price of MSA will further drop in the future when the production capacity will be gradually increased, and more economic processes become operational. Still, MSA will never be as cheap as sulfuric acid, so it is not realistic to propose MSA as a general acid in hydrometallurgy that will comprehensively replace sulfuric acid in all hydrometallurgical applications. However, in those cases where metallurgical engineers are faced with serious problems due to the low solubility of metal sulfates, such as in lead hydrometallurgy, it is worth considering MSA as an alternative for tetrafluoroboric acid ore hexafluorosilicic acid. Also, in electrorefining processes, where a high concentration of metal salts is required, MSA technology is an option. Although the number of experimental studies on possible applications of MSA in hydrometallurgy is steadily increasing, there are still significant gaps in our knowledge of the physicochemical properties of the methanesulfonate salts and their aqueous solutions. Research should focus on those areas, as such information is essential for flowsheet modelling of MSA-based hydrometallurgical processes.
References
Free ML (2013) Hydrometallurgy: fundamentals and applications. Wiley, New York
Gupta GK, Mukherjee TK (2019) Hydrometallurgy in extraction processes, volume I and II. CRC Press, Boca Raton, Florida, USA
Schlesinger M, King M, Sole K, Davenport W (2011) Extractive metallurgy of copper. Elsevier, Amsterdam
Demopoulos GP, Li Z, Becze L et al (2008) New technologies for HCl regeneration in chloride hydrometallurgy. World Metall-ERZMETALL 61:89–98
Verhulst D, Lakshmanan VI (2011) Hydrometallurgy of chlorides: a review of recent developments. In: EPD Congress 2011. Wiley, pp 398–413
Van Weert G (1989) Chloride and nitrate systems in hydrometallurgy applications and opportunities. PhD thesis, TU Delft
Gernon M, Wu M, Buszta T, Janney P (1999) Environmental benefits of methanesulfonic acid: comparative properties and advantages. Green Chem 1:127–140. https://doi.org/10.1039/a900157c
Billeter OC (1905) V. Entstehung von Anhydriden der Sulfonsäuren durch Einwirkung von Sulfochloriden auf cyansaures Silber. Ber Dtsch Chem Ges 38:2015–2020. https://doi.org/10.1002/cber.190503802136
Proell WA, Adams CE, Shoemaker BH (1948) Properties and uses of alkanesulfonic acids. Ind Eng Chem 40:1129–1132. https://doi.org/10.1021/ie50462a028
Rosenstein C (1990) Methane sulfonic acid as an electrolyte for tin, lead and tin-lead plating for electronics. Met Finish 88:17–21
BASF (2016) Lutropur®–the friendly acid. The purest form of methanesulfonic acid (MSA) made by BASF. BASF, Ansan
Roitman D, McAlister J, Oaks F (1994) Composition characterization of methanesulfonic-acid. J Chem Eng Data 39:56–60. https://doi.org/10.1021/je00013a016
Lafitte J-A, Monguillon B (2013) Use of alkane sulphonic acid for rust removal. US patent 8,574,370
BASF (2019) Mining solutions SolutrixTM E: the efficient descaling solution for the mining industry. BASF, Ansan
Bertkau W, Hatscher S, Frenzel S, Ossmer U (2015) Process for dissolving deposits comprising alkaline earth metal sulfates (US Patent US20150175872)
Walsh FC, de Leon CP, Berlouis L et al (2015) The development of Zn-Ce hybrid redox flow batteries for energy storage and their continuing challenges. ChemPlusChem 80:288–311. https://doi.org/10.1002/cplu.201402103
Ewe T, Scheible W (2017) Der Griff zum Heiligen Gral. Bild der Wiss 62–70
McCoy M (2016) German firm claims new route to methanesulfonic acid. C&EN Glob Enterp 94:10–10. https://doi.org/10.1021/cen-09426-notw7
Diaz-Urrutia C, Ott T (2019) Activation of methane: a selective industrial route to methanesulfonic acid. Science 363:1326–1329. https://doi.org/10.1126/science.aav0177
Kappenthuler S, Olveira S, Wehrli J, Seeger S (2018) Environmental assessment of alternative methanesulfonic acid production using direct activation of methane. J Clean Prod 202:1179–1191. https://doi.org/10.1016/j.jclepro.2018.07.284
Weil ED, Sandler SR, Gernon M (2006) Sulfur compounds. Kirk-Othmer Encycl Chem. https://doi.org/10.1002/0471238961.1921120623050912.a01.pub2Technology
Clarke J, Woodward L (1966) Raman spectrophotometric determination of degrees of dissociation of methanesulphonic acid in aqueous solution at 25 degrees C. Trans Faraday Soc 62:2226–2233. https://doi.org/10.1039/tf9666202226
Guthrie J, Gallant R (2000) Thermodynamics of methanesulfonic acid revisited. Can J Chem-Rev Can Chim 78:1295–1298. https://doi.org/10.1139/cjc-78-10-1295
Zhang X, Zhang C, Zheng F et al (2020) Alkaline electrochemical leaching of Sn and Pb from the surface of waste printed circuit board and the stripping of gold by methanesulfonic acid. Environ Prog Sustain Energy. https://doi.org/10.1002/ep.13324
Zhang X, Guan J, Guo Y et al (2017) Effective dismantling of waste printed circuit board assembly with methanesulfonic acid containing hydrogen peroxide. Environ Prog Sustain Energy 36:873–878. https://doi.org/10.1002/ep.12527
Gernon M (1996) Preparation of a precious metal salt of a nonoxidizing acid by direct reaction. US Patent 5,491,247
Finsgar M, Milosev I (2010) Corrosion behaviour of stainless steels in aqueous solutions of methanesulfonic acid. Corros Sci 52:2430–2438. https://doi.org/10.1016/j.corsci.2010.04.001
Baker S, Kelly D, Murrell J (1991) Microbial-degradation of methanesulfonic-acid—a missing link in the biogeochemical sulfur cycle. Nature 350:627–628. https://doi.org/10.1038/350627a0
De Marco P, Murrell J, Bordalo A, Moradas-Ferreira P (2000) Isolation and characterization of two new methanesulfonic acid-degrading bacterial isolates from a Portuguese soil sample. Arch Microbiol 173:146–153. https://doi.org/10.1007/s002039900124
Hopf J, Weigelt A, Bombach H et al (2022) Investigations of the leaching of raw silver granules with methanesulfonic acid (MSA). World Metall ERZMETALL 75:94–99
Palden T, Onghena B, Regadio M, Binnemans K (2019) Methanesulfonic acid: a sustainable acidic solvent for recovering metals from the jarosite residue of the zinc industry. Green Chem 21:5394–5404. https://doi.org/10.1039/c9gc02238d
Palden T, Regadio M, Onghena B, Binnemans K (2019) Selective metal recovery from Jarosite residue by leaching with acid-equilibrated ionic liquids and precipitation-stripping. ACS Sustain Chem Eng 7:4239–4246. https://doi.org/10.1021/acssuschemeng.8b05938
Capelato M, Nobrega J, Neves E (1995) Complexing power of alkanesulfonate ions—the lead methanesulfonate system. J Appl Electrochem 25:408–411. https://doi.org/10.1007/BF00249661
Belova EV, Krasnov VS, Ilyukhin AB, Uspenskaya IA (2018) Solid-liquid phase equilibrium in the water-Zn(II) methanesulfonate and water-Cu(II) methanesulfonate systems. Thermochim Acta 668:46–57. https://doi.org/10.1016/j.tca.2018.08.004
Belova EV, Finkelshteyn DI, Maksimov AI, Uspenskaya IA (2019) Water-zinc (copper) methanesulfonate systems: thermodynamic properties and phase equilibria. Russ J Phys Chem A 93:2117–2122. https://doi.org/10.1134/S0036024419110050
Novikov AA, Belova EV, Uspenskay IA (2019) Phase equilibria and thermodynamic properties in the zinc chloride-zinc methanesulfonate-water system. J Chem Eng Data 64:4230–4238. https://doi.org/10.1021/acs.jced.9b00292
Palitzsch W, Loser U (2015) Inexpensive and environmentally friendly recycling of photovoltaic scrap. In: 2015 IEEE 42nd Photovoltaic Specialist Conference (PVSC). IEEE, pp 1–4
Palitzsch W (2013) Innovative Recycling Strategies for Rare Earths. In: Presentation at the Rare Earth Elements and Compounds Conference (REEC), Münster, Germany, 10–12 September 2013
Loser U, Palitzsch W (2015) Hydrometallurgisches Verfahren zur Verwertung von Seltenerdmetalboridabfällen (German patent DE 10 2014 112 952)
Lu J, Dreisinger D (2021) Lead electrowinning from methane sulfonic acid. Hydrometallurgy 203:105623. https://doi.org/10.1016/j.hydromet.2021.105623
Li M, Yang J, Liang S et al (2019) Review on clean recovery of discarded/spent lead-acid battery and trends of recycled products. J Power Sourc 436:226853. https://doi.org/10.1016/j.jpowsour.2019.226853
Tan S, Payne DJ, Hallett JP, Kelsall GH (2019) Developments in electrochemical processes for recycling lead–acid batteries. Curr Opin Electrochem 16:83–89. https://doi.org/10.1016/j.coelec.2019.04.023
Fingland JJ (1930) The Betts electrolytic lead refining process in practice. Trans Am Electrochem Soc 57:177–204. https://doi.org/10.1149/1.3492149
González-Domínguez JA, Peters E, Dreisinger DB (1991) The refining of lead by the Betts process. J Appl Electrochem 21:189–202. https://doi.org/10.1007/BF01052570
Prengaman R (1995) Recovering lead from batteries. JOM J Miner Met Mater Soc 47:31–33. https://doi.org/10.1007/BF03221127
Olper M (1998) Fluoborate technology-a new challenging way for primary and. In: Zinc and Lead Processing: Proceedings of an International Symposium on Zinc and Lead Processing, Calgary, Canada, August 16–19, 1998. Canadian Institute of Mining Metallurgy and Petroleum, pp 185–198
Ojebuoboh F, Wang S, Maccagni M (2003) Refining primary lead by granulation-leaching-electrowinning. JOM 55:19–23. https://doi.org/10.1007/s11837-003-0082-2
Jin B, Dreisinger D (2016) A green electrorefining process for production of pure lead from methanesulfonic acid medium. Sep Purif Technol 170:199–207. https://doi.org/10.1016/j.seppur.2016.06.050
Fassbender S, Dreisinger D, Wu ZH (2016) Recovering lead from a mixed oxidized material. US Patent 9,322,104
Fassbender S, Dreisinger D, Wu ZH (2016) Recovering lead from a lead material including lead sulfide. US Patent 9,322,105
Wu Z, Dreisinger D, Urch H, Fassbender S (2014) Fundamental study of lead recovery from cerussite concentrate with methanesulfonic acid (MSA). Hydrometallurgy 142:23–35. https://doi.org/10.1016/j.hydromet.2013.10.018
Wu Z, Dreisinger DB, Urch H, Fassbender S (2014) The kinetics of leaching galena concentrates with ferric methanesulfonate solution. Hydrometallurgy 142:121–130. https://doi.org/10.1016/j.hydromet.2013.10.017
Dreisinger D, Baxter K, Worland A, et al (2020) Lead metal production at Paroo station mine using leach-electrowinning process in methane sulfonic acid solution. In: Siegmund A, Alam S, Grogan J, Kerney J, Shibata E (ed) 9th International Symposium on Lead and Zinc Processing, PBZN 2020. Minerals, Met & Mat Soc; MetSoc; GDMB; MMIJ; NFSoc, pp. 135–163
Feng Q, Wen S, Zhao W et al (2015) Leaching of copper from malachite with methane-sulfonic acid. Solvent Extr Res Dev Jpn 22:159–168. https://doi.org/10.15261/serdj.22.159
Barton IF, Hiskey JB (2022) Chalcopyrite leaching in novel lixiviants. Hydrometallurgy 207:105775. https://doi.org/10.1016/j.hydromet.2021.105775
Ahn J, Wu J, Lee J (2019) Investigation on chalcopyrite leaching with methanesulfonic acid (MSA) and hydrogen peroxide. Hydrometallurgy 187:54–62. https://doi.org/10.1016/j.hydromet.2019.05.001
Wu J, Ahn J, Lee J (2021) Kinetic and mechanism studies using shrinking core model for copper leaching from chalcopyrite in methanesulfonic acid with hydrogen peroxide. Miner Process Extr Metall Rev 42:38–45. https://doi.org/10.1080/08827508.2020.1795850
Hidalgo T, Kuhar L, Beinlich A, Putnis A (2018) Kinetic study of chalcopyrite dissolution with iron(III) chloride in methanesulfonic acid. Miner Eng 125:66–74. https://doi.org/10.1016/j.mineng.2018.05.025
Hidalgo T, Kuhar L, Beinlich A, Putnis A (2019) Kinetics and mineralogical analysis of copper dissolution from a bornite/chalcopyrite composite sample in ferric-chloride and methanesulfonic-acid solutions. Hydrometallurgy 188:140–156. https://doi.org/10.1016/j.hydromet.2019.06.009
Ahn J, Wu J, Lee J (2022) A comparative kinetic study of chalcopyrite leaching using alternative oxidants in methanesulfonic acid system. Miner Process Extr Metall Rev 43:390–401. https://doi.org/10.1080/08827508.2021.1893719
Urch H, Rein C, Fitzmaurice NJ, et al (2018) Removal of radionuclides from mixtures. Patent application US 2018/0010208
Feng Q, Wen S, Zhao W et al (2015) Dissolution regularities of smithsonite in methane sulfonic acid. Russ J Non-Ferrous Met 56:365–371. https://doi.org/10.3103/S1067821215040033
Zhang Q, Wen S, Feng Q et al (2019) Dissolution kinetics of hemimorphite in methane sulfonic acid. Physicochem Probl Miner Process 55:1–9. https://doi.org/10.5277/ppmp18105
Nikkhou F, Kartal M, Xia F (2021) Ferric methanesulfonate as an effective and environmentally sustainable lixiviant for Zn extraction from sphalerite (ZnS). J Ind Eng Chem 96:226–235. https://doi.org/10.1016/j.jiec.2021.01.017
Hopf J, Weigelt A, Bombach H et al (2021) Refining of precious metal bearing materials from secondary sources-methanesulfonic acid leaching of raw silver granules as a promising approach towards a green way of silver refining. Materials 14:6095. https://doi.org/10.3390/ma14206095
Rodriguez N, Onghena B, Binnemans K (2019) Recovery of lead and silver from zinc leaching residue using methanesulfonic acid. ACS Sustain Chem Eng 7:19807–19815. https://doi.org/10.1021/acssuschemeng.9b05116
Binnemans K, Jones P, Blanpain B et al (2015) Towards zero-waste valorisation of rare-earth-containing industrial process residues: a critical review. J Clean Prod 99:17–38. https://doi.org/10.1016/j.jclepro.2015.02.089
Ait Brahim J, Merroune A, Boulif R et al (2022) Efficient leaching process of rare earth, alkali and alkaline earth metals from phosphogypsum based on methanesulfonic acid (MSA) as green & eco-friendly lixiviant. RSC Adv 12:30639–30649. https://doi.org/10.1039/D2RA04124C
Banerjee R, Chakladar S, Mohanty A et al (2022) Leaching characteristics of rare earth elements from coal ash using organosulphonic acids. Miner Eng 185:107664. https://doi.org/10.1016/j.mineng.2022.107664
Crane RA, Sapsford DJ (2018) Towards Greener Lixiviants in value recovery from mine wastes: efficacy of organic acids for the dissolution of copper and arsenic from legacy mine tailings. Minerals 8:383. https://doi.org/10.3390/min8090383
Bevandić S, Xanthopoulos P, Muchez P (2021) Chemical leaching of sulfidic mining waste, Plombières tailings pond, eastern Belgium: insights from a mineralogical approach. J Sustain Metall 7:1444–1455. https://doi.org/10.1007/s40831-021-00445-0
Cagno S, Gijsemans L, Tyrpekl V et al (2017) Use of triflic acid in the recycling of Thoria from nuclear fuel production scrap. J Sustain Metall 3:659–667. https://doi.org/10.1007/s40831-017-0136-2
Lyczko K, Lyczko M, Walo M, Lipkowski J (2012) Conversion of thorium(IV) oxide into thorium(IV) trifluoromethanesulfonate: crystal structure of thorium(IV) trifluoromethanesulfonate dihydrate. Inorg Chem Commun 24:234–236. https://doi.org/10.1016/j.inoche.2012.07.024
Tyrpekl V, Lommelen R, Wangle T et al (2019) Studies on the thoria fuel recycling loop using triflic acid: effects of powder characteristics, solution acidity, and radium behavior. J Sustain Metall 5:118–126. https://doi.org/10.1007/s40831-018-0205-1
Clarke RL, King MJ, Dougherty B, Hurwitz MD (2020) Electrodeposited lead composition, methods of production, and uses. US Patent 10,689,769
Clarke RL, Dougherty B, Mohanta S (2021) Systems and methods for recovery of lead from lead acid batteries. US Patent 11,028,460
Chang C, Yang S, Li Y et al (2023) Green hydrometallurgical extraction of metallic lead from spent lead paste in the methanesulfonic acid system. Sep Purif Technol 306:122592. https://doi.org/10.1016/j.seppur.2022.122592
Wang B, Lin X-Y, Tang Y et al (2019) Recycling LiCoO2 with methanesulfonic acid for regeneration of lithium-ion battery electrode materials. J Power Sourc 436:226828. https://doi.org/10.1016/j.jpowsour.2019.226828
Binnemans K, Jones PT, Blanpain B et al (2013) Recycling of rare earths: a critical review. J Clean Prod 51:1–22. https://doi.org/10.1016/j.jclepro.2012.12.037
Binnemans K, Jones PT (2014) Perspectives for the recovery of rare earths from end-of-life fluorescent lamps. J Rare Earths 32:195–200. https://doi.org/10.1016/S1002-0721(14)60051-X
Gijsemans L, Forte F, Onghena B, Binnemans K (2018) Recovery of rare earths from the green lamp phosphor LaPO4:Ce3+, Tb3+ (LAP) by dissolution in concentrated methanesulphonic acid. RSC Adv 8:26349–26355. https://doi.org/10.1039/c8ra04532a
Binnemans K, Jones PT (2017) Solvometallurgy: an emerging branch of extractive metallurgy. J Sustain Metall 3:570–600. https://doi.org/10.1007/s40831-017-0128-2
Rodriguez NR, Grymonprez B, Binnemans K (2021) Integrated process for recovery of rare-earth elements from lamp phosphor waste using methanesulfonic acid. Ind Eng Chem Res 60:10319–10326. https://doi.org/10.1021/acs.iecr.1c01429
Forte F, Yurramendi L, Aldana JL et al (2019) Integrated process for the recovery of yttrium and europium from CRT phosphor waste. RSC Adv 9:1378–1386. https://doi.org/10.1039/C8RA08158A
Palitzsch W, Loser U (2017) Integrated PV-recycling—more efficient, more effective. In: 2017 IEEE 44th Photovoltaic Specialist Conference (PVSC), pp. 2272–2274
Palitzsch W, Killenberg A, Schoenherr P, Loser U (2018) Photovoltaic recycling with the help of water and light—it does not get greener. In: 2018 IEEE 7th World Conference on Photovoltaic Energy Conversion (WCPEC). IEEE, pp. 2465–2466
Loser U, Palitzsch W (2016) Method for concentrating metals from scrap containing metal (Patent US 2016/0053343)
Palitzsch W, Loser U (2013) Systematic photovoltaic waste recycling. Green 31:79–82. https://doi.org/10.1515/green-2013-0008
Yang E-H, Lee J-K, Lee J-S et al (2017) Environmentally friendly recovery of Ag from end-of-life c-Si solar cell using organic acid and its electrochemical purification. Hydrometallurgy 167:129–133. https://doi.org/10.1016/j.hydromet.2016.11.005
Lee J-K, Lee J-S, Ahn Y-S, Kang G-H (2018) Efficient recovery of silver from crystalline silicon solar cells by controlling the viscosity of electrolyte solvent in an electrochemical process. Appl Sci 8:2131. https://doi.org/10.3390/app8112131
Lee J-K, Lee J-S, Ahn Y-S, Kang G-H (2019) Restoring the reactivity of organic acid solution used for silver recovery from solar cells by fractional distillation. Sustainability 11:3659. https://doi.org/10.3390/su11133659
De Greef R, Janssen L (2001) Electrochemical dissolution of tin in methanesulphonic acid solutions. J Appl Electrochem 31:693–702. https://doi.org/10.1023/A:1017534220761
Lahl U, Hawxwell KA (2006) REACH—the new European chemicals law. Environ Sci Technol 40:7115–7121. https://doi.org/10.1021/es062984j
Hefter G (2013) Some highs and lows (and in-betweens) of solubility measurements of solid electrolytes. Pure Appl Chem 85:2077–2087. https://doi.org/10.1351/PAC-CON-12-11-06
Pitzer K (2018) Activity coefficients in electrolyte solutions, 2nd edn. CRC Press, Boca Raton
Canales M, Aleman C (2014) Molecular dynamics simulation study of methanesulfonic acid. J Phys Chem B 118:3423–3430. https://doi.org/10.1021/jp500817s
Canales M, Guardia E (2016) A comparative molecular dynamics study of sulfuric and methanesulfonic acids. J Mol Liq 224:1064–1073. https://doi.org/10.1016/j.molliq.2016.10.097
Wills B (2011) Hydrometallurgy- is it ‘bucket chemistry’? https://min-eng.blogspot.com/2011/01/hydrometallurgy-is-it-bucket-chemistry.html. Accessed 22 Aug 2022
Binnemans K, Jones PT (2023) The twelve principles of circular hydrometallurgy. J Sustain Metall. https://doi.org/10.1007/s40831-022-00636-3
Vossenkaul D, Birich A, Mueller N et al (2017) Hydrometallurgical processing of eudialyte bearing concentrates to recover rare earth elements via low-temperature dry digestion to prevent the silica gel formation. J Sustain Metall 3:79–89. https://doi.org/10.1007/s40831-016-0084-2
Holler HD, Peffer EL (1916) The density of aqueous solutions of copper sulfate and sulfuric acid. J Am Chem Soc 38:1021–1029. https://doi.org/10.1021/ja02262a008
Balaji R, Pushpavanam M (2003) Methane sulphonic acid in electroplating related metal finishing industries. Trans Inst Met Finish 81:154–158. https://doi.org/10.1080/00202967.2003.11871526
Acknowledgements
The authors thank the Industrial Research Fund (IOF) and the LRD Division RARE3 of KU Leuven for financial support. The authors acknowledge Paul McGuiness for the text editing and the layouting/redrawing of the figures. Finally, the authors are indebted to SOLVOMET researchers (Thomas Abo Atia, Brecht Dewulf, Jong Won Choi) for their much-appreciated inputs and suggestions.
Funding
The funding was provided by Industrial Research Fund, KU Leuven and LRD Division RARE3, KU Leuven.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interest to declare.
Additional information
The contributing editor for this article was Yongxiang Yang.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Binnemans, K., Jones, P.T. Methanesulfonic Acid (MSA) in Hydrometallurgy. J. Sustain. Metall. 9, 26–45 (2023). https://doi.org/10.1007/s40831-022-00641-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-022-00641-6