
Abstract
The recoveries of rare earth elements (REEs), nickel, and cobalt from hybrid electric vehicle batteries by sulfation, selective roasting, and water leaching have been studied. The cathode and anode materials of a Panasonic Prismatic Module nickel metal hydride (NiMH) battery were used in the study. The optimal conditions for each step of the process were determined by performing lab-scale experiments. It was found that 8 mol/L of sulfuric acid was sufficient for the sulfation with a solid-to-liquid ratio of 1/5. The optimal roasting conditions was determined to be 850 °C for 2 h. Under optimal conditions, 96% of the REEs could be obtained in the aqueous phase with negligible contamination of Ni and Co. The Ni and Co remained in solid phase as oxides together with traces of aluminum, zinc, and iron oxides. This method provides a way for the separation of the REEs from nickel, cobalt, and other elements present in the NiMH battery, into a leachate suitable for further processing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nickel metal hydride (NiMH) batteries are commonly used in many applications in our daily life. They are available in many sizes and forms from AA batteries to all-electric plug in vehicles in hybrid electric vehicle (HEV) applications. The big companies in the automobile industry, such as Toyota, Honda, Ford, and Chevrolet have been using NiMH batteries in their HEVs since the early 1980s. Upon the end of the batteries life cycle, the need to recycle them has been gaining importance. A NiMH battery is a source of rare earth elements (REEs) as well as of nickel and cobalt. The European Commission has declared REEs as critical elements due to their supply risk where Asia is dominating the market [1]. Moreover, to decrease the landfill and follow the European legislations for handling waste [2], recycling of NiMH batteries is an inevitable requirement for both economic and environmental reasons.
There are many studies on how to recycle batteries, from small-sized household batteries to the large-sized batteries used in vehicles [3,4,5,6,7,8]. Several techniques have been applied for the recovery of REEs and other valuable elements from NiMH batteries including acid leaching with various acids [3, 4, 7, 9,10,11,12,13,14,15,16], precipitation and solvent extraction methods with various reagents [5, 17,18,19,20,21,22], and pyrometallurgical methods [6, 23,24,25].
The objective of this study is to investigate a method that first transforms all the constituent elements of active material in a NiMH battery into their sulfate form by mixing with sulfuric acid and then roasting the dried mixture at a specific temperature for a certain amount of time. The resultant solid is a mixture of REE sulfates and the other elements in their oxide forms. Leaching the solid phase with water at ambient temperature will allow for the dissolution of rare earth sulfates and leave the rest of the elements in the leach residue as oxides. In the proposed method, the REEs are separated from the other elements present and the key process parameters are easy to control. Similar methods have been proposed to process nickel sulfide concentrates in a two-step process of oxidation–sulfation [26, 27] and to process limonitic nickel laterites with a sulfation–roasting–leaching process [28]. Selective sulfation with SO2 followed by water leaching has also been proposed for extraction of manganese from manganese oxide ore with high iron content [29]. Recent efforts have also been made to recover REEs from bauxite residue and from NdFeB magnets by sulfation, selective roasting, and water leaching [30, 31]. The key outcome in these processes is a total dissolution of the rare earth sulfates in water, while iron, nickel, cobalt, and other elements remain in their solid oxide form.
The Panasonic Prismatic Module 6.5 Ah nickel metal hydride (NiMH) 7.2 V plastic casing HEV battery (used in the present study) has been characterized [10, 32]. Solid-phase characterization of the active electrode materials was done using powder X-ray diffraction (XRD) and scanning electron microscopy with energy dispersive spectroscopy (SEM–EDS), and the chemical compositions of the active materials were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis after total dissolution in a mixture of acids. The anode-active material consists of Ce0.47 La0.34 Nd0.14 Pr0.05 Ni3.56 Co0.75 Al0.29 Mn0.4 (01-076-7719), and LaNi5. The cathode-active material consists of nickel hydroxide with a substitution of cobalt, yttrium oxide, and metallic nickel and cobalt.
The main reactions during the sulfation process are presented below, where M represents any metal.
All through the treatment, SO2 gas evolution is expected due to the strong oxidizing properties of the concentrated sulfuric acid. The kinetics is controlled by the temperature and possibly by the formation of sulfate or oxide layers on the surface of the metal particles and their resistance to dissolve in the acidic and high-temperature surroundings created by the exothermic reaction.
The thermal stability of the metal sulfates during the decomposition is a critical step to be considered in the selective roasting. Thermal decompositions of the Ni, Co, and REE sulfates have been studied extensively [31, 33,34,35,36,37,38]. Solubility data of different REE, Ni, and Co sulfate hydrates and stability constants for the formation of different REE, Ni, and Co sulfate complexes are available in the literature [39,40,41]. An important aspect is the formation of intermediate products during the selective roasting, such as rare earth oxysulfates. The solubilities of REE oxysulfates are limited in water; however, they dissolve under more acidic conditions. Most metals in oxide forms are almost insoluble in water. This shows the importance of the roasting temperature in order to obtain a high yield for the recovery of REEs in the aqueous phase and Ni and Co in the solid phase in their oxide forms after water leaching [42,43,44].
Experimental Procedure
The battery was manually disassembled, and the anode- and cathode-active materials were grinded separately. The active materials were mixed with concentrated sulfuric acid at room temperature to facilitate the sulfation process. The mixture was then dried and afterward roasted in a ventilated furnace at a fixed temperature for a certain amount of time. The roasted solid mixture was collected and leached with water at ambient temperature. During the selective water-leaching step, liquid samples were collected for analysis with ICP-OES, and at the end of the leaching, the solid residue was separated for powder XRD and SEM–EDS analyses. The complete set of experiments is presented in Table 1.
Material Characterization
The elemental compositions of the cathode and anode materials were determined by performing a total dissolution of dry samples of the respective materials using a strong acid cocktail of perchloric acid, hydrochloric acid, and nitric acid at 85 °C. The solution was then allowed to reach room temperature before dilution and elemental analysis by ICP-OES.
Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) at a heating rate of 10 °C/min under O2 atmosphere and 100 mL/min dry air flow were used to investigate the thermal decomposition behaviors of the materials (Mettler-Toledo, model-TGA/DSC 1). The samples used in the DSC/TGA analyses were prepared by mixing an equal weight mixture of anode- and cathode-active materials with 12 mol/L of sulfuric acid with solid-to-liquid (s/L) ratio of 1/5. The sample was then dried at 150 °C for 24 h and thereafter grinded into powder form.
For the characterization of the roasted materials before and after water leaching, scanning electron microscope was used with EDS capabilities (Hitachi S-3700N) as well as powder XRD (CuKα radiation, Siemens, Model D5000) for phase analyses.
Sulfation and Selective Roasting
The anode-active material was first grinded and sieved to obtain a powder with particle sizes < 38 µm. In the majority of the experiments, anode material was used since the REEs are concentrated in the anode-active material. Based on the performance of the separation of REE from the anodic material, the same conditions should be applicable to the cathode material and/or their mixture. The conditions for the roasting experiments are presented in Table 1. Acidic solutions were prepared by adding analytical grade sulfuric acid to deionized water. The solid-to-liquid ratios 1/5, 1/10 and 1/20 were studied to compare the effect of amount of acid versus acid concentration. Weighed samples of the dry solid material (1 g) were mixed with the aqueous phases in individual alumina crucibles using a silica fume glass rod. During this step gas evolution was observed. Constant stirring with the silica fume glass rod was important in order to avoid losing material due to bubbling of the gas evolved. The bubbling should be prevented as much as possible otherwise active battery material could escape from the crucible carried on the surface of the bubbles since the particle size is fine. Stirring also helped to avoid possible sintering of the material due to the heat evolved from the reaction. With the help of the silica fume glass rod, the bottom of the crucible was constantly swiped to force the reaction of all solid material with the acid.
A sharp decrease in intensity of the bubbling was observed with the decreasing acid concentration in experiments R4–R9. After the gas evolution stopped, the samples were dried in a furnace with ventilation. The drying was conducted at 150 °C for 24–72 h, until no difference in the sample weight over time was detected. The importance of the drying process before the roasting is to give the slurry enough time to complete the metal–acid reactions as well as to prevent instant boiling and loss of material due to splashing and liquid gas movement just after placing the crucible in the preheated furnace. Mixtures with lower initial acid concentrations (2M, 4M, 6M, and 8M) were sufficiently dried at 150 °C for 24 h. The slurries prepared with high acid concentrations (10M, 12M, and 14M) required 48 to 72 h of drying.
The dried samples were then roasted in an electrical furnace at 800, 850, or 900 °C. The residence time was varied from 1 to 3 h. This roasting process allowed the Ni and Co sulfate solid phases to decompose into their oxide forms while the REE remained in their sulfate forms. The temperature was selected based on available literature data on the thermal decomposition temperature range of the sulfates of REEs, nickel and cobalt [31, 33,34,35,36,37,38, 45].
Water Leaching
The roasted material obtained from the selective roasting stage was carefully scraped off from the crucible and put in polyethylene (PE) bottles for water leaching using deionized water (50 mL for 1 g of solid material). Part of the water was used to wash the crucibles before being transferred into HDPE bottles for leaching. The weight of the crucibles was noted before roasting and after water washing to know whether there was a material loss or not. The bottles were placed in a water bath held at 25 °C ± 0.1 °C. Each sample was continuously stirred (500 rpm) using magnetic bars. Samples of the aqueous phase were taken after 1, 24 and 72 h by using a syringe and solid material was immediately separated from the aqueous phase by using a syringe filter (0.2 μm). The leach residues were separated from the leach liquor after 72 h of leaching by using filter paper (0.2 μm). All aqueous phases were stored in HDPE bottles prior to ICP-OES analysis. Solid phases were analyzed with SEM–EDX and powder XRD. The pH after water leaching under optimal conditions was measured by using an Orion ROSS electrode, which was calibrated by Thermo Fisher Scientific standard solutions at pH 2, 4, and 7. The recovery percentages of the elements were calculated based on the measured aqueous concentrations of the elements after water leaching in relation to the total composition of the original materials.
Results and Discussion
Material Characterization
The weight percentages of the elements in the anode- and cathode-active material are presented in Table 2. The leach residue was 3.5% of the initial weight. An attempt to dissolve the residue in a fresh portion of acid mixture was not successful.
The data from the TGA is presented in Fig. 1. The first major weight loss starts at 25 °C and continues until 119 °C; this weight loss is due to a physical dehydration of the material. As seen in Fig. 1, the physically and chemically bonded water removal is completed around 286 °C, which agrees with previous work for materials of similar composition [31, 46, 47]. Initial decomposition temperatures measured by TGA normally vary depending on the characteristics of the solids (particle size, shape, quantity), the sample holder, the heating rate and the composition of the gas phase [35]. Lanthanum and cerium makes up the major part of the REE fraction of the sample (see Table 2). According to literature data decomposition of cerium sulfate hydrate occurs from about 75 °C with complete dehydration at about 300 °C and the dehydration of lanthanum sulfate hydrate is observed between 175 and 300 °C [35, 47, 48]. The existence of endothermic peaks at 78, 105 and 209 °C (see Fig. 1) also indicates dehydration since all these reactions are endothermic. The endothermic peak located at 586 °C results from the decomposition of the nickel, cobalt and other minor element sulfates. The decompositions have been reported to occur at temperatures of 810 and 916 °C for nickel sulfate and cobalt sulfate, respectively [35, 37, 48, 49]. The thermal decomposition of nickel sulfate into nickel oxide is designated by the massive weight loss located in the center of the graph by considering the high mass percentage of nickel with respect to the rest of the elements. At temperatures greater than about 900 °C, rare earth sulfates are expected to decompose into rare earth oxysulfates, which are practically insoluble in water [42]. Lanthanum oxysulfate was identified in the XRD pattern of the solid residues after water leaching in the roasting experiment R11, performed at 900 °C, Fig. 5b. Based on these results, a temperature interval from 800 to 850 °C was selected for the selective roasting in order to obtain a maximum yield of REE(III) in the water-leaching step.

DSC/TGA analysis of a mixture of anode and cathode material (color figure online)
Sulfation and Selective Roasting
The EDS result of mapping analysis of a particle in experiment R6 after sulfation but before roasting is given in Table 3. The atomic ratio of the sulfur and oxygen indicates that the elements are mainly present as sulfates after the sulfation step.
The results from SEM–EDS mapping analyses of the solid phases obtained after roasting at 850 °C for 2 h (R6) are presented in Fig. 2. The results show the distribution of different elements within a selected region of the sample. The original image of the selected area is shown in the upper left corner with the scale of 20 micrometers. The rare earth sulfates have a glassy appearance compared to its surrounding. The mapping results show that the REEs are associated with S and O in the form of the sulfate ions and that Ni and Co are associated with O within the oxides. By investigating the small rare earth sulfate cluster in the upper right corner of the scanned area in Fig. 2, it can be seen that lanthanum is the dominating REE. This is in accordance with the overall composition of the anode material (see Table 2) where lanthanum is the REE present at the highest concentration. The same results were obtained by mapping analyses of other selected areas of the sample.

Mapping analysis of the material after selective roasting at 850 °C during 2 h (R6). Scale marker = 20 µm (Color figure online)
The powder XRD diffractogram of the solid phase obtained after roasting at 850 °C during 2 h (R6) is presented in Fig. 3a. Peaks belonging to nickel and cobalt oxides and to rare earth sulfates were identified.

a XRD patterns of the solid phase after sulfation using 8 mol/L of H2SO4 and selective roasting at 850 °C (R6), b XRD patterns of the solid phase after selective water leaching (R6) (color figure online)
After water leaching, the leach residue in the experiment R6 consisted of nickel and cobalt oxides together with traces of the other minor elements, as shown in Fig. 3b. Both the nickel and cobalt were in oxide form as well as the aluminum, zinc, and a small amount of iron. All the peaks belonging to the REE solid phases, which were present in the powder diffractogram before water leaching (Fig. 3), disappeared after leaching.
Effect of Acid Concentration in the Sulfation Process
The elemental recovery percentages after water leaching in experiments where the active material was roasted for 2 h at 850 °C are presented in Table 4 and Fig. 4. The results reflect the influence of using different concentration of sulfuric acid in the sulfation step while keeping the solid-to-liquid ratio constant. The recovery percentages of the rare earths (La, Ce, Pr, Nd, and Y) are high as expected.

Percentage recovery of REE after 1 h of water leaching versus acid concentration in the sulfation step (exp. R2, R5, R6, R7, R8, R9). All samples have been roasted for 2 h at 850 °C (color figure online)
The stoichiometric need of sulfate ions for all elements to react and form sulfates can be obtained from a solution with 3.5 mol/L sulfuric acid concentration with the given solid-to-liquid ratio (1/5). The main objective was to separate the REEs from the other elements present, and high recovery percentages of rare earths have been achieved even with low initial acid concentration of 2 mol/L as readily seen in Table 2. However, in order to reach the maximum recovery of REE after 1 h of water leaching, a sulfuric acid concentration of at least 8 mol/L (exp. R6) is needed for a solid-to-liquid ratio of 1/5.
For the highest molarity of sulfuric acid (14 mol/L, exp. R12), the recovery percentages of the REE are lower than those in the experiments where 8 or 10 mol/L of sulfuric acid was used in the sulfation step; this is due to the formation of water-insoluble REE oxysulfates at high sulfuric acid concentration. Traces of REEs were found in the leach residue in experiment R12 (sulfation using 14 mol/L of sulfuric acid) by SEM–EDS (see Table 5), which indicates the presence of insoluble REE oxysulfates. Traces of REE oxysulfates in the solid residue left after water leaching in experiment R12 were also identified by powder XRD analysis (see Fig. 5a). The recovery percentage of the heavy REE yttrium is lower than the recovery percentages of the light REE under all conditions studied. This can be explained by a higher tendency of yttrium to form oxysulfates compared to the lighter REE. Based on the results, a sulfuric acid concentration of 12 mol/L or higher should be avoided to prevent the formation of undesired oxysulfate phases.

a XRD patterns of the leach residue after water leaching in experiment R12 with sulfation using 14 mol/L of H2SO4 and roasting at 850 °C; b XRD patterns of the leach residue after water leaching in experiment R11 with sulfation using 12 mol/L of H2SO4 and roasting at 900 °C (color figure online)
To further investigate the effect of the sulfuric acid concentration in the sulfation step, a series of experiments were conducted at different acid concentrations and by varying the solid-to-liquid ratio, while keeping the moles of sulfate ions constant (experiments R13–R15). As seen in Table 6, with the decreasing acid concentration, the elemental recovery of REE in the water leaching decreases. Thus, the concentration of acid in the investigated range and, not only the stoichiometric need of acid, is important in order to allow complete sulfation of all reactants.
Effect of Roasting Duration
The effect of the duration of roasting was studied by performing roasting experiments at 850 °C for 1, 2, and 3 h after sulfation using 12 mol/L of sulfuric acid, as reported in Table 1. The dissolution percentages of nickel, cobalt, manganese, and aluminum after 1 h of roasting followed by water leaching at 25 °C for 1, 24, and 72 h are presented in Fig. 6a. The results show that nickel, cobalt, manganese, and aluminum dissolve in the aqueous phase during the water leaching. After 72 h, the dissolution of nickel was 62.8%. This means that 1 h of roasting at 850 °C is not enough to completely turn the nickel and cobalt sulfates into their insoluble oxide forms. In the experiments where the samples were roasted for 2 and 3 h (R2 and R3), the dissolutions of Ni and Co were much lower (0.08% Ni and 0.65% Co and 0.05% Ni and 0.39% Co after 2 and 3 h of water leaching, respectively), which shows that 2 h is enough to complete the transformation of Ni and Co into oxides.

a Percentages of dissolution of Ni, Co, Mn, and Al after 1, 24, and 72 h of water leaching in experiments where the roasting duration was 1 h (R1). In all experiments, the sulfation was performed using 12 mol/L sulfuric acid, and the roasting temperature was 850 °C; b percentage recoveries of REE after 1-hour water leaching in experiments where the roasting durations were 1 h (R1), 2 h (R2), and 3 h (R3). In all the experiments, the sulfation was performed using 12 mol/L sulfuric acid, and the roasting temperature was 850 °C (color figure online)
The leaching percentage recoveries of the light REE La, Ce, Pr, Nd, and Y are presented in Fig. 6b. The recovery of La is > 93% already after 1 h of water leaching for 1 and 2 h of roasting. However, it can be seen that the recoveries of the REE, most notably of Y, decrease as the time of roasting increases. A possible explanation for this observation can be that part of the REE, given enough time, turns into water-insoluble REE oxysulfates also at 850 °C. The optimal roasting time is thus 2 h, which is sufficient to turn the Ni and Co sulfates into oxides, while it is short enough to prevent the REE sulfates to turn into water-insoluble oxysulfates.
Effect of Roasting Temperature
Figure 7 shows the recovery percentages after 1, 24, and 72 h of water leaching in experiments R2 and R4 using a roasting temperature of 800 and 850 °C, respectively (see Table 1). The results show that nickel, cobalt, and manganese dissolve to a higher degree in the water-leaching step after roasting at 800 °C than when roasting at 850 °C. The results indicate that the nickel and cobalt sulfates did not fully transform into water-insoluble oxides during the roasting step at 800 °C. After 72 h of leaching time, the recovery percentages of yttrium are 98% after roasting at 800 °C (R4) and 81% after roasting at 850 °C (R2). These experiments were performed using 12 mol/L of sulfuric acid in the sulfation step. The lower recovery of yttrium after roasting at 850 °C could be due to the formation of yttrium oxysulfate, which is favored for higher concentration of sulfuric acid in the sulfation step and at higher roasting temperatures. The time to completely dissolve yttrium is shorter in experiment R2 (850 °C roasting) than that in the experiment R4 (800 °C roasting); this can be attributed to yttrium forming mixed Co and Ni sulfates making the time to completely dissolve Y longer than for the case when Ni and Co are completely transformed into oxides. The leaching recoveries for the light REE, represented by La, after 1 h of water leaching were > 92% and > 94% after roasting for 2 h at 800 and 850 °C, respectively (R2 and R4). The water-leaching recovery of REE in experiment R11, where the roasting was performed at 900 °C, was less than 5%. The solid residue after water leaching in experiment R11 was investigated by powder XRD, and the diffractogram indicated the formation of oxysulfates, as seen in Fig. 5b, which would explain the low REE recovery percentages in the water-leaching step. In addition, a pure sample of yttrium sulfate hydrate crystals was synthesised and analyzed separately by TGA/DSC (see, Supplementary Figure S1), indicating the formation of yttrium oxysulfates above about 900 °C.

Percentage recoveries of Ni, Co, and Mn after different durations of water leaching at 25 °C. The samples have been roasted for 2 h at 800 °C (R4) or 850 °C (R2) after sulfation using 12 mol/L of sulfuric acid (color figure online)
Water Leaching
The results from water-leaching step are presented in Figs. 4, 6a, b, and 7. The results show that 1 h of water leaching is sufficient to dissolve nearly all of the REE sulfates with only a small increase in the total recovery percentage after 72 h. In those cases, where the samples contain Ni and Co sulfates after the roasting step, the concentrations of Ni and Co in the aqueous phase increase substantially between 24 and 72 h of water leaching, as seen in Figs. 6a and 7. No precipitation was observed in any of the leaching experiments.
Optimal Conditions
Based on the conducted experiments the optimal conditions for each step was determined. The optimal conditions consist of sulfation using 8 mol/L of sulfuric acid with a solid-to-liquid ratio of 1/5. The sample is then dried at 150 °C for 24 h. Roasting is performed for 2 h at 850 °C. Water leaching is performed at 25 °C with a solid-to-liquid ratio of 1/50 for 1 h. The optimal conditions were selected for an experiment using a mixture of both anode and cathode material (R10). The recovery percentages and the concentration of the main elements present in the battery material in the aqueous phase after water leaching, following the process under optimal conditions, is presented in Table 7. The total REE recovery was 96%. The pH in the aqueous phase was 5.6. The total concentration of REE(III) in the aqueous phase is limited by the solubility of the respective REE sulfate hydrates in the slightly acidic aqueous phase. The solid residue after water leaching consists of a mixture of nickel and cobalt oxides with minor amounts of impurities in oxide form.
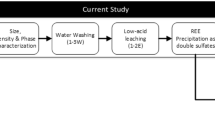
The flow sheet of the complete process is presented in Fig. 8. The main aim of the process is to selectively separate the REEs from the rest of the impurities which mainly consist of Ni and Co. The selection of acid concentration in the sulfation step is based on the recovery percentages of the economically valuable REE. The equipment must resist strong acid, and the evolving gases (SO2 and SO3) must be scrubbed from the vent and recycled as sulfuric acid.

Flowsheet of separation of REEs from HEV battery active material via selective roasting and water leaching under optimal conditions (color figure online)
Conclusion
Sulfation, selective roasting and water leaching is shown to be a viable option for separation of REEs from cobalt and nickel present in the cathode and anode materials of NiMH HEV batteries. Both the amount of acid and the acid concentration are important for achieving an efficient sulfation of the material. When the concentration of acid is high (≥ 10 mol/L) or the roasting temperature is high (≥ 900 °C), the recovery of REE decreases due to the formation of REE oxysulfates that will remain in the solid phase together with Ni and Co in the water-leaching step. The optimal sulfuric acid concentration in the sulfation step was determined to be 8 mol/L for a solid-to-liquid ratio of 1/5. The optimal condition for the roasting step was keeping the temperature at 850 °C for 2 h. Under these conditions Ni and Co turn into oxides while the REE remain in sulfate form. The REE sulfates are easily leached by water to obtain a slightly acidic relatively pure aqueous phase containing the REE suitable for further processing. The total recovery of rare earths from the anode and cathode mixture was 96%. At the optimal conditions negligible amounts of nickel and cobalt appeared in the REE stream.
References
European Commission (2010). Critical raw materials for the EU. Report of the ad-hoc working group on defining critical raw materials. European Commission, Raw Materials Supply Group, 30 July 2010, pp 85
European Commission Directive 2006/66/EC: Directive 2006/66/EC of the European Parliament and of the council of 6 September 2006 on batteries and accumulators and waste batteries and accumulators and repealing Directive 91/157/EEC. OJ, L 266 26/09/2006, pp 1–14
Al-Thyabat S, Nakamura T, Shibata E, Iizuka A (2013) Adaptation of minerals processing operations for lithium-ion (LiBs) and nickel metal hydride (NiMH) batteries recycling: critical review. Miner Eng 45:4–17. https://doi.org/10.1016/j.mineng.2012.12.005
Bernardes AM, Espinosa DCR, Tenório JAS (2004) Recycling of batteries: a review of current processes and technologies. J Power Sources 130(1–2):291–298. https://doi.org/10.1016/j.jpowsour.2003.12.026
Bertuol DA, Bernardes AM, Tenório JAS (2009) Spent NiMH batteries—the role of selective precipitation in the recovery of valuable metals. J Power Sources 193(2):914–923. https://doi.org/10.1016/j.jpowsour.2009.05.014
Müller T, Friedrich B (2006) Development of a recycling process for nickel-metal hydride batteries. J Power Sources 158(2):1498–1509. https://doi.org/10.1016/j.jpowsour.2005.10.046
Pietrelli L, Bellomo B, Fontana D, Montereali M (2005) Characterization and leaching of NiCd and NiMH spent batteries for the recovery of metals. Waste Manag 25(2):221–226. https://doi.org/10.1016/j.wasman.2004.12.013
Espinosa DCR, Bernardes AM, Tenório JAS (2004) An overview on the current processes for the recycling of batteries. J Power Sources 135(1–2):311–319. https://doi.org/10.1016/j.jpowsour.2004.03.083
Gharabaghi M, Irannajad M, Azadmehr AR (2013) Leaching kinetics of nickel extraction from hazardous waste by sulphuric acid and optimization dissolution conditions. Chem Eng Res Des 91(2):325–331. https://doi.org/10.1016/j.cherd.2012.11.016
Larsson K, Ekberg C, Odegaard-Jensen A (2013) Dissolution and characterization of HEV NiMH batteries. Waste Manag 33(3):689–698. https://doi.org/10.1016/j.wasman.2012.06.001
Meshram P, Pandey BD, Mankhand TR (2015) Leaching of base metals from spent Ni–metal hydride batteries with emphasis on kinetics and characterization. Hydrometallurgy 158:172–179. https://doi.org/10.1016/j.hydromet.2015.10.028
Meshram P, Pandey BD, Mankhand TR (2016) Process optimization and kinetics for leaching of rare earth metals from the spent Ni–metal hydride batteries. Waste Manag 51:196–203. https://doi.org/10.1016/j.wasman.2015.12.018
Nan J, Han D, Yang M, Cui M, Hou X (2006) Recovery of metal values from a mixture of spent lithium-ion batteries and nickel-metal hydride batteries. Hydrometallurgy 84(1–2):75–80. https://doi.org/10.1016/j.hydromet.2006.03.059
Pietrelli L, Bellomo B, Fontana D, Montereali MR (2002) Rare earths recovery from NiMH spent batteries. Hydrometallurgy 66(1–3):135–139. https://doi.org/10.1016/S0304-386X(02)00107-X
Rodrigues LEOC, Mansur MB (2010) Hydrometallurgical separation of rare earth elements, cobalt and nickel from spent nickel–metal–hydride batteries. J Power Sources 195(11):3735–3741. https://doi.org/10.1016/j.jpowsour.2009.12.071
Zhang P, Yokoyama T, Itabashi O, Wakui Y, Suzuki TM, Inoue K (1998) Hydrometallurgical process for recovery of metal values from spent nickel-metal hydride secondary batteries. Hydrometallurgy 50(1):61–75. https://doi.org/10.1016/S0304-386X(98)00046-2
Fernandes A, Afonso JC, Dutra AJB (2013) Separation of nickel(II), cobalt(II) and lanthanides from spent Ni-MH batteries by hydrochloric acid leaching, solvent extraction and precipitation. Hydrometallurgy 133:37–43. https://doi.org/10.1016/j.hydromet.2012.11.017
Innocenzi V, Vegliò F (2012) Recovery of rare earths and base metals from spent nickel-metal hydride batteries by sequential sulphuric acid leaching and selective precipitations. J Power Sources 211:184–191. https://doi.org/10.1016/j.jpowsour.2012.03.064
Larsson K, Ekberg C, Ødegaard-Jensen A (2012) Using Cyanex 923 for selective extraction in a high concentration chloride medium on nickel metal hydride battery waste. Hydrometallurgy 129–130:35–42. https://doi.org/10.1016/j.hydromet.2012.08.011
Mubarok MZ, Lieberto J (2013) Precipitation of Nickel Hydroxide from Simulated and Atmospheric-leach Solution of Nickel Laterite Ore. Procedia Earth Planet Sci 6:457–464. https://doi.org/10.1016/j.proeps.2013.01.060
Yang X, Zhang J, Fang X (2014) Rare earth element recycling from waste nickel-metal hydride batteries. J Hazard Mater 279:384–388. https://doi.org/10.1016/j.jhazmat.2014.07.027
Zhang P, Yokoyama T, Itabashi O, Wakui Y, Suzuki TM, Inoue K (1999) Recovery of metal values from spent nickel–metal hydride rechargeable batteries. J Power Sources 77(2):116–122. https://doi.org/10.1016/S0378-7753(98)00182-7
Feng X-L, Long Z-Q, Cui D-L, Wang L-S, Huang X-W, Zhang G-C (2013) Kinetics of rare earth leaching from roasted ore of bastnaesite with sulfuric acid. Trans Nonferr Met Soc China 23(3):849–854. https://doi.org/10.1016/S1003-6326(13)62538-8
Tang K, Ciftja A, van der Eijk C, Wilson S, Tranell G (2013) Recycling of the rare earth oxides from spent rechargable batteries using waste metallurgical slags. J Min Metall Sect B Metall 49(2):233–236. https://doi.org/10.2298/jmmb120808004t
Yang Z, Li H-Y, Yin X-C, Yan Z-M, Yan X-M, Xie B (2014) Leaching kinetics of calcification roasted vanadium slag with high CaO content by sulfuric acid. Int J Miner Process 133:105–111. https://doi.org/10.1016/j.minpro.2014.10.011
Yu D, Utigard TA, Barati M (2014) Fluidized bed selective oxidation-sulfation roasting of nickel sulfide concentrate: part I. Oxidation roasting. Metall Mater Trans B 45(2):653–661. https://doi.org/10.1007/s11663-013-9958-x
Yu D, Utigard TA, Barati M (2014) Fluidized bed selective oxidation-sulfation roasting of nickel sulfide concentrate: part II. Sulfation roasting. Metall Mater Trans B 45(2):662–674. https://doi.org/10.1007/s11663-013-9959-9
Guo XY, Li D, Park KH, Tian QH, Wu Z (2009) Leaching behavior of metals from a limonitic nickel laterite using a sulfation-roasting-leaching process. Hydrometallurgy 99(3–4):144–150. https://doi.org/10.1016/j.hydromet.2009.07.012
You ZX, Li GH, Zhang YB, Peng ZW, Jiang T (2015) Extraction of manganese from iron rich MnO2 ores via selective sulfation roasting with SO2 followed by water leaching. Hydrometallurgy 156:225–231. https://doi.org/10.1016/j.hydromet.2015.05.017
Borra CR, Mermans J, Blanpain B, Pontikes Y, Binnemans K, Van Gerven T (2016) Selective recovery of rare earths from bauxite residue by combination of sulfation, roasting and leaching. Miner Eng 92:151–159. https://doi.org/10.1016/j.mineng.2016.03.002
Önal M, Borra C, Guo M, Blanpain B, Van Gerven T (2015) Recycling of NdFeB magnets using sulfation, selective roasting, and water leaching. J Sustain Metall 1(3):199–215. https://doi.org/10.1007/s40831-015-0021-9
Korkmaz K, Forsberg KM, Alemrajabi M, Rasmuson ÅC (2016) Sustainable hydrometallurgical recovery of valuable elements from spent nickel-metal hydride HEV batteries. In: IMPC 2016: XXVIII International Mineral Processing Congress Proceedings
Pandher R, Thomas S, Yu D, Barati M, Utigard T (2011) Sulfate formation and decomposition of nickel concentrates. Metall Mater Trans B 42(2):291–299. https://doi.org/10.1007/s11663-010-9466-1
Pelovski Y, Petkova V (1997) Mechanism and kinetics of inorganic sulphates decomposition. J Therm Anal 49(3):1227–1241. https://doi.org/10.1007/bf01983679
Poston JA, Siriwardane RV, Fisher EP, Miltz AL (2003) Thermal decomposition of the rare earth sulfates of cerium(III), cerium(IV), lanthanum(III) and samarium(III). Appl Surf Sci 214(1–4):83–102. https://doi.org/10.1016/s0169-4332(03)00358-1
Straszko J, Możejko J, Olszak-Humienik M (1995) Kinetics of thermal decomposition of nickel sulfate hexahydrate. J Therm Anal 45(5):1109–1116. https://doi.org/10.1007/bf02547483
Straszko J, Olszak-Humienik M, Możejko J (2000) Study of the mechanism and kinetic parameters of the thermal decomposition of cobalt sulphate hexahydrate. J Therm Anal Calorim 59(3):935–942. https://doi.org/10.1023/a:1010186628054
Tomaszewicz E, Kotfica M (2004) Mechanism and kinetics of thermal decomposition of nickel(II) sulfate(VI) hexahydrate. J Therm Anal Calorim 77(1):25–31. https://doi.org/10.1023/B:JTAN.0000033184.32714.7f
Lide DR (2004) CRC handbook of chemistry and physics. CRC Press, New York
Lokshin EP, Tareeva OA, Ivlev KG, Kashulina TG (2005) Solubility of double alkali metal (Na, K) rare-earth (La, Ce) sulfates in sulfuric-phosphoric acid solutions at 20°C. Russ J Appl Chem 78(7):1058–1063. https://doi.org/10.1007/s11167-005-0449-y
The National Physical Laboratory (1995) 3.2 Properties of inorganic compounds. Tables of physical & chemical constants (16th edition 1995). 2.1.4 Hygrometry. Kaye & Laby Online. Version 1.0 (2005) from http://www.kayelaby.npl.co.uk/chemistry/3_2/3_2.html
Dixini PVM, Celante VG, Lelis MFF, Freitas MBJG (2014) Recycling of the anode from spent Ni-MH batteries for synthesis of the lanthanide oxysulfide/oxysulfate compounds used in an oxygen storage and release system. J Power Sources 260:163–168. https://doi.org/10.1016/j.jpowsour.2014.03.006
Patnaik P (2002) Handbook of inorganic chemicals. McGraw-Hill, New York
Todorovsky DS, Milanova MM, Minkova NL, Balarev C (1993) Solubility of some lanthanide sulfates in polycomponent systems containing H2SO4. Monatshefte für Chemie (Chem Mon) 124(6–7):673–679. https://doi.org/10.1007/BF00817302
Niinistö L, Saikkonen P, Sonninen R (1982) Thermal decomposition of rare earth sulfate and selenate hydrates. In: McCarthy GJ, Silber HB, Rhyne JJ, Kalina FM (eds) The rare earths in modern science and technology, vol 3. Springer, Boston, pp 257–263
Nathans MW, Wendlandt WW (1962) The thermal decomposition of the rare-earth sulphates. J Inorg Nucl Chem. https://doi.org/10.1016/0022-1902(62)80108-0
Wendlandt WW (1958) The thermal decomposition of yttrium and the rare earth metal sulphate hydrates. J Inorg Nucl Chem 7(1):51–54. https://doi.org/10.1016/0022-1902(58)80026-3
Kolta GA, Askar MH (1975) Thermal decomposition of some metal sulphates. Thermochim Acta 11(1):65–72. https://doi.org/10.1016/0040-6031(75)80038-4
Tagawa H (1984) Thermal decomposition temperatures of metal sulfates. Thermochim Acta 80(1):23–33. https://doi.org/10.1016/0040-6031(84)87181-6
Acknowledgements
The authors would like to acknowledge the Swedish Energy Agency (Energimyndigheten) for funding the project (nr 37724-1) and Chalmers University of Technology, Department of Chemistry and Chemical Engineering for supplying the battery modules.
Author information
Authors and Affiliations
Corresponding author
Additional information
The contributing editor for this article was T. Hirato.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Korkmaz, K., Alemrajabi, M., Rasmuson, Å. et al. Recoveries of Valuable Metals from Spent Nickel Metal Hydride Vehicle Batteries via Sulfation, Selective Roasting, and Water Leaching. J. Sustain. Metall. 4, 313–325 (2018). https://doi.org/10.1007/s40831-018-0169-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40831-018-0169-1