
Abstract
The recently developed fluorolytic sol–gel route to metal fluorides opens a very broad range of both scientific and technical applications of the accessible high surface area metal fluorides, many of which have already been applied or tested. Specific chemical properties such as high Lewis acidity and physical properties such as high surface area, mesoporosity and nanosize as well as the possibility to apply metal fluorides on surfaces via a non-aqueous sol make the fluorolytic synthesis route a very versatile one. The scope of its scientific and technical use and the state of the art are presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although nanomaterials have played a role in science and technology for many years, it was only in the 1990s that a consensus was established among the scientific and industrial community that nanoscience will start a new kind of industrial and technological revolution. Not surprisingly, a drastic jump in the number of publications dealing in general with nanomaterials can be observed.
A short excursion into the nano world
Nanotechnology is science, engineering and technology conducted at nanoscale, which is about 1–100 nm.
The ideas and concepts behind nanoscience and nanotechnology started with a talk given by the physicist Richard Feynman (1918–1988) at an American Physical Society meeting at the California Institute of Technology (CalTech) on December 29, 1959. The title of his talk was “There’s Plenty of Room at the Bottom” and this was long before the term nanotechnology was used. More than a decade later, Professor Norio Taniguchi created the term nanotechnology. It is hard to imagine how small nano really is. One nanometre is a billionth of a metre, 10–9 m. A few examples will illustrate this:
-
The diameter of a hair is about 50 µm = 50,000 nm.
-
A bacterium is about 100 nm.
-
A virus is about 10 nm.
-
A protein is about 1 nm.
-
A molecule is about 0.1 nm.
Thus, nanoscience and nanotechnology involve the ability to see and to control individual atoms and molecules. But something as small as an atom is impossible to see with the naked eye. In fact, it is impossible to see it with microscopes typically used in school education. It was not until 1981, with the development of the scanning tunneling microscope (STM), that one could “see” individual atoms, and shortly after that the atomic force microscope (AFM), that the age of nanotechnology was born.
Because of making materials smaller and smaller up to the nanoscale, their chemical and physical properties may change dramatically. An atom at the surface of a solid is no longer perfectly coordinated by other counterparts, meaning it is undercoordinated and thus more reactive. In the case of a classical, macroscopic particle fewer than a billionth of all atoms are surface atoms; most atoms are bulk atoms with full coordination. In the case of a nanoparticle, the reverse is true: many more atoms lay at the surface and just a minor part constitutes the bulk. It is obvious that such particles have a quite higher energetic level and thus a higher reactivity than their macroscopic analogues, making them interesting materials for applications in chemical reactions, catalysis, adsorption processes etc. This effect is known as the surface effect in the nanosciences. Even characteristic properties like the melting point are no longer constant but decrease with decreasing particle size. There is a second nano effect, which is called the quantum size effect, which means that some physical properties may change abruptly with decreasing size of the materials. For instance, magnetization of small particles may differ fundamentally from macroscopic analogues, or a semiconducting material may become isolating when the particle size decreases because of changes in the band gap structure.
As a consequence of all these differences, materials of the same chemical composition may exhibit fundamentally different properties when they are small enough. However, the challenge is to explore synthesis strategies that allow access to nanoscopic materials since thermodynamics forces reactions to form well-crystallized macroscopic materials because they are more stable than their nanoscopic counterparts. Consequently, chemists were and still are searching for effective powerful synthesis routes toward nanoscopic materials.
Although modern nanoscience and nanotechnology are quite new, nanoscale materials have been used for centuries. Alternate-sized gold and silver particles created colours in the stained-glass windows of medieval churches hundreds of years ago. The artists back then just did not know that the process they used to create these beautiful works of art led to changes in the composition of the materials they were working with.
Inorganic nanoparticles
Inorganic materials chemistry is almost the whole time characterized by the application of metal-containing compounds. Metal oxides represent the most important class of inorganic materials being used for many centuries in industry. As a result of increasingly fast industrial development and explosive increase in demand for materials with specific properties, binary oxides no longer could fulfil all requirements of modern materials. Specifically tuned properties by introducing additional metal components in stoichiometric amounts (ternary, quaternary metal oxides, oxometallates etc.) or just in small amounts (doped systems) resulted in new materials with better performance, higher efficiency and new fields of application [1,2,3]. Thus, metal oxides are used in fields like processor and sensor fabrication, catalytic large-scale production in the chemical industry, energy storage, thin-layer applications etc. [4,5,6].
The composition and properties can be tuned not only by modifying the cationic components inside of metal oxides but also by partial or total replacement of oxide anions by other anions, resulting in new materials with new properties and consequently new options for applications. Thus, moving from metal oxides to the respective metal fluorides may result in significant changes e.g. of thermal and optical properties. Owing to their attractive perspectives, metal fluorides are of interest in application areas like catalysis, optics, photonics, optical amplifiers, optoceramics, composite materials, biosensing and biolabelling.
Since fluorine carries the highest electronegativity, it is the strongest electron-withdrawing element and thus induces the highest electron deficiency at any counter atom, making fluorides extremely strong Lewis acids depending on the counter element. Consequently, metal fluorides are found among the strongest Lewis acids, making them ideal for Lewis acid catalysed reactions [7, 8]. Moreover, metal fluorides are also of interest as optical materials e.g. in solid-state lasers, luminophores, scintillators and antireflective coatings. Another, outstanding burst of interest arose recent years over their use for energy storage applications [9,10,11].
Inspired by the great potential of industrial applications, which other nanomaterials, especially metal oxides, had already gained, there has been increasing development of new materials based on nano metal fluorides. The growing interest in nanoscopic metal fluorides can clearly be evidenced by checking Elsevier’s Scopus database for the keyword “nano” in combination with “fluoride materials” (Fig. 1) [12]. Starting at around 2000 with just very few publications, in 2010 about 200 publications can be counted, whereas in 2019 around 900 publications appeared.

Published items in each year for the past 25 years in the Scopus database using the keywords “fluoride materials” and “nano” [12]
Note that the first book emphasizing synthesis and applications of new nano metal fluorides appeared in 2010 [7], thus perfectly reconfirming the conclusion that can be drawn from the numbers given in Fig. 1.
In contrast to metal oxides, there were no satisfactory synthesis routes toward nano metal fluorides until the early 1990s. Consequently, material scientists were strongly engaged in developing synthesis routes allowing access to nano metal fluorides in a similar elegant way as had already been successfully realized for nano metal oxides a long time ago.
Fortunately, in the early 2000s a non-aqueous—so-called fluorolytic—sol–gel synthesis was developed which is similarly universal and powerful as the classical Stöber process-based aqueous sol–gel synthesis for metal oxides [13] (Werner Stöber 1973–2019). Thus, the intention of this review is to provide an overview on mechanistic aspects of the non-aqueous fluorolytic sol–gel synthesis that yields for the first time direct access to novel nanoscaled metal fluorides.
As stated above, nanomaterials have become very attractive targets over the past 25 years because the properties of nanoscopic compounds usually differ significantly from their classically prepared analogues. Among other factors, the high potential of nanomaterials for industrial applications is the driving force for research and development of new synthesis routes toward these materials. Consequently, a variety of new synthesis techniques have been developed over the past decades, opening the door to the fascinating world of nanomaterials. It is worth mentioning that, especially in this context, the main focus was on metal oxides with their extremely wide area of applications. Among many new synthesis routes toward nanoscopic metal oxides, the sol–gel synthesis is one of the most powerful approaches in terms of diversity of synthesis variations and technical applications. However, although it is in fact very complex, this sol–gel synthesis is based on a very simplified description of the hydrolytic reaction of a metal alkoxide (often silicon alkoxide) with water.
However, motivated by new developments for applications of such nanoscopic materials, non-aqueous sol–gel synthesis approaches like atomic layer deposition (ALD) and others have been developed recently, thus extending access to new nanoscopic materials.
Synthesis strategies toward nano metal fluoride-based materials
As a rule, two synthesis strategies towards nanoparticles exist, namely top-down and bottom-up synthesis. Top-down synthesis starts from macroscopic crystalline materials, which are crushed down to smaller particles by mechanochemical treatment, usually milling. Bottom-up synthesis usually means to react molecular precursors in a suitable way ending up with nanoscopic small particles (NPs) but not with macroscopic crystalline materials. Both synthesis approaches will be briefly introduced here.
Top-down synthesis of nano metal fluorides
Milling of solids results in an apparent disruption of particles. Depending on the mechanical device and introduced energy, particle sizes might be reduced down to the nanometre scale. Basically it starts with a so-called mechanical activation, meaning that new, fresh surfaces are generated from the mechanical impact, thus resulting in a reduction of particle size [14]. In the same way, the surface area increases. This milling process increases defect concentrations, leading to changes of the reactivity of the solid. Especially when high-performance mills are used, materials will be obtained with altered chemical and physical properties, like high catalytic activity, increased solubility, improved sorption capacity or higher chemical reactivity. An example that illustrates nicely the effect of different kinds of energy input is the thermal versus mechanochemical decomposition of solid silver chloride [15,16,17]. Introduction of thermal energy causes melting in the first step followed, at higher temperature, by evaporation of AgCl. Mechanical treatment, however, results into the formation of metallic silver and evolution of chlorine gas. Today, this approach is used to fabricate silver NPs. Although less is known about the mechanism of mechanochemical activation and synthesis of NPs, respectively, it is generally accepted that according to the magma-plasma model the particles that collide with high speed release large amounts of energy that enables the formation of plasma-like conditions [18, 19]. Alternative mechanistic proposals may be found in Baláz et al. [16].
In recent years, mechanochemical synthesis of fluorides has gained increasing attraction. A number of papers appeared describing synthesis and properties of binary, ternary, and even more complex nano metal fluorides. More detailed information about all these systems is reviewed by Gudrun Scholz [20].
However, the mechanochemical method is limited because the very high energy input causes the activation barrier to be reached, at which point small metastable particles will react back to form larger, energetically more stable particles. Although particle dimensions of 100 nm and below can be achieved, e.g. by high energy ball milling, single nanoparticles on the low digit nanometre scale are only available by bottom-up synthesis.
Another even more interesting aspect of milling is to initiate solid-state reactions, which usually need high temperatures; under milling conditions, the reactions can be performed at room temperature, resulting very often also in the formation of nanosized materials. Especially for the synthesis of different kinds of metal fluorides, the mechanochemical synthesis has been successfully applied over the past years. For more details, we refer again to the review by Gudrun Scholz [20].
Bottom-up synthesis of nano metal fluorides: mechanistic overview
Bottom-up synthesis routes start from atomic or molecular precursors. Larger units are built up by classic chemical reactions. In principle, particles with diameters ranging from the nanometre to millimetre scale might be formed. The main challenge here is size control, i.e. to run the reaction under conditions allowing the formation of single particles at the nanometre scale instead of macrocrystalline bulk materials. From a thermodynamic point of view, nature intends to reach the lowest energy level, meaning that formation of well-crystallized large particles/crystallites is evidently the energetically driven process. Consequently, synthesis of nanoparticles is against the thermodynamics and it needs kinetic control over the synthesis to overcome the thermodynamic driving forces. However, the classical Stöber process for the sol–gel synthesis of nano silica particles gives an excellent example how this can be achieved [13]. In simple words, an alkoxysilane is reacted with nearly stoichiometric amounts of water in an excess of an organic solvent, e.g. ethanol or methanol. Since the solubility of SiO2 that is formed in the course of the hydrolysis reaction in organic solvents is far lower than in water, a fast nucleation and fast crossing of the extremely low solubility product causes a strong limitation of particle grow. The so-called LaMer mechanism (Fig. 2a) predicts that by combining two dissolved reactants, an oversaturation is needed until a critical concentration is reached that results in spontaneous nucleation. Further growth of these nuclei reduces the overcritical concentration of free ions to the value of equilibrium solubility concentration and further particle growth. If the solubility of the respective compounds is extremely low, as can be achieved by working in organic solvents, there are insufficient dissolved ions that may cause further precipitation onto the already formed particles. Thus, the choice of a suitable solvent is a very crucial parameter to obtain small particles. If all the other reaction conditions like temperature, concentration etc. are also optimized, particles with diameters in the nanometre dimension can be obtained. According to the LaMer model, further particle growth may occur because smaller particles—owing to their higher surface energy—exhibit higher solubility, and thus slowly dissolve, thereby providing ions that can precipitate onto the larger particles with lower surface energy [21]. The latter process is also known as Ostwald ripening [22] (Wilhelm Ostwald 1853–1932). As a result, larger particles grow at the cost of smaller particles which finally disappear.

LaMer mechanism of particle formation shown schematically for a aqueous reaction medium and b alcoholic reaction medium. Note that the final particle size in alcohol is much smaller than in water because of a shorter growth time caused by reduced solubility [21]
Working in e.g. alcohol instead of water has the big advantage that the solubility of the addressed compounds is several orders of magnitude smaller; thus, nucleus formation and particle growth proceed on a very short timescale because the consumption of dissolved building blocks happens very fast. For those compounds that exhibit extremely low solubility in organic solvents, dissolution and precipitation are suppressed; thus, Ostwald ripening under these conditions cannot take place.
Trifluoroacetic acid (TFA) route
The TFA route was mainly developed by Fujihara et al. and is based on the reaction of a metal alkoxide or acetate with TFA in an organic, mainly alcoholic, solvent (Eq. 1, M = metal, n = metal valence) [23, 24].
TFA is a strong acid as a result of the electron-withdrawing CF3 group, and the trifluoroacetate substitutes the acetate or alkoxide group; thus, a metal trifluoroacetate sol might be formed which can further be processed, e.g. for coating or powder materials. Thermal decomposition of the trifluoroacetate sols and gels at temperatures up to 400 °C results in the formation of crystalline metal fluorides. Oxide fluorides and oxides might be formed as additional phases if temperatures exceed 400 °C [25]. Gaseous fluorine-containing species liberated under these conditions are for example (CF3CO)2O, CF3COF, COF2, and COx which were detected by MS and IR spectroscopy [26]. Evidence of difluoromethylene diradicals (:CF2) as intermediate species of the decomposition of TFA was reported; these radicals play a major role as fluorinating agent during the thermal decomposition [23]. The general overall reaction scheme for lanthanide trifluoroacetates is given in Eq. 2 (Ln = Y, La–Lu):
The general pathway of the TFA synthesis is shown in Fig. 3 starting from metal alkoxides or acetates, which are reacted with TFA in solution to form a metal trifluoroacetate precursor sol, which can be used for the preparation of thin-film coatings or xerogels. The final step is the thermal decomposition of the fluoroorganic compound to give the corresponding metal fluoride. Thus, substrates to be coated are limited to those materials that are stable under the given reaction conditions. Additionally, this method is limited to fluorides of rather hard cations (according to the hard soft acid base (HSAB) concept). The synthesis e.g. of CuF2 is not possible, because the thermal decomposition of the corresponding trifluoroacetate leads to oxidic species (Cu2O, CuO) only.

Thin film and xerogel metal fluoride preparation via metal fluoroacetate sol–gel formation followed by thermal decomposition. Reproduced from Dalton Trans. 2015, 44, 19411–19431 (Ref. [27]) with permission of the Royal Society of Chemistry
Fluorolytic sol–gel synthesis
Introduction to fluorolytic sol–gel synthesis
Unlike the TFA route, the fluorolytic sol–gel approach is a direct sol–gel synthesis. This synthesis approach resembles somewhat the classical sol–gel synthesis of metal oxides. As briefly stated above, the classical sol–gel synthesis consists in the hydrolysis reaction of a sensitive precursor under defined conditions followed by the condensation reaction of the initially formed metal–OH moieties, finally resulting in the formation of M–O–M bridges. The fluorolytic sol–gel synthesis uses anhydrous hydrogen fluoride, HF, instead of water. Since instead of hydrolysis, bond cleavage by fluorolysis (fluorination) is the crucial reaction step, it is in principle a non-aqueous sol–gel synthesis that was discovered in 2003 and introduced in the literature as fluorolytic sol–gel synthesis [27,28,29].
The general reaction principle is represented by Eq. 3. An appropriate metal precursor is reacted with anhydrous solvated hydrogen fluoride in a non-aqueous solvent (M = metal, n = metal valence, X = monovalent anion):
This simple reaction scheme describes not only the fluorolytic sol–gel synthesis but also classical precipitation of an insoluble fluoride from aqueous solution. The main difference lays in the reaction kinetics. Size control is achieved by different solvents. Reduction of the solubility of the fluoride by appropriate choice of the solvent results in smaller particles (cf. Fig. 2). The main effect for the size control in the fluorolytic sol–gel synthesis is the exclusion of water. The reaction medium is usually a solvent like alcohol or ether. Hydrogen fluoride is to be added in a well-defined quantity as a solution in another non-aqueous solvent, allowing accurate stoichiometric dosing using volumetric methods.
The exact reaction conditions, i.e. precursor material, solvent, reaction temperature, additives etc., must be adjusted for every fluoride system. The synthesis ends up in water-clear transparent dispersions of nanoparticles, when the particle diameter is below ca. 20 nm. Provided appropriate reaction conditions are established, a whole bunch of metal fluorides may be synthesized. In the simplest case, these are the binary fluorides like LiF, MgF2, CaF2, SrF2, ZnF2, AlF3, YbF3 [30,31,32,33,34,35,36,37], but nanoparticles consisting of several multiphase systems like MgF2–CaF2, ZnF2–FeF3 and others have also gained interest for several applications [38, 39].
The synthesis of complex metal fluorides like KMgF3, CaAlF5, MgAl2F8 and solid solutions like Ca1−xSrxF2 and Sr1−xLnxF2+x (Ln = Y, La–Lu) is a more challenging task. The main challenge here is to prevent the possible formation of mixed binary fluorides, i.e. a multiphase system instead of a one-phase system. These obstacles were successfully circumnavigated and hence the synthesis of such systems is also possible [40,41,42,43,44]. Figure 4 shows some typical examples of the appearance and particle analysis of different binary metal fluoride sols.

Optical appearance, particle size distribution and TEM images of three typical sols
The main advantage of the fluorolytic sol–gel synthesis over all other synthesis methods towards metal fluoride nanoparticles is the capability of upscaling the reactions and a reduction of by-products during synthesis. Although HF is a hazardous agent, the synthesis is environmentally friendly. Since any commercially available fluorine compound is synthesized from HF, the direct application of HF for the fluorolytic sol–gel synthesis saves chemicals and reduces waste. The synthesis of hundreds of litres of sol, containing several kilograms of particles, is possible in one batch.
Choice of precursor and solvent
As already mentioned, the exact reaction conditions need to be adjusted for the synthesis of every particular metal fluoride. Of course, thermodynamics must not be neglected in the fluorolytic sol–gel chemistry. Although nanoparticles are not in a thermodynamic equilibrium, thermodynamic considerations are helpful to understand the synthesis. Without a thermodynamic driving force, the fluorination reaction will not take place, even in the case when no thermodynamic equilibrium is reached. Thus, the combination of precursors must be chosen in such a way that:
-
(i)
The reaction with HF is thermodynamically allowed.
-
(ii)
The precursor is preferably soluble in the solvent.
-
(iii)
The by-product HX can be tolerated.
Table 1 illustrates the reaction enthalpy of different solid metal precursors (chlorides, acetates and hydroxides) upon reaction with gaseous HF. The fluorination reaction is carried out in a solvent, but these data show some clear trends which can be transferred to the corresponding sol–gel reaction.
The reaction of metal chlorides with HF is only exothermic when the cation is small and a hard Lewis acid according to the HSAB concept. HCl formed during the reaction is a very strong Brønsted acid, much stronger than HF. Thus, the driving force of the reaction is the formation of the fluoride phase.
For hydroxides, the reaction is always exothermic, even for a metal ion that is a soft Lewis acid. The major part of the reaction’s driving force is the protonation of the basic hydroxide anion.
Acetates and other carboxylates are somewhere between both of these extremes, because the acid strengths of HF and acetic acid are similar.
Although thermodynamically favoured, hydroxides are not very suitable precursors, because water is formed during the fluorination reaction. Another drawback is their usually low solubility in organic solvents. Metal alkoxides are much better here. The reaction with HF is still more exothermic than in the case of hydroxides, and alcohol is formed as the only by-product. Hence, in principle metal alkoxides would be the best precursors for the fluorolytic sol–gel synthesis. Indeed, the alkoxides of alkali metals and other electropositive metals like Mg and Al are very suitable precursors for the fluorolytic sol–gel synthesis [34, 39, 46,47,48,49]. However, for many of the other metals, this is not the case. Either the alkoxides are very costly (e.g. Y) or not commercially available at all (e.g. Cu), or the alkoxides are very oxygen-sensitive (e.g. Ca, Sr) or they are insoluble.
Thus, metal carboxylates may in some cases be a better choice for the synthesis. They are either commercially available or can be synthesized comparatively easily. A disadvantage of carboxylates is the formation of carboxylic acids during the fluorination reaction, which usually have to be removed before any application. On the other hand, free carboxylic acid slowly reacts with the solvent alcohol to form ester and water. The water may cause gelation and Ostwald ripening and hence sols made from carboxylate precursors are not stable on a long-term scale [31, 50].
The synthesis of nano metal fluorides from chloride precursors is only successful for LiF, MgF2, CaF2 and AlF3 [30, 32, 35, 51]. These sols contain large amounts of anhydrous HCl and are therefore very corrosive. Although such sols perform well for different applications on a laboratory scale, there are limits in their industrial application.
There may be other thermodynamic obstacles due to possible other side reactions. For instance, although the reaction of K(CH3COO) with one equivalent HF to KF is thermodynamically allowed, the reaction results in a mixture of KHF2 and unreacted precursor, because the formation of KHF2 is thermodynamically even more strongly enforced. Only the application of potassium alkoxides, K(OR), prevents the formation of KHF2 since the latter is not stable in the presence of a strong base. Another example is the fluorination of SrCl2 with two equivalents of HF, which results in a mixture of SrF2, SrClF and unreacted HF, and therefore SrCl2 is an inappropriate precursor [36].
The choice of solvents is also of importance due to the different solubility of precursors. A very good solvent for a lot of reactions is methanol. In the case of MgF2, the soluble precursor magnesium methoxide, Mg(OCH3)2, can be straightforwardly reacted with HF leading to transparent sols [34, 46]. Removal of methanol is easy owing to its low boiling point of 65 °C. Unfortunately, methanol is a toxic substance and therefore such sols are limited in their application. Another very good solvent for most metal carboxylates is ethylene glycol. It is successfully applied for the synthesis of pure and rare-earth doped CaF2 and SrF2; but removal of this high boiling solvent (197 °C) is challenging [43, 44].
Transferring the fluorination of magnesium alkoxides to other alcohols, e.g. ethanol, is not straightforward, because these precursors are insoluble. Fluorination of magnesium ethoxide, Mg(OC2H5)2, is possible, but ends up with agglomerated precipitates of MgF2 [39]. This obstacle is elegantly circumnavigated by the fluorination of a mixture of magnesium alkoxide, Mg(OR)2, and magnesium chloride, MgCl2, in a one-pot reaction (Fig. 5). MgCl2 is soluble in most alcohols and will react rapidly with HF to form MgF2 nanoparticles. Anhydrous HCl is formed as by-product, which immediately reacts with the insoluble basic Mg(OR)2 to form soluble MgCl2 again, which reacts with HF again. In this way, a catalytic process is established, ending up with a homodisperse MgF2 nanoparticle-containing sol. The HCl content of such MgF2 sols is much lower than that of sols derived from the fluorination of pure MgCl2 and hence the former sols are more suitable for application on a larger scale [52].

Formation of nano-MgF2 in ethanol from ethoxide precursor (x = 0.05–0.3) [39]
Mechanistic aspects
General overview
The mechanism of the hydrolytic sol–gel process of oxide nanoparticles, e.g. SiO2, TiO2, ZrO2, Al2O3, from metal alkoxide precursors and water is a well-understood process [13, 53]. Briefly, alkoxide groups undergo hydrolysis in the first step leading to hydroxide nuclei (Eq. 4), followed by condensation reactions to build up larger units (Eqs. 5a, 5b; M = metal, n = metal valence).
According to the Bogush–Zukoski model, larger aggregates are formed during this process, which after some time break down to form larger stable particles (Fig. 6a) [54]. By appropriate choice of the reaction conditions (temperature, concentration and process time), the size and morphology of the formed metal oxide nanoparticles can be adjusted.

a Nanoparticle formation according to the Bogush–Zukoski model for oxides [54]. b Nanoparticle formation for fluorides
Although the brutto reaction scheme for the fluorolytic sol–gel synthesis looks very similar (cf. Eq. 3), the mechanistic details are different. Condensation reactions as in the case of OH groups (Eqs. 5a and 5b) cannot take place in the case of fluorides. Before going into detail, a brief and simplified description of the processes during the fluorolytic sol–gel synthesis will be given (M = metal, n = metal valence).
In the first step, an alkoxide group of the precursor undergoes fluorolysis and is replaced by fluoride (Eq. 6). Usually, a fluoride ion bound to a metal strongly tends to bridge to another neighbouring metal ion. Note that only very few examples of solid fluorides with non-bridging terminal fluoride ions are known. Thus, the formed intermediates are not stable and coordinate to other precursor entities via bridging fluoride ions. These other precursors may be fluorinated (Eq. 7a) or not (Eq. 7b). In consecutive reaction steps, more and more alkoxide groups are replaced by fluoride followed by aggregation, forming increasingly larger units, until finally a nearly stoichiometric metal fluoride is formed. This process is equivalent to the so-called explosive nucleation in the Bogush–Zukoski model for the hydrolytic sol–gel synthesis (Fig. 6).
Larger agglomerates of primary particles are formed during this first nucleation step. Contrary to the hydrolytic sol–gel synthesis, these agglomerates do usually not grow to larger primary particles. Under appropriate conditions, which will be discussed later, an increasing process time leads to deagglomeration of these agglomerates under prevention of the primary particle size. The agglomerates become smaller and smaller, finally resulting in ultrasmall, finely dispersed primary particles. When these primary particles have a mean diameter below 20 nm, a transparent dispersion is formed (Fig. 4).
Mechanism of fluorination
Usually, metal alkoxides are not monomeric molecules, but are somehow aggregated. Soluble alkoxides occur as multinuclear clusters, while insoluble alkoxides form 3D networks. Upon fluorination, the alkoxide groups of these clusters are attacked consecutively by HF, while simultaneously rearrangement reactions and nucleation occur. Equations 6, 7a and 7b give a very simplified overview. The details of this fluorination mechanism were investigated for the fluorination of aluminium alkoxides and to a lesser extent for magnesium alkoxides. Characterization of the intermediate products is a challenging process. Crystalline intermediates can be found only by chance [55, 56]. Addition of donor molecules like pyridine, C5H5N, sometimes even polar solvent molecules, may trap intermediate products and prevent them from consecutive reaction, thus yielding crystal structures [47, 57]. Mass spectrometry revealed the presence of numerous differently fluorinated intermediates and therefore crystallisation from such reactions systems is suppressed [58]. Hence, just a few fluorinated intermediates were obtained in the form of single crystals. As a rule, intermediates and final products mostly are amorphous. However, liquid- and solid-state NMR were extensively applied to characterise these amorphous intermediates and led to a deeper insight into the related phenomena for MgF2 and AlF3 [34, 48, 49, 59,60,61,62]. It is generally expected that the underlying mechanisms are more or less analogous for all other systems.
A short outline will be given here using the structural features of fluorinated intermediates derived from aluminium isopropoxide, Al(OiPr)3. Figure 7 shows a more detailed illustration of the structural processes which are derived from real single crystals of intermediates that have been isolated from reaction mixtures of different degree of conversion [27]. Aluminium isopropoxide usually occurs as a tetrameric cluster Al[Al(OiPr)4]3 bearing bridging and terminal isopropoxide groups (structure 1). The attack of HF starts at a bridging isopropoxide group, yielding a trinuclear Al3(OiPr)8F·HOiPr unit bearing one terminal fluoride ion, i.e. F:Al = 1:3 (structure 2). Further reaction with HF leads to the formation of a tetrameric [Al4(OiPr)7F4(µ4-O)]− unit bearing four terminal fluoride ions, i.e. F:Al = 1:1 (structure 3).

Schematic representation of known structural elements occurring during the fluorination of aluminium isopropoxide
In principle, these first reactions are those described by the simplified Eq. 6. However, the whole process is more complex. Although introduced fluoride ions still occupy terminal positions only, rearrangement of the clusters takes place. Additionally, bridging oxygen starts to occur at this stage of the reaction. The source of this oxygen is not completely clarified, but most probably it derives from traces of water, which are either intrinsically present or formed by the acidic dehydration of isopropanol. Therefore, the reactions described by Eq. 5b for the hydrolytic sol–gel synthesis will also take place to a minor extent. The occurrence of minor oxygen species is very pronounced for AlF3, but they are also detected in traces by NMR spectroscopy for other systems like MgF2 and CaF2. It is speculated that these bridging oxygen moieties in particular cause mainly the high degree of structural distortion inside the metal fluoride nanoparticles, which are finally formed.
Increasing amounts of HF, i.e. F:Al > 1, quickly lead to the formation of larger structures with bridging fluoride ions (structures 4, 5 and 6). This process is described by the simplified Eqs. 7a and 7b. The majority of mechanistic and structural information derives from NMR spectroscopy, but a few of these structures are known from X-ray diffraction of single crystals which fully reconfirm structural conclusions from NMR. The more the F:Al ratio approaches the stoichiometric ratio of 3, the less terminal fluoride is present. Finally, the result of the reaction is a product with the approximate stoichiometry Al(iOPr)0.1–0.2F2.8–2.9·nHOiPr (n = 0.5–2.0). Residual isopropoxide groups are still present, and large amounts of solvent are still loosely bound. Investigation of the fluorination of aluminium sec-butoxide in THF solution fortified this interpretation of the mechanism. In this case, well-defined intermediates could be also identified by mass spectrometry [58].
Full fluorination of aluminium precursors cannot be achieved in liquid phase even with an excess of HF. In the worst case, crystalline AlF3 is obtained instead of nanoscopic AlF3. However, full fluorination without destruction of the nanostructure is achieved by an additional gas-phase post-fluorination with suitable fluorinating reagents like CCl2F2, CHClF2 or related compounds at temperatures between 180 and 240 °C, ending up with an almost stoichiometric AlF3 possessing a high surface area and an extraordinarily high Lewis acidity [28].
In the case of the alkaline earth metal fluorides MgF2, CaF2 and SrF2, a nearly stoichiometric fluorination is less challenging than in the case of AlF3. However, as a general principle for these systems, comparatively large amounts of coordinated residual organic molecules are always present at the particle surface. Those are usually solvent molecules or the protonated precursor anions released during the fluorination, i.e. acetic acid in the case of acetate precursors, lactic acid in the case of lactate precursors and so on. These absorbed molecules fulfil the important task of kinetic stabilisation of the nanoparticles, preventing them from agglomeration.
Deagglomeration and gelation
The nucleation process described in the previous section does not always lead directly to isolated primary nanoparticles. Often agglomerates with diameters of 100–200 nm are formed, which results in turbid sols. Ageing of such sols under appropriate reaction conditions, just by stirring in the simplest case, leads to a continuous breakdown of these agglomerates, finally resulting in transparent sols of homodisperse, ultrasmall metal fluoride nanoparticles (cf. Fig. 6b). This behaviour of metal fluoride nanoparticles is strikingly different from that of metal oxide nanoparticles. In the latter case, the agglomerates grow into larger particles.
The details of agglomeration and deagglomeration are best studied for MgF2 [31, 34]. The fluorination of magnesium methoxide in methanol leads to the formation of a turbid sol. The diameter of the initially formed agglomerates is around 130 nm. Within a few days, the diameter dropped below 40 nm, and after 1 month it dropped further to 10 nm, resulting in a transparent sol of low viscosity (Fig. 8). The size of the nanocrystallites, i.e. the size of the nanoparticles, does not change during this time, strongly supporting the model of deagglomeration discussed above.

Reproduced from J. Mater. Chem. 2011, 21, 15015–15021 (Ref. [34]) with permission of the Royal Society of Chemistry
Hydrodynamic diameter of a MgF2 sol (0.25 M) at different stages of ageing beginning 6.5 h after HF addition to 1 month as size distribution (a) and mean diameter vs. ageing time (b) obtained from dynamic light scattering (DLS) measurements.
The nature of the forces binding the primary particles inside the agglomerates has been a field of speculation. The processes during the nucleation (Eqs. 7a, 7b and Fig. 7) will also lead to weakly bound particle surfaces, which may be “stuck” together by precursor anions (alkoxides, carboxylates etc.) and traces of unreacted HF bound to unsaturated metal centres at the particle surface. Upon ageing, these groups become replaced by solvent molecules and the last traces of HF react to form fluorides, weakening the forces between the particles. Unlike in the case of metal oxides, the surface zeta potential does not play any role here and remains nearly constant during the whole process. For most metal fluoride nanoparticles, the zeta potential is between + 20 and + 40 mV regardless of solvent or pH value. Thus, electrostatic effects can be ruled out as an explanation for the deagglomeration. This is another striking difference between metal fluoride and metal oxide nanoparticles.
Further investigation of ageing processes led to the conclusion that water also plays an important role in this process [31, 50]. Traces of water are always present in these systems and can be tolerated, but water may form bridging OH groups linking particles via their surfaces (Fig. 9). This process seems to be reversible as long as the amount of water is small. Larger amounts of water quickly increase the viscosity of the sol and finally lead to gelation. This is a significant problem for MgF2 sols synthesized from magnesium acetate instead of magnesium methoxide. Magnesium acetate is commercially available as the tetrahydrate, Mg(OAc)2·4H2O. This compound is not suitable for the sol–gel synthesis because of the large amount of water of crystallisation. Removal of the water of crystallisation in a vacuum is possible (but not at ambient pressure), leading to the anhydrous salt Mg(OAc)2 [62, 63], which is fortunately soluble in ethanol. During the fluorolytic sol–gel synthesis, large amounts of acetic acid are formed, which slowly undergoes esterification with the solvent ethanol (Eq. 8).

Schematic representation of water-catalysed agglomeration of metal fluoride nanoparticles. Presence of more water will lead to gelation and finally to precipitation due to Ostwald ripening
As the reaction proceeds, increasing amounts of water are formed, leading to agglomeration of the particles, increasing the viscosity and finally resulting in the formation of a gel. Thus, MgF2 sols synthesized from acetates or other carboxylates are not stable even on a medium timescale, but can be used for a few weeks. This is a remarkable difference to MgF2 sols synthesized from magnesium methoxide, which keep their low viscosity for more than 1 year.
Several approaches exist to accelerate the deagglomeration process. Most promising is the addition of small amounts of a strong acid. Appropriate acids are fluorinated carboxylic acids, phosphonic or sulfonic acids. Addition of ca. 10 mol% of phenylphosphonic acid, C6H5PO3H2, or vinylphosphonic acid, C2H3PO3H2, to a turbid sol of MgF2 prepared from magnesium methoxide, Mg(OCH3)2, leads to deagglomeration down to diameters of 5 nm and below within 1 day instead of 1 month without the addition of acid (cf. Fig. 8).
Two effects are responsible for this remarkable effect (Fig. 10). First, the strong acid protonates bridging OH groups leading to the release of water and thus bonds between particles are broken. Second, the acid anions may bind to the particle surface, blocking reactive surface sites and hence preventing the formation of new bonds between the particles. HF itself or carboxylic acids are too weak to enforce this effect. A drawback of this approach is the fact that strong acids catalyse the esterification reaction (Eq. 8), which quickly increases the amount of water in the sol, leading to additional agglomeration. Both effects act in opposition to each other and therefore addition of this additional stronger acid requires caution. While in the case of MgF2 sols synthesized from methoxide 10–15 mol% of phenylphosphonic acid, C6H5PO3H2, or TFA is appropriate for deagglomeration, this is far too much for MgF2 sols synthesized from acetate. Here 2–5 mol% of TFA is sufficient. Less acid will not lead to fast deagglomeration, whereas more acid will increase the esterification reaction. But anyway, such sols may be stabilized to a certain extent by these small amounts of TFA.

Schematic representation of acid-catalysed deagglomeration of metal fluoride nanoparticles
The second approach to increase deagglomeration utilises the fact that the metal fluoride nanoparticles always have a slightly positive surface zeta potential of + 20 to + 40 mV. The reverse conclusion is that adjustment of this zeta potential during or immediately after the reaction will a priori reduce agglomeration and increase deagglomeration. Using slightly less HF than stoichiometrically necessary for the full fluorination will leave unreacted metal cations at the particle surface. Alternatively, the addition of small amounts of suitable electrolytes (a metal salt or an easily hydrolysing metal compound) after the synthesis has a similar effect. The latter option leaves plenty of space for adjustment, because salts of any other metal can also be used.
Additionally, the reactivity of water and surface sites is influenced by the solvent. As a rule of thumb, the more hydrophilic a solvent is, the less is the tendency of the nanoparticles to form agglomerates. This effect is very distinct for ethylene glycol, less for methanol and further decreases for ethanol and isopropanol. Small amounts of water are effectively solvated by dihydric alcohols, i.e. glycols, reducing their reactivity towards the particle surface. Additionally, ethylene glycol can better bind to the particle surface then monohydric alcohols and hence blocks the reactive surface site more efficiently. Similar effects occur when a hydroxycarboxylic acid, e.g. lactic acid, CH3CH(OH)COOH, is present. Contrary to acetic acid, lactic acid is bound more strongly to the particle surface owing to a chelate-like effect, preventing agglomeration even for larger amounts of water. As an example, CaF2 and SrF2 sols in ethylene glycol containing lactic acid can be diluted with water without the occurrence of cloudiness or gelation [43, 44].
The main bottleneck of adaption of the fluorolytic sol–gel synthesis to any new systems is the deagglomeration behaviour. It is a complex interaction between solvent, precursor anion, additives and reaction conditions. Prediction of the optimum is not straightforward; however, some general trends were outline in this article.
In conclusion, all these parameters do not disturb the formation of primary small metal fluoride nanoparticles, but may cause agglomeration of these nanoparticles. That might be a drawback if clear, homodisperse metal fluoride sols are desired e.g. for coatings of surfaces or preparation of composite materials.
Applications of nano metal fluorides
Catalysis
Solid metal fluorides obtained after drying the respective sols exhibit extremely high surface areas in comparison to their crystalline analogues and are in the range of 200–300 m2 g−1 or even higher. Because of their high surface areas these metal fluorides are usually defined as HS-MFn (HS = high surface). As a result of the strong electron-withdrawing effect of the fluorine anions in combination with the high degree of structural distortion and the large surface area, these metal fluorides exhibit extraordinarily high Lewis acidity. Thus, HS-AlF3 is one of the strongest Lewis acids and comparable to SbF5 and aluminium chloride fluoride (ACF) [28]. CO adsorption FTIR investigations revealed the strongest Lewis acid sites ever measured at solid surfaces [64]. Figure 11 illustrates the shift of the IR frequency of carbon monoxide adsorbed onto HS-AlF3, thus indicating extremely strong Lewis acid sites at the surface that have never been observed at any solid Lewis acid before.

Reproduced from Cat. Sci. Tech. 2015, 5, 786–806 (Ref. [65]) with permission of the Royal Society of Chemistry
IR difference spectra of stepwise CO adsorption on sol–gel derived HS-AlF3. ν(CO) = 2150 cm−1—physisorbed CO; ν(CO) = 2175 cm−1—medium strong Lewis acid sites; ν(CO) = 2240–2220 cm−1—very strong Lewis acid sites.
As a consequence, these new metal fluorides are exciting solid catalysts for Lewis acid catalysed reactions and outperform even the best homogenous catalysts in several reactions [37, 66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81]. A comprehensive overview on the catalytic properties of this new class of catalysts and their wide applications in different fields of catalysed reactions can be found in recently published reviews [65, 82, 83]. Therefore, just a few highlights will be given here.
Dehydrohalogenation reactions of chlorofluorocarbons yield fluoro(chloro)olefins which are very interesting synthons for fluoropolymers. These kinds of reactions usually proceed in the presence of solid Lewis acids, but need temperatures between 500 and 600 °C in order to give high conversions. Since the C–Cl bond is weaker than the C–F bond, all classically known solid Lewis acidic catalysts also enable dehydrochlorination reactions. However, HS-AlF3 was found to catalyse the dehydrofluorination reaction of 3-chloro-1,1,1,3-tetrafluorobutane (Eq. 9) with greater than 99% conversion and 100% selectivity exclusively toward the dehydrofluorination product at as low as just 200 °C [76].
Interestingly, with nanoscopic high surface BaF2 as solid catalyst, exclusively the dehydrochlorination reaction (Eq. 10) was observed, giving ca. 98% conversion and 100% selectivity regarding the dehydrochlorination product.
Mechanistically, the contrary catalytic behaviour was discussed on the basis of the HSAB concept; the very hard Lewis acid Al3+ preferentially interacts with the very hard fluorine atom (Fig. 12a) of the CFC whereas the softer Lewis acidic Ba2+ surface sites preferentially interact with the weak chlorine atoms of the CFC (Fig. 12b).

Reproduced from Cat. Sci. Tech. 2017, 7, 773–796 (Ref. [83]) with permission of the Royal Society of Chemistry
Proposed catalytic mechanism for the dehydrohalogenation of 3-chloro-1,1,1,3-tetrafluorobutane. a dehydrofluorination with AlF3 and b dehydrochlorination with BaF2. Note, for simplicity reasons, products resulting here will be the terminal alkenes and not the bond isomerised products as given in Eqs. 9 and 10.
Another exciting example for the superior catalytic power of metal fluoride nanoparticles is the catalytic cleavage of C–F bonds which became possible for the first time without any precious metal and surprisingly under room temperature conditions. Thus, this became possible by creating silylium-like species at the surface of aluminium chloride fluoride (ACF = AlClxF3−x with x ≈ 0.05–0.2) and HS-AlF3, respectively, as was evidenced by 1H MAS NMR spectroscopy as shows in Fig. 13 [67].

Reproduced from Cat. Sci. Tech. 2015, 5, 786–806 (Ref. [65]) with permission of the Royal Society of Chemistry
a 1H MAS NMR spectrum which shows the signals for surface-bound Et3SiH (νrot = 10 kHz) with the Si–H resonance at δ = 3.45 ppm. For Et3SiH in C6D6 solution the resonance is observed at δ = 3.85 ppm. b Schematic representation of ACF···H–SiEt3.
On treatment of CH3F, CH2F2 and CHF3 with these aluminium fluoride catalysts in C6D6 in the presence of Et3SiH at room temperature at 1 atm, vigorous reactions and the evolution of gaseous products were observed (Fig. 14). In all these cases, formation of fully hydrogenated methane was observed for the first time under heterogeneous conditions, at low temperature and in the absence of a precious metal catalyst.

Reproduced from Angew. Chem. Int. Ed. 2013, 52, 5328–5332 (Ref. [67]) with permission of Wiley
ACF-catalysed C–F activation reactions of fluoromethanes and trifluorotoluene.
Although classically prepared MgF2 is a rather neutral compound, nanoscopic MgF2 obtained via the fluorolytic sol–gel synthesis exhibits very interesting catalytic properties. Post-fluorination of the dried xerogels at 120 °C with HF yields nearly X-ray amorphous magnesium fluoride (HS-MgF2) with a specific surface area in the range of 200–300 m2 g−1 [84]. The acid–base properties of the sol–gel MgF2 were determined by FTIR spectroscopy of different adsorbed probe molecules. While the adsorption of carbon monoxide shows medium to weak Lewis acid sites due to coordinatively unsaturated magnesium sites, we explored the basicity of surface fluorine atoms by pyrrole adsorption for the first time [85]. The exciting catalytic properties of nanoscale MgF2 have been recently reviewed [82].
By combination of two or even more metals, ternary, quaternary or even more complex metal fluorides can be obtained which allow adjustment of the optimized surface Lewis acidity over a very wide range [86, 87].
A further extension of this new approach results from the combination of the fluorolytic with the hydrolytic sol–gel reaction. In the first step the fluorination is performed with substoichiometric amounts of HF resulting in an alkoxide fluoride according to Eq. 11 (M = metal, n = metal valence).
The remaining OR groups can be reacted with water in the second step, resulting in the formation of hydroxide fluorides (Eq. 12).
In this way, metal hydroxide fluorides are accessible in which the metal site is coordinated by anions, hydroxide and fluoride, thus presenting real hydroxide fluorides which are almost inaccessible via any other synthesis route [88]. By varying the F to OH ratio, the whole series of M(OH)xFn−x ranging from M(OH)n to MFn can be synthesized. As a result, the Lewis to Brønsted acid site ratio of these materials can be tuned over a wide range, resulting in optimised solid catalysts that are of great interest for any kind of acid–base-catalysed reaction [65, 82].
A slightly modified synthesis approach is the application of stoichiometric amounts of HF but in the presence of water, such that the fluorolysis of the methoxide is in direct competition with the hydrolysis. As a consequence of different reaction kinetics, magnesium hydroxide fluoride particles with a MgF2-like structure were obtained when the sol–gel synthesis was performed with aqueous hydrofluoric acid and a F/Mg ratio of 2:1 [80, 89]. The 19F MAS NMR spectra show a major signal at − 198 ppm corresponding to the rutile structure of MgF2 and a broad shoulder in the lower field (− 184 ppm), which evidences an oxygen-rich coordination sphere around [FMg3] sites. FTIR adsorption studies using CO and lutidine (2,6-dimethylpyridine) have identified Lewis acid sites of medium strength and Brønsted acidic Mg–OH sites. The latter can be explained by the electron-withdrawing effect of the magnesium fluoride backbone on the hydroxyl group at the particle’s surface. Different MgF2−x(OH)x phases with varying ratio of Lewis to Brønsted acidic surface sites can be obtained by variation of the HF concentration in water [89].
Owing to their high surface areas in combination with adjustable acid–base properties, these new materials also have great potential as catalyst support materials, which have been reported in several recent publications [65, 83].
Optical applications
Optical properties overview
Metal fluorides possess unique optical properties compared to metal oxides and other solid inorganic materials (Table 2). Their transparency extends further into the UV region than any other material as a result of the high ionic character of the solids, resulting in a large gap between valence and conduction bands. Transparency also extends further into the IR region than for metal oxides as a result of weaker Coulomb interaction, resulting in lower phonon energy. Very importantly, they have lower refractive indices than metal oxides and most other inorganic materials because of the low polarisability of the fluoride anion.
These properties make metal fluorides interesting materials for optical applications. Their wide transparency is applied in UV optics, their low refractive index is used for antireflective coatings and similar applications, and the low phonon energy makes them interesting as host materials for luminescent rare-earth metal ions.
The availability of these materials as nanoparticles offers new fields of applications, which are not accessible by using bulk materials.
Antireflective coating
Light passing through the interface between different optical transparent media, e.g. air and glass, is partially reflected, even when each single component is fully transparent. Float glass has a refractive index of ca. 1.52, resulting in a reflection loss of 4% at the phase interface [91]. Thus, the transparency of an untreated panel is 92% only. This effect sums up drastically for stacks of several glass panels. For instance, a standard composite window consisting of three untreated glass panels has a transparency of only 78% (\(0.96^{6} \approx 0.78\)). Reduction of reflection loss is highly desired to reduce the cost for indoor illumination.
Reflection loss may be reduced by coating glass with an antireflective λ/4 layer with a thickness of ca. 125 nm and a refractive index between those of air (η ≈ 1) and glass (η ≈ 1.52). For an ideal antireflective effect, the refractive index of the coating should be \(\sqrt {1.52} \approx 1.23\). Bulk materials offering this refractive index do not exist. However, coating/deposition of a dense MgF2 layer with a refractive index of 1.38 already reduces reflection and hence increases optical transmission, although not totally. Such dense layers are manufactured by chemical vapour deposition (CVD) or magnetron sputtering and possess an adequate mechanical stability for industrial application [92, 93].
Porous antireflective layers possess lower refractive indices than their dense counterparts. Silica layers with 50% porosity have a refractive index of ca. 1.25. Indeed, such layers provide better antireflective properties than dense MgF2 layers. Unfortunately, the high porosity decreases the mechanical stability of such layers, limiting their application [52].
The application of MgF2 nanoparticles can overcome this gap by combining both effects. Dip-coating of glass panels with MgF2 sols in ethanol produces a porous layer of MgF2 instead of a dense layer [35, 39, 50]. As a result of the lower refractive index of MgF2 compared to SiO2 (Table 2), a porosity of 35% is sufficient for appropriate optical properties. Therefore, these porous MgF2 layers have a higher mechanical stability than the highly porous SiO2 layers.
Layer thickness is adjusted by variation of the withdrawal speed and porosity is adjusted by the annealing temperature (350–500 °C). Under appropriate conditions, MgF2 layers with 35% porosity are obtained that have a refractive index of 1.23–1.25 [35, 39, 50]. Figure 15a illustrates the enhanced optical transparency of the coated glass. The SEM image in Fig. 15b clearly shows the porosity of the MgF2. The residual reflection of coated glass panels is far below 1% for float glass and around 1% for borosilicate glass (Fig. 15c). These MgF2 layers are mechanically much more stable than porous SiO2 layer. MgF2 even resists scratching with coarse steel wool [52]. Although CaF2 has a slightly higher refractive index than MgF2, mechanically stable antireflective porous CaF2 layers are also accessible via the fluorolytic sol–gel synthesis [32].

Picture b reproduced from J. Mater. Chem. 2012, 22, 18535–18541 (Ref. [35]) with permission of the Royal Society of Chemistry
The synthesis of MgF2 sols for coating is easily upscaled to the amounts needed for the coating of larger objects. Successful coating of glass panels with a size of 1 m × 1 m was demonstrated. The application of the dip-coating process has another advantage, because coating of irregularly shaped objects like lenses and tubes is possible, while CVD is limited to flat substrates.
Luminescence host materials
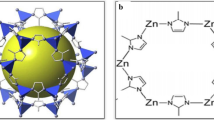
Luminescent nanoparticles based on rare-earth element doped inorganic fluorides possess a wide field of applications [94,95,96,97]. The luminescence of the trivalent ions of the rare-earth elements covers a certain spectral range ranging from UV to IR. As a brief outline, Eu3+ and Tb3+ show intense emission of red or green light, respectively, upon excitation in the UV region and are used phosphors; Nd3+ and Er3+ show emission in the near-IR range and are used for lasers and amplifiers; and the pairs Yb3+/Ho3+, Yb3+/Er3+ and Yb3+/Tm3+ show the effect of photon upconversion, i.e. the emission of visible light upon excitation with IR radiation.
Compared to metal oxides or other inorganic materials, metal fluorides are superior host matrices for luminescent rare-earth element ions owing to their low phonon energy, which reduces non-radiative relaxation. Appropriate matrix compounds are the non-luminescent YF3, LaF3 and LuF3, complex fluorides like LiYF4, NaYF4 and their relatives, and finally also the alkaline earth metal fluorides CaF2, SrF2 and BaF2. Out of those, SrF2 has proven to be an excellent host matrix.
Doping of the luminescent trivalent rare-earth element ions into SrF2 nanoparticles transfers their luminescent properties to the nanoparticles. Up to 40 mol% of Ln3+ (Ln = Y, La–Lu) can be incorporated into SrF2 without disturbing the cubic crystal structure, ending up with nanoparticles of the stoichiometry Sr1−xLnxF2+x (x = 0–0.4). This holds both for single crystals [98] and nanoparticles [43].
The fluorolytic sol–gel synthesis of rare-earth metal doped SrF2 is straightforward. Combinations of different rare-earth metal ions with a total content of up to 40% are possible. Figure 16a illustrates the optical appearance of Eu3+ and Tb3+ doped SrF2 sols, showing intense red and green luminescence. Compared to similar systems, these nanoparticles derived from the fluorolytic sol–gel synthesis particles are unique in their luminescence properties. Figure 16b shows the emission spectra of Eu3+ doped SrF2 with Eu doping rates. These are the typical emission spectra of Eu3+ caused by 5D0 → 7FJ transitions inside the 4f energy level with three intensive bands centred at 590 nm (J = 1), 610 nm (J = 2) and 698 nm (J = 4). Surprisingly, the emission intensity increases up to a Eu content of 40%. This behaviour is somehow unexpected, because the usually observed effect is a decrease of the emission intensity above 10–15% doping rate because of non-radiative cross relaxation. Obviously, cross relaxation is a minor effect in these particles, underlining their unique potential for further application.

Reproduced from Dalton Trans. 2017, 46, 2925–2936 (Ref. [43]) with permission of the Royal Society of Chemistry
a Sols of SrF2 nanoparticles doped with 10% Tb3+ and Eu3+ in daylight and under UV excitation. b Photoluminescence emission spectra of SrF2 nanoparticles with different doping rates of Eu3+ ranging from 0.1% to 40%. Inset: total integrated emission intensity.
Further tuning and improvement of the luminescent properties is possible by the synthesis of core–shell particles, increasing the quantum yield and facilitating tailor-made systems [44]. Significant increase of the luminescence intensity is achieved by co-doping with Ce3+ or by coupling of organic antenna ligands to the particle’s surface [43, 99]. Particles co-doped with Yb3+ and Er3+ simultaneously are capable of photon upconversion, showing emission of photons in the visible range upon laser excitation at a wavelength of 980 nm [43]. Figure 17 shows a few examples illustrating the nearly unlimited flexibility of these systems.

a Sol of SrF2:Ce3+,Gd3+,Ln3+ excited at 254 nm. Emission colour is varied by variation of the element Ln. b Sol of SrF2:Yb3+,Er3+ excited at 980 nm showing emission of visible light (photon upconversion).
Biocidal products
Timber decay caused by fungi and fire is estimated to cost $300 million per year in the USA [100]. In the northern hemisphere, fungal decay is associated with brown rot fungi which predominantly attack conifer trees, a major source for timber used for construction [101]. Brown rot fungi such as Coniophora puteana (Cp) and Rhodonia placenta (Rp) attack wood by sequential decomposition of cellulose and hemicellulose [102].
Usually, copper- and chromium-based compounds are very active wood preservation agents but there are environmental reservations against the use of such compounds [103]. An alternative set of compounds known for their toxicity against insects and fungi are fluorides such as sodium fluoride and fluosilicates [104, 105]. Unfortunately, similar to copper, these water-soluble compounds need to be combined with fixatives such as chromium to reduce their leaching into the environment as higher concentration of fluorides in water can be toxic to the environment [106]. Instead of using fixatives, fluorides with low water solubility would be interesting alternatives since they should not undergo leaching or only to a low extent [104]. However, to introduce such low soluble fluorides into cellulose, small nanoparticles are needed which might be able to penetrate into wood and may create a “fluoride reservoir” inside the wood cells from which there is a controlled release of fluoride ions over a long time period dependent on the solubility of the fluoride compounds. In this way, uncontrolled environmental pollution can be reduced to an extremely low level.
Thus, nanoscopic MgF2 and CaF2 prepared according the fluorolytic sol–gel approach were tested as wood preservation agents [107]. Inclusion of nano metal fluorides in the wood specimens was confirmed by back-scattered electron microscopy and energy-dispersive X-ray spectrometry (EDX) (Fig. 18). Aggregates of MgF2 and CaF2 nanoparticles appear as bright particles in back-scattered images (Fig. 18a, d). The corresponding EDX maps confirm the distribution of Mg, Ca and F inside the wood cells (Fig. 18b, c, e, f).

Back-scattered electron (BSE) images and elemental distribution maps of wood (cross-cut section) specimens after impregnation. a BSE image of MgF2-treated wood and corresponding EDX map, b Mg—yellow and c F—red; d BSE image of CaF2-treated wood and corresponding EDX map, e Ca—blue and f F—red. Scale bar 60 µm
After characterization of the treated wood specimens, their resistance against decay by brown rot fungi (Cp and Rp) was assessed in accordance with the European standard EN 113 [108]. Differences in mass loss between treated and control specimens exposed to fungi are depicted in Fig. 19. The mass loss caused by Cp in treated wood was below 3%, while the control specimens had a significantly higher mass loss of 38%. For Rp the mass loss in treated wood was lower than 15%, while the control specimens showed a mass loss of 34%. The mass loss in CaF2- and MgF2-treated specimens does not differ significantly. For Cp, the mass loss of treated wood specimens was 1% (MgF2) and 2% (CaF2), respectively. Similarly, for Rp the mass loss of treated wood specimens was 12% (MgF2) and 14% (CaF2), respectively. The minor differences in mass loss between MgF2- and CaF2-treated specimens are due to the differences in their solubility; with a free fluoride concentration of 16 mg/L for CaF2 and 130 mg/L for MgF2, the latter provides a larger F− concentration [109]. Hence, MgF2 is more active because of the slightly higher fluoride ion concentration in the wood specimens. Notwithstanding, the lower water solubility of CaF2 makes it attractive for long-term treatment because it would remain longer within the wood cells.

Average mass losses of pine sapwood blocks treated with nanoscopic MgF2 and CaF2 after exposure to Coniophora puteana and Rhodonia placenta
The change in the appearance of the wood specimens after exposure to Cp and Rp is shown in Fig. 20. It is evident that the control specimens are severely damaged, while the fluoride-treated specimens had minimal damage for specimens exposed to Cp. Thus, the nano metal fluorides protect the wood specimens from extensive fungal deterioration. Even though treated specimens exposed to Rp show cracks on their surfaces, their damage is less compared to the control samples which have shrunk remarkably because of cellulose degradation.

Wood specimens after exposure to fungi, narrow and wide longitudinal surface. a, b MgF2-treated after exposure to Cp; c, d CaF2-treated after exposure to Cp; e, f MgF2-treated after exposure to Rp and g, h CaF2-treated after exposure to Rp
Most importantly it has to be highlighted that these nano metal fluoride particles were tested without any fixatives; thus, their effectivity against fungi is promising. Previous papers reported that soluble fluorides can also be effective against termites [110,111,112]. Thus, nano MgF2 and CaF2 were tested against termites [113]. These studies showed that specimens treated with nano metal fluorides are less damaged than the severely attacked controls when exposed to termites. Over the duration of 8 weeks, while the termite resistance test with other nano treatments such as nano boron and nano zinc borate reported in the literature were performed just for 4 weeks or less, nano metal fluoride-treated samples provide longer-term protection compared to any other nano formulations (Fig. 21).

Termite mortality in specimens treated with nano metal fluorides synthesized in ethanol as solvent
Susceptibility to leaching is significantly reduced with nano metal fluorides compared to NaF. The lower water solubility of MgF2 and CaF2 means that they can be used without fixatives, making them more environmentally friendly and sustainable alternatives for wood protection against termites, too.
Organic–inorganic composites
Introduction to composites
Organic–inorganic composites are a class of materials finding more and more interest for different applications. They possess properties which differ from the single components alone. These properties can be tuned by variation of the organic or inorganic component, or both. The availability of homodisperse metal fluoride nanoparticles opens new fields of applications.
Out of many possible combinations, composites of metal fluorides and polyacrylates are of increasing interest. Transparent polyacrylates find a wide field of application. Transparency is not only desired for the final product but is also a necessary precondition for UV polymerisation of the monomers even in those cases where the transparency of the final polymer is irrelevant. An opaque polymer can only polymerise by thermal activation, limiting the field of application. As already demonstrated, sols of metal fluoride nanoparticles are transparent when the mean particle size is below 20 nm (Figs. 4, 16). This is a good premise for obtaining transparent composite materials using these inorganic materials. For that reason it is necessary to prevent agglomeration of the nanoparticles during the transfer from the originally synthesised sol to the acrylates.
As already discussed previously, the equilibrium between agglomeration and deagglomeration strongly depends on the solvent and furthermore may be influenced by different additives. Many acrylates are hydrophobic compounds, while the metal fluoride nanoparticles are synthesised in hydrophilic solvents. Therefore, normally some kind of adaption is necessary to ensure compatibility between the organic and the inorganic component of the composite. This goal is achieved by different additives, which adjust the surface properties of the particles (cf. Fig. 18). Non-reactive surface modification adjusts the surface polarity to the acrylate, making the particles more hydrophobic. Reactive surface modification additionally introduces polymerisable groups, which bind directly to the backbone of polyacrylate (Fig. 22). In the simplest case, the latter goal is already achieved by addition of a certain fraction of a more hydrophilic acrylate bearing hydroxyl groups like HEMA (2-hydroxyethyl methacrylate), HBMA (2-hydroxybutyl methacrylate) or bis-GMA (bisphenol A-glycidyl methacrylate) [49, 114, 115].

Different type of possible surface modifications of metal fluoride nanoparticles
Dispersion of the nanoparticles in acrylate monomers is achieved either by mixing of the metal fluoride sols with the monomer followed by evaporation of the solvent, or by drying of the sol followed by re-dispersion of the particles in the acrylate. Provided that no agglomeration of the nanoparticles occurs during the polymerization, transparent nanocomposites with high filling grades are obtained. Figure 23 shows transparent volume composites of MgF2 and polyHEMA. Transparency is kept even at high fraction of the inorganic filler. The properties of the composites change with increasing content of the filler. For instance, the glass transition temperature increases with increasing MgF2 content.

Pure polyHEMA and composites with MgF2 nanoparticles. Tg glass transition temperature, HEMA 2-hydroxyethyl methacrylate
Variation of the composite’s mechanical properties is not as straightforward as is might appear at first glance. The hardness of the composite is not just a mean value between the hardnesses of the organic and inorganic material. The whole subject is more complex, because dynamic phenomena like crack propagation must not be neglected. Nevertheless, an increase of the hardness of the polymer by introduction of metal fluorides is in general possible.
Figure 24 illustrates the Martens hardness of two different composites, polyHDDMA filled with MgF2 nanoparticles and polyBDDMA filled with AlF3 nanoparticles. In the case of MgF2/polyHDDMA, the composites show a distinct increase of hardness compared to the pure polymer even at low MgF2 fractions. At 10% inorganic content, the hardness has nearly doubled from 100 to 200 N mm−2. Further increase of the MgF2 fraction has little effect on the material’s hardness. In contrast, there is nearly no increase of the hardness of AlF3/polyHDDMA up to 15% AlF3 content. At even higher filling contents up to 25%, only a slight increase of the hardness occurs.
However, these examples illustrate that tuning of the mechanical properties of the composite materials is possible. Further research has to be done. A promising approach is the addition of reactive surface modifications (cf. Fig. 18), which ensure a direct binding between the organic and the inorganic component.

Optical applications
Refractive index
Metal fluorides possess outstanding optical properties, in which they differ from nearly all other materials (Table 2). In particular, their low refractive indices between 1.35 and 1.45 make them very interesting materials for optical composites. The refractive index of most polyacrylates ranges between 1.49 and 1.55. Transparent organic materials with lower refractive indices are very rare. Indeed, polyacrylates bearing perfluorinated alkyl groups offer significantly lower refractive indices, but these compounds are environmentally critical because of their long persistence in nature and their use will be phased out soon [117].
A novel approach to the abandonment of fluorinated organic compounds is the transfer of the properties of inorganic metal fluoride nanoparticles to transparent composite materials. MgF2 is very suitable for that purpose owing to its very low refractive index of 1.38 paired with low solubility and toxicity. The resulting refractive index of the composite material consists of the sum of the refractive indices of the single components multiplied by their volume fraction.
Figure 25a illustrates the variation of the refractive index of MgF2/polyBDDMA composites. The refractive index decreases linearly with increasing MgF2 as theoretically expected. However, the refractive index is additionally influenced by the additives ensuring compatibility of the organic and inorganic component (cf. Fig. 18). Figure 25b demonstrates this behaviour for AlF3/polyBDDMA. The refractive index of the composites decreases with increasing AlF3 content, but the two different composites show slightly different refractive indices due to the different type of functionalisation of the particle surface.

Refractive index of different transparent composites. a MgF2/polyHDDMA composites. b Two AlF3/polyBDDMA composites. Different refractive indices of the AlF3 composites are caused by different functionalisation of the particle surface. BDDMA 1,4-butanediol dimethacrylate, HDDMA 1,6-hexanediol dimethacrylate
As a consequence, transparent polymer composites with low refractive indices are possible without the application of long-chain fluorinated groups. In addition, exact adjustment of the refractive index is possible by variation of the content of the inorganic filler. Therefore, these composite materials are superior over fluorinated organic acrylates, which have fixed refractive indices.
Luminescence
The transfer of optical properties is not limited to tuning of the refractive index. Metal fluoride nanoparticles are excellent host materials for luminescent rare-earth metal ions (cf. Fig. 16). Tuning of their luminescent properties is possible by variation of the rare-earth element, by the synthesis of core–shell systems and by sensitization with organic antenna ligands (cf. Fig. 17) [43, 44]. In particular, the application of antenna ligands not only allows the luminescence properties to be tuned but also opens the possibility of adapting the particle surface to the organic material. By introduction of such particles into organic polymers, transparent luminescent composites are available.
For instance, SrF2:Eu3+ nanoparticles modified with SDBS (sodium dodecylbenzene sulfonate) show intense luminescence and are hydrophobic enough to form transparent composites with hydrophobic polyacrylates [99]. Figure 26 shows several additional examples of composites of differently modified SrF2:Eu3+ nanoparticles and diacrylates (mainly TEGDMA and D3MA) exhibiting deep-red luminescence upon excitation with UV light.

Composites of 10 wt% of SrF2:Eu3+ (10% Eu) and different formulations of polyacrylates. Mod surface modification, TEGDMA triethylene glycol dimethacrylate, D3MA 1,10-decanediol dimethacrylate, BMA butyl methacrylate, LMA dodecyl methacrylate, IBMA isobornyl methacrylate
Thus, new fields of application are opened here. Green luminescent composites are available by replacing Eu3+ by Tb3+, and yellow and orange colours are achieved by mixing. Such systems can be directly used for illumination. Doping with Er3+ broadens the scope to materials capable of amplifying modulated IR radiation (1500 nm) used for fast information transfer in optical fibres.
Dental applications
Fluorides find wide application in dental medicine. Fluoride has an antibacterial effect and hence prevents caries formation. Beside the antibacterial effect, which directly influences caries microorganisms, fluoride enhances remineralisation. Furthermore, hydroxyapatite in tooth enamel is slowly transformed into fluorapatite (Eq. 13), which has enhanced resistance to acids.
For that purpose, many toothpastes contain tiny amounts of fluoride. Additionally, fluoride varnish is applied by dentists for further caries prevention.
The availability of metal fluoride nanoparticle sols of MgF2 and CaF2, which are insoluble in crystalline form, offers new fields of application. Recently it was shown that nano-MgF2 and nano-CaF2 possess superior properties in caries protection and remineralisation [118, 119]. Their superiority is caused by their small particle size paired with low solubility. Nanoparticles with a diameter of less than 20 nm can penetrate dentin tubuli, which have a mean diameter of 400 nm, and hence form a fluoride reservoir. This reservoir very slowly demounts over several years as a result of the residual solubility of MgF2 (130 mg L−1) and CaF2 (16 mg L−1), ensuring a very low concentration of free fluoride. This reservoir is stable against water, teeth brushing and similar influences, which does not hold for soluble sodium fluoride, NaF, or commercially applied amine hydrofluorides.
Nevertheless, there is still one weak point in the application of fluorides. Areas covered by a dental filling are not accessible to fluoride, not for soluble fluorides and not for nanoparticles. Thus, prevention of caries under the filling is a very challenging task. A solution to this problem is the use of dental fillings containing fluoride [120, 121]. Briefly, tiny amounts of fluoride are released at the filling–tooth interface, and this fluoride prevents caries formation even in areas under the filling, which are not accessible otherwise. Figure 27a illustrates this principle schematically.
In recent years, composite fillings consisting of polyacrylates and SiO2 nanoparticles became increasingly popular owing to their easy adaption to an irregularly shaped cavity. They are polymerised in vivo from acrylate monomers or oligomers. Thermal polymerisation inside a living tooth is impossible. Thus, transparency of these composites is urgently required for UV polymerisation. The lack of ultrasmall metal fluoride nanoparticles prevented their application on a large scale until now. The fluorolytic sol–gel synthesis opened up new possibilities for such composite materials.
The most evident metal fluoride for application in dental composites is calcium fluoride, CaF2. It is non-toxic and it provides not only fluoride for caries prevention but also calcium ions for remineralisation. As demonstrated in Fig. 27b, transparent acrylate-based composites with 20 wt% CaF2 and even more are available. Owing to their transparency, volume composites are easily manufactured by UV polymerisation.

a Illustration of the slow fluoride release of a CaF2-containing dental filling. b X-ray contrast (60 µS) of composites of different metal fluoride nanoparticles and polyHEMA. SrF2:Bi30 describes an idealised formula Sr0.7Bi0.3F2.3
For future applications, X-ray opacity of these composites is desired as an additional functionality. The contrast of an acrylate polymer in X-ray imaging is virtually zero due to the low atomic numbers of C and O. Although composite fillings with SiO2 have higher X-ray contrast due to the higher atomic number 14 of Si, the contrast is still low compared to teeth containing calcium with an atomic number of 20. X-ray contrast of the composite filling higher than that of the surrounding tooth is desired without loss of the fluoride-releasing properties. Thus, fluorides of heavier elements are more suitable for that purpose. Strontium has an atomic number of 38 and hence transparent composites of strontium fluoride, SrF2, and polyacrylates show a much more intense contrast in X-ray imaging than composites filled with CaF2 (Fig. 27b). There is still one drawback here. To ensure decent X-ray contrast, comparatively high filling grades of 20 wt% SrF2 and more are necessary.
Fluorides of still heavier elements show good X-ray contrast even at low filling grades. Unfortunately, barium (atomic number 56) fluoride, BaF2, is a toxic compound and not suitable for that purpose. Instead, ytterbium (atomic number 70) and bismuth (atomic number 83) compounds are more promising. Ytterbium fluoride already finds application in dental composites, although these composites are not totally transparent [122, 123]. Transparency is achieved by the application of YbF3 nanoparticles from the fluorolytic sol–gel synthesis [124]. Additionally, tuning of the X-ray opacity is possible by the application of Sr1−xYbxF2+x nanoparticles instead of the costly pure YbF3.
Ultimately, the heaviest stable element is bismuth. Therefore, bismuth fluoride BiF3 would in principle generate the best X-ray contrast among all fluorides. Although bismuth(III) ions, Bi3+, are non-toxic, BiF3 indeed is a toxic compound as a result of its water sensitivity. Upon hydrolysis, free HF is formed, which potentially etches the teeth. An elegant workaround is the application of mixed fluorides with a nominal composition of Sr1−xBixF2+x (x = 0.05–0.3). These compounds are stable towards water and therefore non-toxic. Manufacturing of transparent composites is possible, and these composites exhibit a very decent X-ray contrast [125]. For comparison, a composite of 30 wt% SrF2 and polyHEMA has the same X-ray contrast as a composite of 10 wt% Sr0.7Bi0.3F2.3 and polyHEMA. Thus, variation of the Bi content allows tuning of the X-ray opacity of the composites.
Summary
Nanoscaled metal fluorides have gained enormous interest over the past decade owing to their novel properties in several fields of application. Consequently, much effort has been made in developing new synthesis approaches for nano fluoride-based materials. Although several new synthesis routes have been developed recently, they usually have one or more drawbacks. However, the fluorolytic sol–gel synthesis—like the classical hydrolytic sol–gel synthesis of metal oxides—is so far the most powerful approach for nanoscaled metal fluorides. There are several advantages to this approach: (i) easy scale-up even up to ton scale, (ii) resulting directly in the formation of monodispersed nanoparticles based on a simple one-step process, thus giving access to clear transparent metal fluoride sols, and (iii) possible processing of these sols according to already established technologies for classical metal oxide sols. The use of anhydrous hydrogen fluoride is not a drawback since each fluorine atom in any fluorine-containing compound—no matter if inorganic or organic—has to be introduced at the very beginning of any synthesis by HF even if the synthesis could be performed with elemental fluorine. Thus, the handling of hundreds of kilotons of HF is state of the art for chemists specialized in fluorine chemistry in both academia and industry.
The introduction of a second or even a third metal into these new compounds allows for further functionalization, resulting in unlimited new compounds with high impact on applications in catalysis, optics, sensing, up- and downconversion, electrode materials, and many others.
With the fluorolytic synthesis approach an equally powerful access towards nanoscaled metal fluorides is available that now offers an equally general access to nano metal fluorides as the classical sol–gel synthesis allows for nanoscopic metal oxides.
References
Fierro JLG (2006) Metal oxides: chemistry and applications. CRC, Boca Raton
Janiak C, Meyer H-J, Gudat D, Alsfasser R (2012) Riedel—moderne anorganische chemie. Walter de Gryuter GmbH, Berlin
Yuan C, Wu HB, Xie Y, Lou XW (2014) Mixed transition-metal oxides: design, synthesis, and energy-related applications. Angew Chem Int Ed 53(6):1488–1504
Hering E, Schönfelder G (2012) Sensoren in Wissenschaft und Technik (Sensors in science and technnology). Vieweg+Teubner Verlag, Berlin
Göbel H (2005) Einführung in die Halbleiter-Schaltungstechnik (Introduction into semiconductor circuit technology). Springer, Berlin
Ogale SB, Venkatesan TV, Blamire M (2013) Functional metal oxides: new science and novel applications. Wiley-VCH, Weinheim
Tressaud A (2010) Functionalized inorganic fluorides. Wiley, Weinheim
Kemnitz E, Groß U, Rüdiger S (2006) Humboldt-Spektrum
Amatucci GG, Pereira N, Badway F, Sina M, Cosandey F, Ruotolo M, Cao C (2011) Formation of lithium fluoride/metal nanocomposites for energy storage through solid state reduction of metal fluorides. J Fluorine Chem 132(12):1086–1094
Munnangi AR, Fichtner M (2015) In: Nakajima T, Groult H (eds) Advanced fluoride-based materials for energy conversion, 1st edn. Elsevier, Amsterdam
Amatucci GG, Pereira N (2007) Fluoride based electrode materials for advanced energy storage devices. J Fluorine Chem 128(4):243–262
Scopus. (2019) Elsevier
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26(1):62–69
Kipp S, Šepelák V, Becker KD (2005) Mechanochemie: chemie mit dem Hammer (Mechanochemistry: chemistry with the hammer). Chem Unserer Zeit 39(6):384–392
Takacs L (2013) The historical development of mechanochemistry. Chem Soc Rev 42(18):7649–7659
Balaz P (2008) Mechanochemistry in nanoscience and minerals engineering. Springer, Berlin
Takacs L (2018) Two important periods in the history of mechanochemistry. J Mater Sci 53(19):13324–13330
Thiessen KP, Sieber K (1979) Energetische Randbedingungen tribochemischer Prozesse (Energetic boundary conditions of tribochemical processes). Z phys Chemie (Leipzig) 260(1):417
Heinicke G (1984) Tribochemistry. Akademie-Verlag, Berlin
Scholz G (2019) Mechanochemie fluoridischer Festkörper—von der mechanischen Aktivierung zur mechanisch stimulierten Synthese (Mechanochemistry of fluoride based solids—from mechanical activation towards mechanically stimulated synthesis). Leibniz Online Nr. 37
LaMer VK, Dinegar RH (1950) Theory, production and mechanism of formation of monodispersed hydrosols. J Am Chem Soc 72(11):4847–4854
Ostwald W (1900) Über die vermeintliche Isomerie des roten und gelben Quecksilberoxyds und die Oberflächenspannung fester Körper (On supposedly isomery of red phosphorus and yellow mercury oxides and the surface tension of solids). 34U (1):495
Fujihara S, Tada M, Kimura T (2000) Controlling factors for the conversion of trifluoroacetate sols into thin metal fluoride coatings. J Sol-Gel Sci Technol 19(1):311–314
Fujihara S, Kato T, Kimura T (2003) Sol-gel processing and luminescent properties of rare-earth oxyfluoride materials. J Sol-Gel Sci Technol 26(1):953–956
Tada M, Fujihara S, Kimura T (2011) Sol-gel processing and characterization of alkaline earth and rare-earth fluoride thin films. J Mater Res 14(4):1610–1616
Mosiadz M, Juda KL, Hopkins SC, Soloducho J, Glowacki BA (2011) An in-depth in situ IR study of the thermal decomposition of barium trifluoroacetate hydrate. Thermochim Acta 513(1):33–37
Kemnitz E, Noack J (2015) The non-aqueous fluorolytic sol-gel synthesis of nanoscaled metal fluorides. Dalton Trans 44(45):19411–19431
Kemnitz E, Groß U, Rüdiger S, Shekar CS (2003) Amorphous metal fluorides with extraordinary high surface areas. Angew Chem Int Ed 42(35):4251–4254
Rüdiger S, Kemnitz E (2008) The fluorolytic sol-gel route to metal fluorides-a versatile process opening a variety of application fields. Dalton Trans 1117–1127
Broßke D (2015) Sol-Gel-Synthese von nanoskaligen binären und ternären Metallfluoriden (Sol-gel synthesis of nanscale binary and ternary metal fluorides). Brandenburgische Technische Universität Cottbus, Cottbus
Scheurell K, Noack J, König R, Hegmann J, Jahn R, Hofmann T, Löbmann P, Lintner B, Garcia-Juan P, Eicher J, Kemnitz E (2015) Optimisation of a sol-gel synthesis route for the preparation of MgF2 particles for a large scale coating process. Dalton Trans 44:19501–19508
Rehmer A, Scheurell K, Kemnitz E (2015) Formation of nanoscopic CaF2 via a fluorolytic sol-gel process for antireflective coatings. J Mater Chem C 3(8):1716–1723
Schmidt L, Dimitrov A, Kemnitz E (2014) A new approach to prepare nanoscopic rare earth metal fluorides: the fluorolytic sol–gel synthesis of ytterbium fluoride. Chem Commun 50(50):6613–6616
Noack J, Emmerling F, Kirmse H, Kemnitz E (2011) Sols of nanosized magnesium fluoride: formation and stabilisation of nanoparticles. J Mater Chem 21(38):15015–15021
Noack J, Scheurell K, Kemnitz E, Garcia-Juan P, Rau H, Lacroix M, Eicher J, Lintner B, Sontheimer T, Hofmann T, Hegmann J, Jahn R, Lobmann P (2012) MgF2 antireflective coatings by sol-gel processing: film preparation and thermal densification. J Mater Chem 22(35):18535–18541
Schmidt L, Emmerling F, Kirmse H, Kemnitz E (2014) Sol-gel synthesis and characterisation of nanoscopic strontium fluoride. RSC Adv 4(1):32–38
Guo Y, Wuttke S, Vimont A, Daturi M, Lavalley JC, Teinz K, Kemnitz E (2012) Novel sol-gel prepared zinc fluoride: synthesis, characterisation and acid-base sites analysis. J Mater Chem 22(29):14587–14593
Guo Y, Gaczyński P, Becker KD, Kemnitz E (2013) Sol-gel synthesis and characterisation of nanoscopic FeF3–MgF2 heterogeneous catalysts with bi-acidic properties. ChemCatChem 5(8):2223–2232
Krahl T, Broßke D, Scheurell K, Lintner B, Kemnitz E (2016) Novel aspects in the chemistry of the non-aqueous fluorolytic sol-gel synthesis of nanoscaled homodisperse MgF2 sols for antireflective coatings. J Mater Chem C 4(7):1454–1466
Rehmer A, Scheurell K, Scholz G, Kemnitz E (2017) Sol-gel-synthesis of nanoscopic complex metal fluorides. Nanomaterials 7(11):362
Schütz F, Lange L, Scheurell K, Scholz G, Kemnitz E (2018) Synthesis and characterization of perovskite-type [K1−xNax]MgF3 mixed phases via the fluorolytic sol-gel synthesis. Crystals 8(2):66
Schütz F, Bäthge M, Scheurell K, Scholz G, Feist M, Kemnitz E (2018) Development of complex magnesium fluoro aluminates via the fluorolytic sol–gel synthesis. J Mater Chem C 6(1):72–82
Ritter B, Haida P, Fink F, Krahl T, Gawlitza K, Rurack K, Scholz G, Kemnitz E (2017) Novel and easy access to highly luminescent Eu and Tb doped ultra-small CaF2, SrF2 and BaF2 nanoparticles—structure and luminescence. Dalton Trans 46(9):2925–2936
Ritter B, Haida P, Krahl T, Scholz G, Kemnitz E (2017) Core-shell metal fluoride nanoparticles via fluorolytic sol-gel synthesis—a fast and efficient construction kit. J Mater Chem C 5(22):5444–5450
Wagman DD, Evans WH, Parker VB, Schumm RH, Halow I, Bailey SM, Churney KL, Nuttall RL (1982) The NBS tables of chemical thermodynamic properties—selected values for inorganic and C1 and C2 organic sustances in SI units. J Phys Chem Ref Data 11(Suppl. 2)
Karg M, Scholz G, Konig R, Kemnitz E (2012) Mechanistic insight into formation and changes of nanoparticles in MgF2 sols evidenced by liquid and solid state NMR. Dalton Trans 41(8):2360–2366
Ruediger SK, Groß U, Feist M, Prescott HA, Shekar SC, Troyanov SI, Kemnitz E (2005) Non-aqueous synthesis of high surface area aluminium fluoride—a mechanistic investigation. J Mater Chem 15(5):588–597
König R, Scholz G, Kemnitz E (2010) The fluorolytic sol–gel reaction of aluminium alkoxides: a multinuclear MAS NMR study of structural influences of the synthesis parameters. J Sol-Gel Sci Technol 56(2):145–156
Fritz C, Scholz G, Feist M, Kemnitz E (2012) Preparation and stabilization of aluminium trifluoroacetate fluoride sols for optical coatings. Dalton Trans 41(37):11351–11360
Scheurell K, Kemnitz E, Garcia-Juan P, Eicher J, Lintner B, Hegmann J, Jahn R, Hofmann T, Löbmann P (2015) Porous MgF2 antireflective λ/4 films prepared by sol–gel processing: comparison of synthesis approaches. J Sol-Gel Sci Technol 76(1):82–89
Marshall CP (2020) Aluminium fluoride-based catalysts as heterogeneous Lewis/Brønsted solid acids. Humboldt-Universität zu Berlin
Löbmann P (2017) Antireflective coatings by sol–gel processing: commercial products and future perspectives. J Sol-Gel Sci Technol 83(2):291–295
Schubert U, Hüsing N (2019) Synthesis of inorganic matrials. Wiley-VCH, Weinheim
Bogush GH, Zukoski CF (1991) Uniform silica particle precipitation: an aggregative growth model. J Colloid Interface Sci 142(1):19–34
Dimitrov A, Wuttke S, Troyanov S, Kemnitz E (2008) Mg6F2(OMe)10(MeOH)14—an alkoxide fluoride of an alkaline earth metal. Angew Chem Int Ed 47(1):190–192
Konig R, Scholz G, Veiczi M, Jager C, Troyanov SI, Kemnitz E (2011) New crystalline aluminum alkoxide oxide fluorides: evidence of the mechanism of the fluorolytic sol-gel reaction. Dalton Trans 40(34):8701–8710
Dimitrov A, Koch J, Troyanov SI, Kemnitz E (2009) Aluminum alkoxide fluorides involved in the sol-gel synthesis of nanoscopic AlF3. Eur J Inorg Chem 35:5299–5301
Fritz C (2013) Anorganisch-organische Kompositesysteme auf Basis von nanoskopischem Aluminiumfluorid. Humboldt-Universität zu Berlin, Berlin
König R, Scholz G, Kemnitz E (2009) Local structural changes in aluminum isopropoxide fluoride xerogels and solids as a consequence of the progressive fluorination degree. J Phys Chem C 113(16):6426–6438
König R, Scholz G, Pawlik A, Jäger C, van Rossum B, Kemnitz E (2009) Identification of AlFx(OR)y species in strongly disordered aluminum isopropoxide fluoride solids: a field-dependent MAS NMR study. J Phys Chem C 113(35):15576–15585
Pawlik A, König R, Scholz G, Kemnitz E, Brunklaus G, Bertmer M, Jäger C (2009) Access to local structures of HS-AlF3 and its precursor determined by high-resolution solid-state NMR. J Phys Chem C 113(38):16674–16680
Scheurell K, König R, Troyanov SI, Kemnitz E (2012) Structural chemistry of magnesium acetates. Z Anorg Allg Chem 638(9):1265–1273
Scheurell K, Troyanov SI, Kemnitz E (2015) Structural chemistry of basic magnesium acetates, Mg5(μ3-OH)2(OAc)8·nL and Mg3(μ4-O)(OAc)4. Z Anorg Allg Chem 641(6):1106–1109
Krahl T, Vimont A, Eltanany G, Daturi M, Kemnitz E (2007) Determination of the acidity of high surface AlF3 by IR spectroscopy of adsorbed CO probe molecules. J Phys Chem C 111(49):18317–18325
Kemnitz E (2015) Nanoscale metal fluorides: a new class of heterogeneous catalysts. Cat Sci Tech 5(2):786–806
Agirrezabal-Telleria I, Hemmann F, Jäger C, Arias PL, Kemnitz E (2013) Functionalized partially hydroxylated MgF2 as catalysts for the dehydration of d-xylose to furfural. J Catal 305:81–91
Ahrens M, Scholz G, Braun T, Kemnitz E (2013) Catalytic hydrodefluorination of fluoromethanes at room temperature by silylium-ion-like surface species. Angew Chem Int Ed 52(20):5328–5332
Wuttke S, Negoi A, Gheorghe N, Kuncser V, Kemnitz E, Parvulescu V, Coman SM (2012) Sn-doped hydroxylated MgF2 catalysts for the fast and selective saccharification of cellulose to glucose. Chemsuschem 5(9):1708–1711
Negoi A, Teinz K, Kemnitz E, Wuttke S, Parvulescu VI, Coman SM (2012) Bifunctional nanoscopic catalysts for the one-pot synthesis of (±)-menthol from citral. Top Catal 55(7):680–687
Dobrinescu C, Iorgulescu EE, Mihailciuc C, Macovei D, Wuttke S, Kemnitz E, Parvulescu VI, Coman SM (2012) One-pot hydroacetylation of menadione (vitamin K3) to menadiol diacetate (vitamin K4) by heterogeneous catalysis. Adv Synth Catal 354(7):1301–1306
Machynskyy O, Łomot D, Teinz K, Kemnitz E, Karpiński Z (2012) n-Pentane hydroisomerization catalyzed by metals supported on nanoscopic aluminum trifluoride. Catal Commun 26:235–238
Natalia C, Stefan W, Erhard K, Simona MC, Vasile IP (2012) Replacing benzyl chloride with benzyl alcohol in heterogeneous catalytic benzylation of aromatic compounds. Pure Appl Chem 84(3):427–437
Prechtl MHG, Teltewskoi M, Dimitrov A, Kemnitz E, Braun T (2011) Catalytic C–H bond activation at nanoscale lewis acidic aluminium fluorides: H/D exchange reactions at aromatic and aliphatic hydrocarbons. Chem Eur J 17(51):14385–14388
Troncea SB, Wuttke S, Kemnitz E, Coman SM, Parvulescu VI (2011) Hydroxylated magnesium fluorides as environmentally friendly catalysts for glycerol acetylation. Appl Catal B 107(3):260–267
Protesescu L, Tudorache M, Neatu S, Grecu MN, Kemnitz E, Filip P, Parvulescu VI, Coman SM (2011) Unusual behavior of a novel heterogeneous chiral dimer Cr(III)−Salen complex in the epoxidation/epoxide ring-opening reaction of trans-methylcinnamate ester. J Phys Chem C 115(4):1112–1122
Teinz K, Wuttke S, Börno F, Eicher J, Kemnitz E (2011) Highly selective metal fluoride catalysts for the dehydrohalogenation of 3-chloro-1,1,1,3-tetrafluorobutane. J Catal 282(1):175–182
Candu N, Wuttke S, Kemnitz E, Coman SM, Parvulescu VI (2011) Friedel–Crafts alkylations on nanoscopic inorganic fluorides. Appl Catal A 391(1):169–174
Negoi A, Wuttke S, Kemnitz E, Macovei D, Parvulescu VI, Teodorescu CM, Coman SM (2010) One-pot synthesis of menthol catalyzed by a highly diastereoselective Au/MgF2 catalyst. Angew Chem Int Ed 49(44):8134–8138
Coman SM, Parvulescu VI, Wuttke S, Kemnitz E (2010) Synthesis of vitamin K1 and K1-chromanol by Friedel–Crafts alkylation in heterogeneous catalysis. ChemCatChem 2(1):92–97
Wuttke S, Coman SM, Kröhnert J, Jentoft FC, Kemnitz E (2010) Sol–gel prepared nanoscopic metal fluorides—a new class of tunable acid–base catalysts. Catal Today 152(1):2–10
Coman SM, Patil P, Wuttke S, Kemnitz E (2009) Cyclisation of citronellal over heterogeneous inorganic fluorides—highly chemo- and diastereoselective catalysts for (±)-isopulegol. Chem Commun 4:460–462
Kemnitz E, Wuttke S, Coman SM (2011) Tailor-made MgF2-based catalysts by sol-gel synthesis. Eur J Inorg Chem 31:4773–4794
Krahl T, Kemnitz E (2017) Aluminium fluoride—the strongest solid Lewis acid: structure and reactivity. Cat Sci Technol 7(4):773–796
Wuttke S, Scholz G, Rüdiger S, Kemnitz E (2007) Variation of sol–gel synthesis parameters and their consequence for the surface area and structure of magnesium fluoride. J Mater Chem 17(47):4980–4988
Wuttke S, Vimont A, Lavalley J-C, Daturi M, Kemnitz E (2010) Infrared investigation of the acid and basic properties of a sol–gel prepared MgF2. J Phys Chem C 114(11):5113–5120
Roberts JE (1961) Lanthanum and neodymium salts of trifluoroacetic acid. J Am Chem Soc 83(5):1087–1088
Rillings KW, Roberts JE (1974) A thermal study of the trifluoroacetates and pentafluoropropionates of praseodymium, samarium and erbium. Thermochim Acta 10(3):269–277
Scholz G, Heidemann D, Kemnitz E (2013) Local structure of nanoscopic magnesium hydroxide fluorides studied by natural abundance 25Mg solid state NMR spectroscopy. Z Anorg Allg Chem 639(5):694–701
Wuttke S, Coman SM, Scholz G, Kirmse H, Vimont A, Daturi M, Schroeder SLM, Kemnitz E (2008) Novel sol-gel synthesis of acidic MgF2−x(OH)x materials. Chem Eur J 14(36):11488–11499
Korth Kristalle GmbH (2020). https://www.korth.de/index.php/material.html
Glaubitt W, Löbmann P (2012) Antireflective coatings prepared by sol–gel processing: principles and applications. J Eur Ceram Soc 32(11):2995–2999
Martinů L, Biederman H, Holland L (1985) Thin films prepared by sputtering MgF2 in an rf planar magnetron. Vacuum 35(12):531–535
Jacob D, Peiró F, Quesnel E, Ristau D (2000) Microstructure and composition of MgF2 optical coatings grown on Si substrate by PVD and IBS processes. Thin Solid Films 360(1–2):133–138
Haase M, Schäfer H (2011) Upconverting nanoparticles. Angew Chem Int Ed 50(26):5808–5829
Tu D, Liu Y, Zhu H, Chen X (2013) Optical/magnetic multimodal bioprobes based on lanthanide-doped inorganic nanocrystals. Chem Eur J 19(18):5516–5527
Gai S, Li C, Yang P, Lin J (2014) Recent progress in rare earth micro/nanocrystals: soft chemical synthesis, luminescent properties, and biomedical applications. Chem Rev 114(4):2343–2389
Zhou J, Liu Q, Feng W, Sun Y, Li F (2015) Upconversion luminescent materials: advances and applications. Chem Rev 115(1):395–465
Sobolev BP, Seiranian KB, Garashina LS, Fedorov PP (1979) Phase diagrams of the SrF2-(Y, Ln)F3 systems part I—X-ray characteristics of phases. J Solid State Chem 28(1):51–58
Krahl T, Beer F, Relling A, Gawlitza K, Rurack K, Kemnitz E (2020) Toward luminescent composites by phase transfer of SrF2:Eu3+ nanoparticles capped with hydrophobic antenna ligands. ChemNanoMat 6(7):1086–1095
Côté WA (1968) Biological deterioration of wood. In: Principles of wood science and technology. Springer, Berlin
Arantes V, Goodell B (2014) Current understanding of brown-rot fungal biodegradation mechanisms: a review. In: Deterioration and protection of sustainable biomaterials, vol 1158. ACS Symposium Series, vol 1158. American Chemical Society, pp 3–21
Schwarze F (2007) Wood decay under the microscope. Fungal Biol Rev 21:133–170
Pařil P, Baar J, Čermák P, Rademacher P, Prucek R, Sivera M, Panáček A (2017) Antifungal effects of copper and silver nanoparticles against white and brown-rot fungi. J Mater Sci 52(5):2720–2729
Roark RC (1926) Fluorides vs. fluosilicates as insecticides. Science 63(1634):431–432
Becker J (1973) Fluorine compounds for wood preservation. J Inst Wood Sci 6:51–62
Reinprecht L (2010) Fungicides for wood protection—world viewpoint and evaluation/testing in Slovakia. In: Odile C (ed) Fungicides. Intech, Rijeka, pp 95–122
Usmani SM, Stephan I, Hübert T, Kemnitz E (2018) Nano metal fluorides for wood protection against fungi. ACS Appl Nano Mater 1(4):1444–1449
EN 113. Wood preservatives test method for determining the protective effectiveness against wood destroying basidiomycetes determination of the toxic values (1997). European Committee for Standardization, Brussels
Lide DR (2003) CRC handbook of chemistry and physics: a ready-reference of chemical and physical data. CRC, Boca Raton
Gascón-Garrido P, Oliver-Villanueva JV, Ibiza-Palacios MS, Militz H, Mai C, Adamopoulos S (2013) Resistance of wood modified with different technologies against Mediterranean termites (Reticulitermes spp.). Int Biodeterior Biodegrad 82:13–16
Pan C, Ruan G, Chen H, Zhang D (2015) Toxicity of sodium fluoride to subterranean termites and leachability as a wood preservative. Eur J Wood Wood Prod 73(1):97–102
Pan C, Wang C (2015) Sodium fluoride for protection of wood against field populations of subterranean termites. J Econ Entomol 108(4):2121–2124
Usmani SM, Plarre R, Hübert T, Kemnitz E (2020) Termite resistance of pine wood treated with nano metal fluorides. Eur J Wood Wood Prod 78:493–499. https://doi.org/10.1007/s00107-020-01522-z
Noack J, Schmidt L, Glasel H-J, Bauer M, Kemnitz E (2011) Inorganic-organic nanocomposites based on sol-gel derived magnesium fluoride. Nanoscale 3(11):4774–4779
Mahn S, Kemnitz E (2019) Modification of low-molecular polylactic acid by CaF2 nanoparticles: a new approach to change its material properties. J Appl Polym Sci 136(33):47875
Noack J, Fritz C, Flugel C, Hemmann F, Glasel H-J, Kahle O, Dreyer C, Bauer M, Kemnitz E (2013) Metal fluoride-based transparent nanocomposites with low refractive indices. Dalton Trans 42(16):5706–5710
Knutsen HK, Alexander J, Barregård L, Bignami M, Brüschweiler B, Ceccatelli S, Cottrill B, Dinovi M, Edler L, Grasl-Kraupp B, Hogstrand C, Hoogenboom L, Nebbia CS, Oswald IP, Petersen A, Rose M, Roudot A-C, Vleminckx C, Vollmer G, Wallace H, Bodin L, Cravedi J-P, Halldorsson TI, Haug LS, Johansson N, van Loveren H, Gergelova P, Mackay K, Levorato S, van Manen M, Schwerdtle T (2018) Risk to human health related to the presence of perfluorooctane sulfonic acid and perfluorooctanoic acid in food. EFSA J 16(12):e05194
Eshed M, Lellouche J, Banin E, Gedanken A (2013) MgF2 nanoparticle-coated teeth inhibit Streptococcus mutans biofilm formation on a tooth model. J Mater Chem B 1(32):3985–3991
Zirk M, Schievelkamp S, Kemnitz E, Lausch J, Wierichs RJ, Esteves-Oliveira M, Meyer-Lueckel H (2019) Evaluation of novel nanoscaled metal fluorides on their ability to remineralize enamel caries lesions. Sci Rep 9(1):1942
Hicks J, Garcia-Godoy F, Donly K, Flaitz C (2003) Fluoride-releasing restorative materials and secondary caries. J Calif Dent Assoc 31(3):229–245
Wiegand A, Buchalla W, Attin T (2007) Review on fluoride-releasing restorative materials—fluoride release and uptake characteristics, antibacterial activity and influence on caries formation. Dent Mater 23(3):343–362
Collares FM, Ogliari FA, Lima GS, Fontanella VRC, Piva E, Samuel SMW (2010) Ytterbium trifluoride as a radiopaque agent for dental cements. Int Endod J 43(9):792–797
Carreño N, Oliveira T, Piva E, Leal F, Moncks M, Raubach C, Ogliari F (2012) YbF3/SiO2 fillers as radiopacifiers in a dental adhesive resin. Nano-Micro Lett 4:189–196
Schmidt L, Mahn S, Kemnitz E (2017) Sol–gel synthesis of Sr1−xYbxF2+x nanoparticles dispersible in acrylates. RSC Adv 7(89):56266–56270
Mahn S (2020) PhD Thesis. Humboldt-Universität zu Berlin
Acknowledgments
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kemnitz, E., Mahn, S. & Krahl, T. Nano metal fluorides: small particles with great properties. ChemTexts 6, 19 (2020). https://doi.org/10.1007/s40828-020-00115-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40828-020-00115-w