
Highlights
-
A hybrid co-catalysts system comprising Au nanoparticles (NPs) and PtSAs with different functions was constructed on the polymeric carbon nitride (PCN) surface by double-solvent method.
-
For the dual co-catalysts, the Au NPs absorb relatively long-wavelength light to produce plasmonic hot-electrons, and the adjacent Pt single atoms (PtSAs) can trap the plasmonic hot-electrons effectively for H2 evolution.
-
The PtSAs–Au2.5/PCN exhibits excellent broad-spectrum photocatalytic H2 evolution activity with the H2 evolution rate of 8.8 mmol g−1 h−1 at 420 nm and 264 μmol g−1 h−1 at 550 nm, respectively.
Abstract
Rationally designing broad-spectrum photocatalysts to harvest whole visible-light region photons and enhance solar energy conversion is a “holy grail” for researchers, but is still a challenging issue. Herein, based on the common polymeric carbon nitride (PCN), a hybrid co-catalysts system comprising plasmonic Au nanoparticles (NPs) and atomically dispersed Pt single atoms (PtSAs) with different functions was constructed to address this challenge. For the dual co-catalysts decorated PCN (PtSAs–Au2.5/PCN), the PCN is photoexcited to generate electrons under UV and short-wavelength visible light, and the synergetic Au NPs and PtSAs not only accelerate charge separation and transfer though Schottky junctions and metal-support bond but also act as the co-catalysts for H2 evolution. Furthermore, the Au NPs absorb long-wavelength visible light owing to its localized surface plasmon resonance, and the adjacent PtSAs trap the plasmonic hot-electrons for H2 evolution via direct electron transfer effect. Consequently, the PtSAs–Au2.5/PCN exhibits excellent broad-spectrum photocatalytic H2 evolution activity with the H2 evolution rate of 8.8 mmol g−1 h−1 at 420 nm and 264 μmol g−1 h−1 at 550 nm, much higher than that of Au2.5/PCN and PtSAs–PCN, respectively. This work provides a new strategy to design broad-spectrum photocatalysts for energy conversion reaction.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Photocatalytic water splitting to H2 has been regarded as a promising technology to convert renewable solar energy and address the global energy crisis [1,2,3,4,5,6]. Among various semiconductor photocatalysts, the polymeric carbon nitride (PCN) has attracted tremendous attention since its pioneering study in 2009 [7], owing to the nontoxicity, excellent chemical stability and facile preparation from abundant precursors [8,9,10]. However, the photocatalytic efficiency for pristine PCN is still too low because of the limited visible-light absorption and serious recombination of photogenerated electrons and holes [11]. To expand the visible-light harvest ability for full utilization of solar energy, a variety of strategies have been adopted to engineer the bandgap structure via elevating valance band (VB) and/or reducing the conductor band (CB) edge positions, including elemental doping [12,13,14], defect constructing [15,16,17] and crystallizing [18,19,20]. Unfortunately, the narrow-bandgap PCN with higher VB and/or lower CB edges generally shows lower redox abilities in compared with pristine PCN [21]. Hence, the irreconcilable relationship between extending intrinsic absorption edges and keeping redox ability of PCN is still a very hard nut to crack through simple bandgap engineering.
Recently, based on the localized surface plasmon resonance (LSPR) of metal nanoparticles (NPs, e.g., Au and Ag), integration of Au NPs into PCN has been widely explored to enable light absorption region up to the near infrared (NIR) without sacrificing its CB/VB redox ability [22,23,24,25,26]. As demonstrated by previous reports, the plasmonic Au NPs can be excited under specific light illumination to generate hot-electrons, which can jump over the Schottky barrier and flow into the CB of PCN when composing hybrid structures between Au NPs and PCN [22, 27]. Through tuning the shape, size and composition of Au NPs, the LSPR intensity and resonant photon wavelength of plasmonic metal can be engineered to harvest entire solar light [25]. For instance, the Au nanorods are assembled on PCN nanosheets to achieve near-infrared photocatalytic H2 evolution activity (63.1 μmol g−1 h−1) [28]. On the other hand, the Au NPs can also act as co-catalysts to trap photoelectrons from PCN via the Au/PCN Schottky interface [21, 29]. But it is a pity that the Au nanocrystals usually possess large size due to the nanostructure design and is inactive for H2 evolution reaction, resulting in poor photocatalytic efficiencies under the intrinsic absorption of PCN [28,29,30]. Furthermore, the photocatalytic activity of Au/PCN hybrid photocatalysts offered by the plasmonic effect is still mediocre due to the short diffusion length of hot-electrons and little active sites in PCN surface for H2 evolution reaction [27]. Hence, it is highly desirable as well as challenging to design plasmonic metal/PCN systems for achieving super photocatalytic activity under both short- and long-wavelength light.
In this work, we report the synergetic plasmonic Au NPs and atomically dispersed Pt single atoms (PtSAs) on PCN surface (PtSAs–Au2.5/PCN) to achieve broad-spectrum photocatalytic H2 evolution. In our design, well-dispersed Au NPs with a small size of about 4 nm were firstly anchored on PCN (Au2.5/PCN), in which tuning the particle size and metal–support interaction greatly improve the H2 evolution activity of Au NPs when served as the co-catalysts. The photocatalytic H2 evolution rate reaches 5.2 mmol g−1 h−1 at 420 nm for Au2.5/PCN. Besides, the Au NPs exhibit strong LSPR effect with broad absorption band from 500 to 800 nm. Subsequently, the PtSAs were incorporated into the Au2.5/PCN hybrid to further accelerate charge transfer and photoelectron capture, especially for plasmonic hot-electrons, which elevates the photocatalytic H2 evolution performance driven by LSPR effect over threefold. As a result, as-obtained PtSAs–Au2.5/PCN exhibits excellent photocatalytic H2 evolution activity under whole visible spectral region. The H2 evolution rate reach 8.8 mmol g−1 h−1 at 420 nm and 264 μmol g−1 h−1 at 550 nm, much higher than that of Au/PCN and PtSAs/PCN, respectively. This work provides a strategy to couple wide-bandgap semiconductor with plasmonic metal for broad-spectrum photocatalysis.
2 Experimental Section
2.1 Materials
Gold acid chloride trihydrate (HAuCl4∙3H2O, 99%), chloroplatinic acid (H2PtCl6∙6H2O, 99%), Potassium tetrachloroplatinate (K2PtCl4, 99%), n-hexane (AR) and triethanolamine (TEOA, AR) were all purchased from Aladdin and used without further purification. Urea was purchased from Tianli Chemical Reagent Co., Ltd. Deionized water was used for all the experiment.
2.2 Synthesis of PCN, Aux/PCN and PtSAs–Au2.5/PCN
2.2.1 Synthesis of PCN
Briefly, the covered crucible with 15 g urea was placed into a muffle furnace and then heated at 550 °C for 4 h with a heating rate of 10 °C min−1 under a stable air atmosphere. After cooling down to room temperature, the resultant yellow products were milled into powder in a mortar, named as PCN.
2.2.2 Synthesis of Au x /PCN
Aux/PCN was prepared by double-solvent method. Typically, 48.75 mg PCN was suspended in 15 mL n-hexane with vigorous stirring. 0.2 mL HAuCl4∙3H2O solution (12.5 mg mL−1) was added into the above solution with constant stirring. After consistently stirring at room temperature for 2 h, the resulting solution was then sonicated for another 1 h. Later, the resultant HAuCl4/PCN slurry sank down into the bottom of the beaker. Subsequently, the HAuCl4/PCN slurry was dried at 50 °C after decanting the supernatant and then milled into powder in a mortar. Finally, the powder was placed into a tube furnace and heated at 450 °C for 2 h under H2/Ar atmosphere to yield Aux/PCN. Aux/PCN hybrids with different loading of Au NPs were prepared by changing the amount of PCN and HAuCl4∙3H2O with controlled mass values (PCN/HAuCl4∙3H2O: 49.5/1, 48.75/2.5 and 47.5/5 mg), respectively. The products were therefore denoted as Aux/PCN (x = 1, 2.5 and 5, and corresponds to the wt% of Au NPs). For comparison, the Au2.5-PCN and Pt-PCN were prepared by traditional photo-deposition method. Typically, 48.75 mg PCN and 2.5 mg HAuCl4∙3H2O were dispersed in 15 mL H2O with ultrasonic blending for 30 min. Then, the suspension was evacuated to remove dissolved air completely and irradiated by a 300 W Xe lamp for 1 h to reduce the Au cations. After centrifugal separation, the obtained powder was named as Au2.5-PCN. The Pt-PCN was prepared by similar method with only changing the precursor.
2.2.3 Synthesis of PtSAs–Au 2.5 /PCN
The PtSAs–Au2.5/PCN was prepared with similar to that Au2.5/PCN. Typically, 0.2 mL K2PtCl4 solution (5.3 mg mL−1) was added into the Au2.5/PCN suspension (49.5 mg Au2.5/PCN in 15 mL n-hexane) with vigorous stirring. After consistently stirring for 2 h, the resulting suspension was then sonicated for another 1 h. Subsequently, the K2PtCl4-Au2.5/PCN slurry was obtained by decanting the supernatant. After drying at 50 °C overnight, the K2PtCl4-Au2.5/PCN intermediate was milled into powder in a mortar. Finally, the powder was placed into a tube furnace and heated at 150 °C for 2 h under H2/Ar atmosphere to yield PtSAs–Au2.5/PCN. In addition, with changing the Au2.5/PCN as PCN, the PtSAs–PCN was successfully prepared.
2.3 Photocatalytic H2 Evolution Tests
The photocatalytic H2 evolution experiments were performed using an on-line photocatalytic analysis system (Labsolar IIIAG, Beijing Perfectlight) with a quartz reactor. Ten milligram photocatalysts were dispersed in 100-mL TEOA (20%) aqueous solution. Then, the suspension was evacuated several times to completely remove dissolved air and then irradiated by a 300 W Xe lamp with various filters, such as cutoff filter (λ ≥ 420 nm) and band-pass filters (420, 450, 550, 600 and 650 nm). The amounts of H2 were collected and analyzed every hour by the gas chromatography equipped with a thermal conductive detector (TCD).
3 Results and Discussion
3.1 Synthesis and Characterizations of Aux/PCN
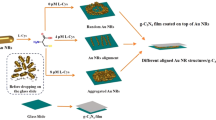
The overall synthesis procedure of Aux/PCN with different Au loading amounts is illustrated in Fig. 1a. Typically, the HAuCl4/PCN intermediates were prepared by a double-solvent method with a subsequent heating treatment at 450 °C to obtain Aux/PCN (x = 1, 2.5 and 5, which represents the loading amount of Au NPs). As shown in Figs. 1b and S1, the transmission electron microscopy (TEM) images of Aux/PCN reveal that the Au NPs are distributed on PCN surface with the average size of about 4 nm (Fig. S2). Clear lattice fringes for select Au NPs in Au2.5/PCN sample (Fig. 1b) are observed in the high-resolution TEM images (Fig. 1c, d), and the lattice distance is calculated to be 0.228/0.229 nm (Fig. 1e, f), corresponding to the (111) planes of fcc structured Au. For comparison, 2.5 wt% of Au NPs were also loaded on PCN by universal photodeposition method (Fig. S3a, named as Au2.5-PCN), in which the average diameter of Au NPs is 13 nm (Fig. S3b), much larger than that of Au2.5/PCN. The size discrepancy of Au NPs in Au2.5/PCN and Au2.5-PCN is further demonstrated by the X-ray diffraction (XRD) patterns, in which significantly reduced full width at half maximum of (111) and (200) diffraction peaks can be found in Au2.5-PCN (Fig. 1g). Besides, the other diffraction peaks at 12.9° and 27.6° in theses samples can be assigned to (100) and (002) crystal planes of PCN, which corresponds to the repeating in-plane packing motif of heptazine units and interlayer stacking of aromatic segments, respectively [31]. This demonstrates that the loading of Au NPs does not destroy the crystalline structure of PCN. Moreover, in the Fourier transform infrared (FTIR) spectroscopy (Fig. 1h), similar characteristic vibrations at 810 cm−1, 1100 to 1700 cm−1 and 3100 to 3300 cm−1 exist in these samples, assigned to vibration of tri-s-triazine units, stretching vibration modes of C-N heterocycles as well as O–H and N–H stretching vibration [32], also suggesting that there is no variation of tri-s-triazine molecular skeleton for PCN.

a Synthesis and structural characterizations of Aux/PCN photocatalysts. a Illustration of the preparation process. b TEM image of Au2.5/PCN. c, d High-resolution TEM images of Au2.5/PCN for the marked rectangular areas in b and e and f the corresponding Au intensity profile. g XRD patterns and h FTIR spectrums and high-resolution XPS spectra: i Au 4f, j N 1s for Au2.5/PCN and reference samples
The X-ray photoelectron spectroscopy (XPS) was performed to investigate the surface compositions and chemical states of PCN and Au NPs-modified PCN samples. As displayed in Fig. S4a, the survey spectra of Au2.5/PCN and Au2.5-PCN show that a new peak appears at around 84 eV correspond to Au 4f signals, which indicates the successful deposition of Au NPs on the PCN surface. For the Au element, compared to those of Au2.5-PCN, the Au 4f peaks of Au2.5/PCN show apparent shift toward lower energy positions by ca. 0.5 eV (Fig. 1i), indicating the increased electron density of Au NPs in Au2.5/PCN sample [33]. The enhanced electron transfer from PCN to Au NPs in Au2.5/PCN confirms a strong electronic metal–support interaction (EMSI) between PCN and Au NPs [34], which is benefit to photogenerated electron transfer at Au-PCN Schottky interface. While for C and N signals, the alteration of peak position is negligible between Au2.5/PCN and reference samples, probably due to the much larger mass content of PCN than that of Au NPs. Besides, the characteristic peaks for triazine units are still overwhelming majority after the loading of Au NPs (Figs. S4b and 1j), including N–C = N sp2 carbon (ca. 288 eV), C–N = C sp2 nitrogen (ca. 398.4 eV) and N-(C)3 sp3 nitrogen (ca. 399 eV) [35], which is in consistent with the above XRD and FTIR results.
3.2 Photocatalytic Properties of Aux/PCN
The ultraviolet–visible diffuse reflectance (UV–vis DR) spectra of these as-prepared samples were acquired to characterize its optical properties (Fig. 2a). As expected, the Au-containing samples show similar absorption feature with pure PCN under UV and short-wavelength visible light, and the absorption edges are around 460 nm without obvious shift, corresponding to the unaltered bandgap energy for PCN (about 2.69 eV) [30]. Compared with pure PCN, an extra broad absorption band appears in the enlarged absorption spectrum of Au-containing samples with a range of 500–800 nm, which originates from the LSPR effect of Au NPs [36,37,38]. Hence, the incorporation of Au NPs could improve the overall light absorption of Au2.5/PCN, especially in the long-wavelength visible light. In addition, the LSPR absorption of Au2.5/PCN with slightly blue-shift is much stronger than that of Au2.5-PCN, which is mainly attributed to the different size and surrounding electric environment of Au NPs [21]. Photochemical tests were carried out to investigate the charge transfer behaviors in Au2.5/PCN, including transient photocurrent response (TPR) and electrochemical impedance (EIS) spectra (Fig. 2b, c). Compared to PCN and Au2.5-PCN, the Au2.5/PCN exhibits highest photocurrent density and smallest impedance arc radius, meaning significantly enhanced electron transfer kinetics in Au2.5/PCN. This highlights the superiority of strong EMSI at Au-PCN interface for charge mobility.

Optics and photocatalytic properties of Aux/PCN photocatalysts. a UV–vis DR spectra of PCN, Au2.5-PCN and Au2.5/PCN. b TPR density and c EIS Nyquist plots of PCN and Au2.5/PCN. d Time-dependent course of H2 evolution and e comparison of the photocatalytic H2 evolution rate for Au2.5/PCN and reference samples. f Photocatalytic H2 evolution under various wavelength for Au2.5/PCN. g The photocatalytic mechanism Au2.5/PCN under different visible-light regions
The photocatalytic water splitting to H2 over Au2.5/PCN and reference samples was evaluated under visible-light irradiation with TEOA as sacrificial reagent via an on-line photocatalytic analysis system (Labsolar IIIAG, Beijing Perfectlight). As shown in Fig. 2d, the pure PCN shows mediocre photocatalytic H2 evolution activity with the rate of only 35 μmol g−1 h−1 (Fig. 2e) under visible light, and its H2 evolution rate increases to 1.36 mmol g−1 h−1 with the loading of Au NPs on PCN surface as co-catalysts via in situ photodeposition way, probably due to the increase of activity sites for H2 evolution as well as enhanced photogenerated carrier separation. Moreover, the photocatalytic H2 evolution activity is further elevated for Aux/PCN with the regulation of Au NPs size and Au-PCN interface, and the highest H2 evolution rate reaches up to 10.01 mmol g−1 h−1 after optimizing the Au loading amount, approximately 7.4 and 17.5 times higher than that of Au2.5-PCN and Pt-PCN.
To further investigate the fundamental role of Au NP in boosting photocatalytic activity of PCN, H2 evolution measurements were conducted under irradiation of different individual wavelength. As shown in Fig. 2f, the Au2.5/PCN displays excellent photocatalytic H2 evolution activity at the intrinsic absorption of PCN (about 400–450 nm). The H2 evolution rate achieves 5.2 mmol g−1 h−1 at 420 nm, suggesting that Au NPs indeed acts as the co-catalysts to trap photogenerated electrons for H2 evolution reaction (Fig. 2g) [29]. In the photocatalytic process, the strong EMSI can accelerate the electron transfer through Au-PCN Schottky interface and the Au NPs with small size can provide abundant active sites for H2 evolution. On the other hand, under the long-wavelength light irradiation (about 500–780 nm), the Au2.5/PCN also exhibits photocatalytic H2 evolution activity, which originates from the LSPR effect of Au NPs, that is, the plasmonic hot-electrons jump over the Schottky barrier and inject into the CB of PCN for H2 evolution (Fig. 2g) [22, 29]. The reproducible photocurrent at 550 nm light irradiation (Fig. S5) further confirms the transfer process of hot-electrons. However, the photocatalytic efficiency purely offered by plasmonic hot electrons is quite poor with the H2 evolution rate of only 64 μmol g−1 h−1 at 550 nm. The major reason for this situation is the short diffusion length of hot-electrons and the lack of active sites near the Au NPs to trap these electrons for H2 evolution.
3.3 Synthesis and Characterizations of PtSAs–Au2.5/PCN
Based on the above analysis, to further improve the photocatalytic activity, the core concept is to incorporate new electron acceptors into PCN plane near the Au NPs for efficient plasmonic hot-electron transfer and capture [39]. Metal SAs with sub-nanometer size have been widely designed as the photocatalytic H2 evolution sites to trap photoexcited electrons from PCN [34, 40,41,42]. Based on its unique size effect, engineering the metal SAs adjacent to Au NPs with several nanometer distance may be an effective way for plasmonic hot-electron capture. As a proof-of-concept demonstration, starting from the Au2.5/PCN sample, the PtSAs are further deposited on PCN surface with similar synthetic process to that of Au2.5/PCN (Fig. 3a, denoted as PtSAs–Au2.5/PCN). As presented in Fig. 3b, the Au NPs are still well dispersed on the surface of PCN with the average size of 4.1 nm (Fig. S6), slightly larger than that of Au2.5/PCN. The high-resolution TEM and HAADF-TEM images show clear lattice fringes with a spacing of 0.23/0.229 nm, which could be attributed to the fcc Au (111) plane (Fig. 3c, e). Moreover, there are numerous bright dots around Au NPs with several nanometer distances (Fig. 3d and Fig. S7), corresponding to the atomically dispersed PtSAs. The elemental distribution is investigated by the energy-dispersive spectroscopy (EDS) elemental mappings and line-scan EDS analysis (Fig. 3e-j). The Au element is contained within the nanocrystalline, while the Pt element distributes across the whole PCN region, especially in the surrounding of Au NPs. The results demonstrate that partial PtSAs are indeed formed near the Au NPs in PtSAs–Au2.5/PCN. For comparison, the PtSAs were also anchored on the surface of PCN (named as PtSAs–PCN, Fig. S8).

Synthesis and structural characterizations of PtSAs–Au2.5/PCN photocatalysts. a Illustration of the preparation process. b TEM and high-resolution TEM (insertion) images of PtSAs-Au2.5/PCN. c HAADF-STEM, d enlarged HAADF-STEM images, e–g EDS elemental mappings and h, i line-scan EDS analysis across a single Au NPs for PtSAs–Au2.5/PCN. j XRD patterns and high-resolution XPS spectra of k Au 4f, i Pt 4f for PtSAs-Au2.5/PCN and reference samples
The crystal structures of as-prepared samples were investigated by powder XRD. As presented in Fig. 3k, the characteristic peaks of PCN at 12.9° and 27.6° exist in these samples without obvious variation, meaning no destruction of crystalline structure for PCN [43, 44], which is in consistent the FTIR results (Fig. S9). Besides, there are other two distinct diffraction peaks at about 38.2° and 44.4° in the XRD patterns of Au2.5/PCN and PtSAs–Au2.5/PCN, indexed to the typical (111) and (200) planes of fcc structured Au. And no peak shift can be found between Au2.5/PCN and PtSAs–Au2.5/PCN, excluding the existence of AuPt alloying during the deposition of PtSAs. The chemical state of surface atoms was analyzed by the XPS (Figs. 3l, m and S10, S11). Obviously, only Ptδ+ peaks (72.8 eV, Pt 4f) are observed for PtSAs–Au2.5/PCN and PtSAs-PCN (Fig. 3k), again excluding the formation of Pt or AuPt NPs in PtSAs–Au2.5/PCN sample. As for the Au 4f signals in Fig. 3l, the Au 4f7/2 peaks at about 83.5 eV for Au2.5/PCN and PtSAs-Au2.5/PCN can be assigned to metallic Au0 species. Compared to that of Au2.5/PCN, Au 4f7/2 peak of PtSAs–Au2.5/PCN shows a negative shift by ca. 0.25 eV, confirming more electron transfer from PCN to Au NPs after the deposition of PtSAs [32]. This strongly indicate an enhanced EMSI between PCN and Au NPs. While for the C 1s and N 1s signals, the peak shift is virtually invisible between Au2.5/PCN and PtSAs–Au2.5/PCN, probably originating from a fairly low mass content of Au NPs in PtSAs–Au2.5/PCN.
To further investigate the valence state and local atomic structure of Pt sites in PtSAs–Au2.5/PCN, the X-ray absorption spectroscopy (XAS) measurements were taken to acquire the X-ray absorption near-edge structure (XANES) and extended X-ray fine structure (EXAFS) spectroscopy. XANES results in Fig. 4a show that the Pt L3-edge white line intensity of PtSAs–Au2.5/PCN is higher than that of PtO2, meaning high oxidation state of Pt sites, which is consistent with the high-resolution XPS spectra Pt 4f. The Fourier transformed (FT) k3-weighted EXAFS spectra of PtSAs–Au2.5/PCN at R space (Fig. 4b) reveals a sharp peak at near 1.6 Å, which can be attributed to the Pt–N/O scattering, testifying the atomically dispersed PtSAs on PCNS surface. Moreover, the Morlet wavelet transforms (WT) of Pt L3-edge EXAFS spectra further confirm the existence of Pt sites in PtSAs–Au2.5/PCN (Fig. 4d-f), which has the maximum WT intensity at about R = 1.6 Å, k = 7.1 Å−1 with compared to the Pt–Pt bond in Pt foil (R = 2.6 Å, k = 13.1 Å−1) [32, 34]. The EXAFS fitting spectrum of Pt foil and PtSAs–Au2.5/PCN was performed to probe quantitative atomic structure of Pt sites as shown in Figs. 4c and S12, S13. The obtained R-space fitting curves of PtSAs–Au2.5/PCN matches well with the experimental data at the range from 1.0 to 2.4 Å, demonstrating the formation of Pt-N6 bonds (Table S1). Hence, the above results provide a quantitative illustration for the existence of PtSAs near Au NPs.

Surface properties of PtSAs-Au2.5/PCN photocatalysts. a Pt L3-edge XANES spectra and the corresponding FT k3-weighted spectra at b R space. c Pt EXAFS fitting curves of PtSAs–Au2.5/PCN at R space. WT of d Pt foil, e PtO2 and f PtSAs–Au2.5/PCN. Difference charge density analysis and two-dimensional average difference charge density diagrams (insertion) for g Au/PCN and f PtSAs-Au/PCN model, yellow and cyan represent electron accumulation and depletion, respectively. i, j The calculated projected density of states (PDOS) for PtSAs-PCN, Au/PCN and PtSAs-Au/PCN model
To identify the influence of PtSAs on the electronic structure of PtSAs–Au2.5/PCN, density functional theory (DFT) calculations were performed on the basis of the above experimental results. The differential charge density diagrams reveal that the electron migration occurs from PCN to Au NPs through the intimate interface for Au/PCN (Figs. 4g and S14a), and the obviously enhanced electron transfer can be observed at the Au-PCN interface with the introduction of PtSAs (Figs. 4h andS14b) [45]. Moreover, the two-dimensional average difference charge density diagrams (insertion of Fig. 4g, h) clearly show that the maximum value of ∆ρ increases from − 0.17 e Å−1 for Au/PCN to − 0.3 e Å−1 for PtSAs–Au2.5/PCN, meaning that the Au NPs acquire more electrons from PCN in PtSAs–Au2.5/PCN. This further confirms the strong electronic metal–support interaction between Au NPs and PCN. On the other hand, in the calculated PDOS profiles (Fig. 4i, j), the decoration of PtSAs into the triangular sub-nanometer cavity near Au NPs greatly varies the energy levels and spatial distributions of Pt 5d and Au 5d orbitals as compared with that of isolated PtSAs or Au NPs, making the Au 5d DOS peaks slightly shift toward the Fermi level, which probably originates from the electron movement between PtSAs/Au NPs and PCN substrate. This not only accelerates the photogenerated charge transfer but also activate the Au NPs for H2 evolution reaction.
3.4 Photocatalytic Properties of PtSAs–Au2.5/PCN
Upon the successful deposition of PtSAs, we set out to explore that whether the PtSAs can facilitate plasmonic hot-electron transfer and improve photocatalytic H2 evolution. Firstly, the optical response properties of samples were characterized by the UV–vis DR spectra (Fig. 5a), the PtSAs–Au2.5/PCN shows similar light absorption to that of Au2.5/PCN with clear plasmonic peak at around 550 nm, which indicates no obvious variation of the LSPR effect of Au NPs after the introduction of PtSAs. Subsequently, to look into the role of PtSAs, the H2 evolution and TPR measurements of PtSAs–Au2.5/PCN and reference samples were conducted under different wavelength light irradiation as shown in Fig. 5b-e. Under the intrinsic absorption of PCN, the PtSAs-PCN shows moderate photocatalytic H2 evolution activity with the rate of 2.6/0.9 mmol g−1 h−1 at 420/450 nm (Fig. 5b), suggesting that the PtSAs can act as co-catalysts to trap the photoelectrons from PCN. Strikingly, the photocatalytic H2 evolution rate of PtSAs-Au2.5/PCN increases to 8.8/4.8 mmol g−1 h−1 under illumination of 420/450 nm light, much higher than that of Au2.5/PCN or PtSAs–PCN and even larger than the sum of H2 production rates of the two samples, which indicates the synergistic effect between Au NPs and PtSAs. Moreover, the enhanced photocurrent intensity further highlights the role of PtSAs on accelerating carrier separation in PtSAs–Au2.5/PCN (Fig. 5c). On the other hand, the photocurrent of PtSAs–Au2.5/PCN is enhanced by about 2.5 times relative to Au2.5/PCN under 550 nm light irradiation (Fig. 5d). This discrepancy is much larger than that of same photocatalysts at 420 nm (about 1.2). It suggests that the PtSAs near the Au NPs can effectively trap the injected plasmonic hot-electrons for H2 evolution, which may be responsible for the significantly enhanced photocatalytic performance of PtSAs–Au2.5/PCN under LSPR region. As displayed in Fig. 5e, the photocatalytic H2 evolution rate of PtSAs–Au2.5/PCN reaches up to 264 μmol g−1 h−1 at the strongest plasmonic-coupling wavelength (550 nm) and remains 55 μmol g−1 h−1 at 650 nm, much higher than that of Au2.5/PCN (about 4.1 times at 550 nm). In the meantime, the amount of photocatalytic H2 evolution is undetected for PtSAs–PCN at 550/600/650 nm, excluding the possibility of PCN as the source of photogenerated electrons. These results evidently demonstrate that the enhanced photocatalytic H2 evolution activity of PtSAs–Au2.5/PCN under LSPR region is attributed to more effective utilization of plasmonic hot-electrons.

Optics and photocatalytic properties of plasmonic PtSAs–Au2.5/PCN. a UV–vis DR spectra of PtSAs–PCN, Au2.5/PCN and PtSAs–Au2.5/PCN. Comparison of the photocatalytic H2 evolution rate between PtSAs-Au2.5/PCN and reference samples under different wavelength light irradiation: b short-wavelength visible light and e long-wavelength visible light. TPR density of PtSAs–Au2.5/PCN and reference samples under different wavelength light irradiation: c λ = 420 nm, d λ = 550 nm and. f Time-dependent course of H2 evolution under visible light (λ ≥ 420 nm). g Comparison of the photocatalytic H2 evolution rate between PtSAs–Au2.5/PCN and reference samples as well as h previous Au-based PCN plasmonic photocatalysts in table S1. i Cycling photocatalytic test for PtSAs-Au2.5/PCN
Hence, benefiting from the photoelectron trapping effect of PtSAs, the PtSAs–Au2.5/PCN exhibits dramatically enhanced photocatalytic H2 evolution performance at both the intrinsic absorption of PCN and the LSPR region. As a result, the photocatalytic H2 evolution rate of PtSAs–Au2.5/PCN is significantly enhanced with compared to Au2.5/PCN (Fig. 5f), achieving 13.7 mmol g−1 h−1 under visible-light region (Fig. 5g), which outperforms a broad range of Au-based PCN plasmonic photocatalysts (Fig. 5h and Table S2). The highest photocurrent response and smallest impedance arc radius further reveal the improved electron transport for H2 evolution in PtSAs–Au2.5/PCN (Fig. S15). In addition, the PtSAs–Au2.5/PCN presents high catalytic stability without obvious decay after 18 h catalytic experiments (Figs. 5i and S16).
Based on the above discussions, the enhanced photocatalytic H2 evolution activity of PtSAs–Au2.5/PCN system under whole visible-light irradiation should be ascribed to two mechanisms as illustrated in Fig. 6. Exposure to short-wavelength light, the PCN is photoexcited to generate electrons on the conduction band due to its intrinsic absorption, and the synergistic Au NPs and PtSAs trap these photoelectrons for H2 evolution. On the other hand, the Au NPs absorb relatively long-wavelength light to produce plasmonic hot-electrons, which can jump over the Schottky barrier and rapidly transfer to the PtSAs for H2 evolution. Therefore, the introduction of PtSAs greatly improve the broad-spectrum photocatalytic H2 evolution activity, especially in the plasma response range of Au NPs.

Photocatalytic mechanism of PtSAs–Au2.5/PCN under visible light
4 Conclusions
In summary, we have successfully designed and fabricated Au NPs and PtSAs on PCN surface to extend the visible-light absorption range as well as expedite plasmonic hot-electron injection and transfer for broad-spectrum photocatalytic H2 evolution. Thanks to the synergetic Au NPs and PtSAs, the as-prepared PtSAs-Au2.5/PCN exhibits excellent photocatalytic performance under whole visible-light irradiation. The H2 evolution rate reaches 13.7 mmol g−1 h−1 under the visible light and 264 μmol g−1 h−1 at 550 nm, much higher than that of Au/PCN and PtSAs/PCN, respectively. Detailed investigation has been carried out to reveal the enhancement photocatalytic mechanism. Briefly, the PCN is photoexcited under short-wavelength light to generate electrons on the conduction band, and the synergistic Au NPs and PtSAs trap these photoelectrons for H2 evolution. While the Au NPs absorb relatively long-wavelength light to produce plasmonic hot-electrons, which can jump over the Schottky barrier and rapidly transfer to the PtSAs for H2 evolution. This work opens up a new avenue to couple wide-bandgap semiconductor with plasmonic metal for broad-spectrum photocatalytic energy conversion applications.
References
X. Tao, Y. Zhao, S. Wang, C. Li, R. Li, Recent advances and perspectives for solar-driven water splitting using particulate photocatalysts. Chem. Soc. Rev. 51(9), 3561–3608 (2022). https://doi.org/10.1039/D1CS01182K
Y. Fang, Y. Hou, X. Fu, X. Wang, Semiconducting polymers for oxygen evolution reaction under light illumination. Chem. Rev. 122(3), 4204–4256 (2022). https://doi.org/10.1021/acs.chemrev.1c00686
C. Feng, Z.-P. Wu, K.-W. Huang, J. Ye, H. Zhang, Surface modification of 2D photocatalysts for solar energy conversion. Adv. Mater. 34(23), 2200180 (2022). https://doi.org/10.1002/adma.202200180
P. Niu, J. Dai, X. Zhi, Z. Xia, S. Wang et al., Photocatalytic overall water splitting by graphitic carbon nitride. InfoMat 3(9), 931–961 (2021). https://doi.org/10.1002/inf2.12219
J. Abdul Nasir, A. Munir, N. Ahmad, T.U. Haq, Z. Khan et al., Photocatalytic Z-scheme overall water splitting: recent advances in theory and experiments. Adv. Mater. 33(52), 2105195 (2021). https://doi.org/10.1002/adma.202105195
C. Gao, J. Low, R. Long, T. Kong, J. Zhu et al., Heterogeneous single-atom photocatalysts: fundamentals and applications. Chem. Rev. 120(21), 12175–12216 (2020). https://doi.org/10.1021/acs.chemrev.9b00840
X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin et al., A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8(1), 76–80 (2009). https://doi.org/10.1038/nmat2317
I.F. Teixeira, E.C.M. Barbosa, S.C.E. Tsang, P.H.C. Camargo, Carbon nitrides and metal nanoparticles: from controlled synthesis to design principles for improved photocatalysis. Chem. Soc. Rev. 47(20), 7783–7817 (2018). https://doi.org/10.1039/C8CS00479J
S. Cao, J. Low, J. Yu, M. Jaroniec, Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 27(13), 2150–2176 (2015). https://doi.org/10.1002/adma.201500033
W.-J. Ong, L.-L. Tan, Y.H. Ng, S.-T. Yong, S.-P. Chai, Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability? Chem. Rev. 116(12), 7159–7329 (2016). https://doi.org/10.1021/acs.chemrev.6b00075
P. Kumar, D. Laishram, R.K. Sharma, A. Vinu, J. Hu et al., Boosting photocatalytic activity using carbon nitride based 2D/2D van der waals heterojunctions. Chem. Mater. 33(23), 9012–9092 (2021). https://doi.org/10.1021/acs.chemmater.1c03166
S. Guo, Z. Deng, M. Li, B. Jiang, C. Tian et al., Phosphorus-doped carbon nitride tubes with a layered micro-nanostructure for enhanced visible-light photocatalytic hydrogen evolution. Angew. Chem. Int. Ed. 55(5), 1830–1834 (2016). https://doi.org/10.1002/anie.201508505
G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin et al., Iodine modified carbon nitride semiconductors as visible light photocatalysts for hydrogen evolution. Adv. Mater. 26(5), 805–809 (2014). https://doi.org/10.1002/adma.201303611
D. Zhao, C.-L. Dong, B. Wang, C. Chen, Y.-C. Huang et al., Synergy of dopants and defects in graphitic carbon nitride with exceptionally modulated band structures for efficient photocatalytic oxygen evolution. Adv. Mater. 31(43), 1903545 (2019). https://doi.org/10.1002/adma.201903545
D. Zhao, Y. Wang, C.-L. Dong, Y.-C. Huang, J. Chen et al., Boron-doped nitrogen-deficient carbon nitride-based Z-scheme heterostructures for photocatalytic overall water splitting. Nat. Energy 6(4), 388–397 (2021). https://doi.org/10.1038/s41560-021-00795-9
H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao et al., Alkali-assisted synthesis of nitrogen deficient graphitic carbon nitride with tunable band structures for efficient visible-light-driven hydrogen evolution. Adv. Mater. 29(16), 1605148 (2017). https://doi.org/10.1002/adma.201605148
Y. Wang, P. Du, H. Pan, L. Fu, Y. Zhang et al., Increasing solar absorption of atomically thin 2D carbon nitride sheets for enhanced visible-light photocatalysis. Adv. Mater. 31(40), 1807540 (2019). https://doi.org/10.1002/adma.201807540
L. Lin, H. Ou, Y. Zhang, X. Wang, Tri-s-triazine-based crystalline graphitic carbon nitrides for highly efficient hydrogen evolution photocatalysis. ACS Catal. 6(6), 3921–3931 (2016). https://doi.org/10.1021/acscatal.6b00922
L. Lin, Z. Yu, X. Wang, Crystalline carbon nitride semiconductors for photocatalytic water splitting. Angew. Chem. Int. Ed. 58(19), 6164–6175 (2019). https://doi.org/10.1002/anie.201809897
L. Lin, Z. Lin, J. Zhang, X. Cai, W. Lin et al., Molecular-level insights on the reactive facet of carbon nitride single crystals photocatalytic overall water splitting. Nat. Catal. 3(8), 649–655 (2020). https://doi.org/10.1038/s41929-020-0476-3
L. Zhang, N. Ding, L. Lou, K. Iwasaki, H. Wu et al., Localized surface plasmon resonance enhanced photocatalytic hydrogen evolution via Pt@Au NRs/C3N4 nanotubes under visible-light irradiation. Adv. Funct. Mater. 29(3), 1806774 (2019). https://doi.org/10.1002/adfm.201806774
W. Cheng, H. Su, F. Tang, W. Che, Y. Huang et al., Synergetic enhancement of plasmonic hot-electron injection in Au cluster-nanoparticle/C3N4 for photocatalytic hydrogen evolution. J. Mater. Chem. A. 5(37), 19649–19655 (2017). https://doi.org/10.1039/C7TA03808A
H. Li, Y. Gao, Z. Xiong, C. Liao, K. Shih, Enhanced selective photocatalytic reduction of CO2 to CH4 over plasmonic au modified g-C3N4 photocatalyst under UV–vis light irradiation. Appl. Surf. Sci. 439, 552–559 (2018). https://doi.org/10.1016/j.apsusc.2018.01.071
D. Ruan, J. Xue, M. Fujitsuka, T. Majima, Ultrafast spectroscopic study of plasmon-induced hot electron transfer under NIR excitation in Au triangular nanoprism/g-C3N4 for photocatalytic H2 production. Chem. Commun. 55(43), 6014–6017 (2019). https://doi.org/10.1039/C9CC02574J
Q. Zhang, X. Liu, M. Chaker, D. Ma, Advancing graphitic carbon nitride-based photocatalysts toward broadband solar energy harvesting. ACS Mater. Lett. 3(6), 663–697 (2021). https://doi.org/10.1021/acsmaterialslett.1c00160
Y. Fu, T. Huang, B. Jia, J. Zhu, X. Wang, Reduction of nitrophenols to aminophenols under concerted catalysis by Au/g-C3N4 contact system. Appl. Catal. B Environ. 202, 430–437 (2017). https://doi.org/10.1016/j.apcatb.2016.09.051
M. Gao, W. Yang, Y. Yu, J. Li, Y. Liu, N-hexane-assisted synthesis of plasmonic au-mediated polymeric carbon nitride photocatalyst for remarkable H2 evolution under visible-light irradiation. J. Colloid Interface Sci. 627, 398–404 (2022). https://doi.org/10.1016/j.jcis.2022.07.056
H. Tian, X. Liu, Z. Liang, P. Qiu, X. Qian et al., Gold nanorods/g-C3N4 heterostructures for plasmon-enhanced photocatalytic H2 evolution in visible and near-infrared light. J. Colloid Interface Sci. 557, 700–708 (2019). https://doi.org/10.1016/j.jcis.2019.09.075
Y. Guo, H. Jia, J. Yang, H. Yin, Z. Yang et al., Understanding the roles of plasmonic au nanocrystal size, shape, aspect ratio and loading amount in Au/g-C3N4 hybrid nanostructures for photocatalytic hydrogen generation. Phys. Chem. Chem. Phys. 20(34), 22296–22307 (2018). https://doi.org/10.1039/C8CP04241A
A. Zada, M. Humayun, F. Raziq, X. Zhang, Y. Qu et al., Exceptional visible-light-driven cocatalyst-free photocatalytic activity of g-C3N4 by well-designed nanocomposites with plasmonic Au and SnO2. Adv. Energy Mater. 6(21), 1601190 (2016). https://doi.org/10.1002/aenm.201601190
Q. Zhou, Y. Guo, Z. Ye, Y. Fu, Y. Guo et al., Carbon nitride photocatalyst with internal electric field induced photogenerated carriers spatial enrichment for enhanced photocatalytic water splitting. Mater. Today 58, 100–109 (2022). https://doi.org/10.1016/j.mattod.2022.06.009
M. Gao, F. Tian, Z. Guo, X. Zhang, Z. Li et al., Mutual-modification effect in adjacent pt nanoparticles and single atoms with sub-nanometer inter-site distances to boost photocatalytic hydrogen evolution. Chem. Eng. J. 446, 137127 (2022). https://doi.org/10.1016/j.cej.2022.137127
J. Ran, H. Wang, H. Jin, C. Ling, X. Zhang et al., Metallic MoN ultrathin nanosheets boosting high performance photocatalytic H2 production. J. Mater. Chem. A 6(46), 23278–23282 (2018). https://doi.org/10.1039/C8TA09358J
P. Zhou, F. Lv, N. Li, Y. Zhang, Z. Mu et al., Strengthening reactive metal-support interaction to stabilize high-density Pt single atoms on electron-deficient g-C3N4 for boosting photocatalytic H2 production. Nano Energy 56, 127–137 (2019). https://doi.org/10.1016/j.nanoen.2018.11.033
Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang et al., Molecule self-assembly synthesis of porous few-layer carbon nitride for highly efficient photoredox catalysis. J. Am. Chem. Soc. 141(6), 2508–2515 (2019). https://doi.org/10.1021/jacs.8b12428
W. Wang, J. Fang, X. Huang, Different behaviors between interband and intraband transitions generated hot carriers on g-C3N4/Au for photocatalytic H2 production. Appl. Surf. Sci. 513, 145830 (2020). https://doi.org/10.1016/j.apsusc.2020.145830
B. Zeng, S. Wang, Y. Gao, G. Li, W. Tian et al., Interfacial modulation with aluminum oxide for efficient plasmon-induced water oxidation. Adv. Funct. Mater. 31(6), 2005688 (2021). https://doi.org/10.1002/adfm.202005688
C.-C. Nguyen, M. Sakar, M.-H. Vu, T.-O. Do, Nitrogen vacancies-assisted enhanced plasmonic photoactivities of Au/g-C3N4 crumpled nanolayers: a novel pathway toward efficient solar light-driven photocatalysts. Ind. Eng. Chem. Res. 58(9), 3698–3706 (2019). https://doi.org/10.1021/acs.iecr.8b05792
S. Bai, X. Li, Q. Kong, R. Long, C. Wang et al., Toward enhanced photocatalytic oxygen evolution: synergetic utilization of plasmonic effect and Schottky junction via interfacing facet selection. Adv. Mater. 27(22), 3444–3452 (2015). https://doi.org/10.1002/adma.201501200
X. Li, W. Bi, L. Zhang, S. Tao, W. Chu et al., Single-atom Pt as co-catalyst for enhanced photocatalytic H2 evolution. Adv. Mater. 28(12), 2427–2431 (2016). https://doi.org/10.1002/adma.201505281
P. Zhou, N. Li, Y. Chao, W. Zhang, F. Lv et al., Thermolysis of noble metal nanoparticles into electron-rich phosphorus-coordinated noble metal single atoms at low temperature. Angew. Chem. Int. Ed. 58(40), 14184–14188 (2019). https://doi.org/10.1002/anie.201908351
Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang et al., Single-atom platinum confined by the interlayer nanospace of carbon nitride for efficient photocatalytic hydrogen evolution. Nano Energy 69, 104409 (2020). https://doi.org/10.1016/j.nanoen.2019.104409
X. Bao, M. Liu, Z. Wang, D. Dai, P. Wang et al., Photocatalytic selective oxidation of hmf coupled with H2 evolution on flexible ultrathin g-C3N4 nanosheets with enhanced n–h interaction. ACS Catal. 12(3), 1919–1929 (2022). https://doi.org/10.1021/acscatal.1c05357
D. Zhao, Y. Wang, C.-L. Dong, F. Meng, Y-C, Huang, et al., Electron-deficient Zn-N6 configuration enabling polymeric carbon nitride for visible-light photocatalytic overall water splitting. Nano-Micro Lett. 14, 223 (2022). https://doi.org/10.1007/s40820-022-00962-x
M. Gao, F. Tian, X. Zhang, Y. Liu, Z. Chen et al., Fast charge separation and transfer strategy in polymeric carbon nitride for efficient photocatalytic H2 evolution: coupling surface Schottky junctions and interlayer charge transfer channels. Nano Energy 103, 107767 (2022). https://doi.org/10.1016/j.nanoen.2022.107767
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51871078 and 52071119), Interdisciplinary Research Foundation of HIT (Grant No. IR2021208), State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (No. 2022TS38) and Heilongjiang Science Foundation (No. LH2020B006).
Funding
Open access funding provided by Shanghai Jiao Tong University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no interest conflict. They have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, M., Tian, F., Zhang, X. et al. Improved Plasmonic Hot-Electron Capture in Au Nanoparticle/Polymeric Carbon Nitride by Pt Single Atoms for Broad-Spectrum Photocatalytic H2 Evolution. Nano-Micro Lett. 15, 129 (2023). https://doi.org/10.1007/s40820-023-01098-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40820-023-01098-2