
Abstract
Background
The antifibrotic drugs, nintedanib and pirfenidone, inhibit the decline in forced vital capacity in patients with idiopathic pulmonary fibrosis (IPF). Nintedanib also inhibits the onset of acute exacerbation and reduces the risk of all-cause mortality. However, their effectiveness in real-world practice remains unclear. Our study aimed to investigate the changes in forced vital capacity, survival period, causes of death, and risk factors for mortality in patients with IPF receiving antifibrotic drugs.
Methods
This retrospective study enrolled Japanese patients who visited Toho University Sakura Medical Center who were diagnosed with IPF and received antifibrotic drugs.
Results
We included 102 patients [mean age ± standard deviation (SD): 71.8 ± 7.5 years], of whom 76 were males. The decline in forced vital capacity (mean ± SD) during the antifibrotic therapy period was − 154 ± 259 mL/year, which was significantly lower than before the antifibrotic therapy period (− 484 ± 589 mL/year; n = 80, p = 0.003). Altogether, 52 deaths were confirmed, and the median survival time from antifibrotic therapy initiation was 38.0 months (95% confidence interval: 25.9–50.1 months). Acute exacerbation accounted for 9.6% of all deaths (95% confidence interval: 1.6–17.6). The decline in forced vital capacity during antifibrotic therapy was a risk factor for mortality.
Conclusions
In actual clinical practice in Japan, antifibrotic drugs suppressed the gradual decline in forced vital capacity, which is a risk factor for mortality. However, the median survival period remained poor at 38 months.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In real-world clinical settings in Japan, antifibrotic drugs were found to suppress the progressive decline in forced vital capacity, which is an important prognostic factor. |
The frequency of acute exacerbation, a major cause of death in IPF patients in Japan, was lower in this study than in previous reports. |
However, the improvement in overall survival through antifibrotic drugs was not observed, highlighting challenges related to case selection, timing of treatment initiation, and management of side effects in real-world clinical practice. |
1 Introduction
Idiopathic pulmonary fibrosis (IPF), a disease of unknown etiology, commonly occurs in middle-aged and older individuals and is characterized by chronic and progressive pulmonary fibrosis leading to irreversible honeycomb lung formation [1, 2]. Patients with IPF have a poor prognosis, with a median survival time of 28–52 months from the time of IPF diagnosis [3,4,5,6].
In IPF, acute exacerbations that rapidly worsen into respiratory failure occur at a frequency of 5–10% per year [7], and their prognosis is poor, with a reported 3-month survival rate of 67% [8].
The antifibrotic drugs pirfenidone and nintedanib inhibit progressive fibrosis in IPF. According to previous clinical trials, both pirfenidone and nintedanib significantly inhibited the loss of forced vital capacity (FVC) in 1 year in the experimental group as compared with the placebo group [9, 10]. Although the effects of these therapies on improving prognosis were not examined in these clinical trials, a pooled analysis of two clinical trials of nintedanib (TOMORROW and INPULSIS trials) revealed a 30% reduction in the risk of death at 52 weeks in the nintedanib group as compared with that of the placebo group [11]. Additionally, a Weibull distribution-based analysis using data from six clinical trials on nintedanib found that nintedanib could improve the survival outcomes [12].
According to the European IPF Registry, which investigated the survival benefit of antifibrotic drugs for IPF in real-world clinical settings in Europe, the median survival times in the groups not using and using antifibrotic agents (pirfenidone or nintedanib) were 68.3 and 123.1 months, respectively, showing a significantly longer survival time in the group using antifibrotic agents [13].
In a study involving patients with IPF in Korea [14], the survival times were 54 and 34 months in patients receiving antifibrotic agents and medications other than antifibrotic agents, respectively. The antifibrotic agent used in the study was mainly pirfenidone, and only 2.2% of patients used nintedanib.
Natsuizaka et al. conducted an epidemiological study involving 553 patients with IPF in Hokkaido, Japan [15]. In their study, 328 patients died during the observation period, and the median survival time from diagnosis was 35 months. The causes of death were acute exacerbation, chronic respiratory failure, concomitant primary lung cancer, pneumonia, and cardiovascular complications in 40%, 24%, 11%, 7%, and 3% patients, respectively.
According to a study from the USA, among 47 IPF patients, 34 died during the follow-up period. The causes of death were acute exacerbation in five patients (15%), respiratory insufficiency other than acute exacerbation in 10 patients (30%), and cardiovascular complications in seven patients (6%) [6].
Compared to reports from the USA, the Hokkaido study found a higher incidence of deaths due to acute exacerbation and a lower incidence of deaths due to cardiovascular complications, suggesting ethnic differences in the causes of death among IPF cases. Additionally, the suppression of acute exacerbation is considered more important for improving the prognosis of these patients in Japan.
The clinical trials of nintedanib, i.e., INPULSIS-1 and INPULSIS-2 trials, investigated the acute exacerbation-suppressing effect of nintedanib. In the INPULSIS-1 trial, the incidence of acute exacerbation was not significantly different between the placebo and nintedanib groups. However, in the simultaneously conducted INPULSIS-2 trial, the nintedanib group exhibited a significantly lower rate of acute exacerbation.
A pooled analysis of these two trials revealed that the incidences of acute exacerbation through a central review were 4.9% and 7.6% in the nintedanib and placebo groups, respectively, with a lower tendency of acute exacerbation in the nintedanib group [16]. The proportion of Asians in INPULSIS-1 and -2 were 21.4% and 38.9%, respectively, which may have influenced the differences in results between the two trials.
As described above, the administration of antifibrotic drugs to IPF patients is expected to inhibit the progressive decline in lung capacity, reduce the incidence of acute exacerbation, and potentially extend survival duration. However, reports on the outcomes in Japan in real-world clinical settings are limited. Therefore, this study aimed to examine the changes in FVC, incidence of acute exacerbation, causes of death, survival time, and risk factors for mortality among IPF patients receiving antifibrotic drug therapy in real-world clinical settings in Japan.
2 Methods
2.1 Study Design and Participants
In the present retrospective study, the study participants were patients who visited Toho University Sakura Medical Center and were diagnosed with IPF through the medical expense assistance program received the Certificate of Medical Benefit (CMB) and underwent treatment with antifibrotic drugs from 1 December 2008 to 31 December 2021.
The Japanese Ministry of Health, Labor, and Welfare established the medical expense assistance program for certain intractable diseases to support patients with diseases of unknown etiologies with no established treatments. Patients with a confirmed diagnosis are granted CMB and provided medical expense assistance. To obtain the CMB, physicians responsible for diagnosing IPF provide the necessary clinical information, including medical history data, high-resolution computed tomography (HRCT) scans, and results of lung function test, blood gas analysis, and 6-min walk test, to the administrative authorities. A panel of pulmonologists specializing in interstitial lung diseases then reviews the submitted information and confirms IPF diagnosis. In cases where surgical lung biopsy is not performed, the presence of honeycombing and a subpleural distribution of shadows on HRCT, which indicates a usual interstitial pneumonia (UIP) pattern, is required for the diagnosis. This comprehensive diagnostic process follows an approach similar to multidisciplinary discussion (MDD) across multiple facilities and is consistent with recent clinical guidelines from ATS/ERS/JRS/ALAT [2, 17].
Acute exacerbation was defined in accordance with the Japanese diagnostic criteria described in the Hokkaido study [18]. Patients with a history of acute exacerbation at the initiation of antifibrotic therapy or with a history of or concurrent lung cancer were excluded from the study.
This study and consent procedure were approved by the Ethics Committee of Toho University Sakura Medical Center (approval number: S21084; approval date: 11 October 2022). We utilized an opt-out method to obtain patient consent regarding the use of their personal information, by publishing the research content and our policy on personal information on the hospital website.
2.2 Data Collection
Participant data were extracted from their medical records, including the lung function test results [FVC, percent of predicted forced predicted vital capacity(%FVC), and diffusing capacity of the lung for carbon monoxide % predicted (%DLCO)], the modified Medical Research Council (mMRC) scale ([19], Table 1), Japanese disease severity classification (JSC) ([20], Table 2), the GAP stage ([21], Table 3), blood gas analysis, 6-min walk test results (walk distance and minimum SpO2), serum lactate dehydrogenase and Krebs von den Lungen-6 levels, and surfactant protein-D levels at the start of antifibrotic therapy. In our facility, patients suspected of having IPF undergo pulmonary function tests every 3 months starting from their initial visit. These tests are performed continuously even after a definitive diagnosis is made and antifibrotic therapy is initiated. Among these pulmonary function tests, we collected the oldest data from the tests conducted 6–12 months prior to treatment initiation, as well as the most recent data from the tests conducted 6–12 months after treatment initiation, to calculate the annual changes in FVC.
Additionally, information about the date of starting oxygen therapy, status and onset date of acute exacerbation, concomitant lung cancer, time and date of death, cause of death, and outcome were obtained from the medical records. For patients transferred to another medical institution, information was obtained by contacting the relevant medical institution. The outcome was evaluated based on the status as of 31 October 2022. Death occurring during hospitalization due to acute exacerbation, or within 3 months following an acute exacerbation, was referred to as “death caused by acute exacerbation.”
2.3 Statistical Analyses
Unless otherwise indicated, the values are presented as mean ± standard deviation or mean (95% confidence interval). The survival time is presented as the median survival time (95% confidence interval) according to the Kaplan–Meier method. For analyzing factors contributing to the survival time, we used the Cox proportional hazards model, and annual changes in vital capacity before and after antifibrotic therapy were analyzed using the two-way analysis of variance. The statistical software used was SPSS version 25.0 (IBM CO., Armonk, NY, USA).
3 Results
3.1 Baseline Characteristics
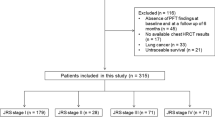
From 1 December 2008 to the end of December 2021, 109 patients who visited Toho University Sakura Medical Center were diagnosed with IPF under the medical expense assistance program and received antifibrotic drug therapy. This study does not include cases that underwent surgical lung biopsy, but the presence of the UIP pattern on HRCT has been confirmed through the evaluation by multiple specialists, as part of the medical expense assistance program. Three patients with concurrent lung cancer and four patients with a history of acute exacerbation at the initiation of antifibrotic therapy were excluded. Finally, 102 patients were included in the analysis. The background characteristics of these patients at the start of antifibrotic drug therapy are presented in Tables 4 and 5. Of these 102 patients, 32 were treated with pirfenidone, 81 with nintedanib, and 11 with both drugs at different times. The average duration of antifibrotic drug therapy in these patients was 22.9 ± 20.1 months.
3.2 Effect of Antifibrotic Agents on Changes in FVC Over Time
Among the 102 patients included in our analyses, changes in FVC over a 6–12-month period before and during antifibrotic therapy were confirmed in 80 patients (61 men and 19 women; mean age: 70.7 ± 7.3 years) at the start of antifibrotic therapy. Of these patients, 31 received pirfenidone and 60 received nintedanib. Before starting antifibrotic therapy, the decline in FVC was − 484 ± 589 mL/year, whereas during the antifibrotic therapy period, it was − 154 ± 259 mL/year, indicating a significantly lower decline during the antifibrotic therapy period (p = 0.003).
Among 80 cases, a two-factor analysis of variance of the decline in FVC before and during antifibrotic treatment was performed in 20 cases treated with pirfenidone alone and in 49 cases treated with nintedanib alone. Compared with the data before antifibrotic treatment, the decline during the antifibrotic therapy period was significantly lesser both in the pirfenidone (− 371 ± 676 and − 105 ± 309 mL/year) and nintedanib (− 574 ± 595 and − 189 ± 257 mL/year) groups (p < 0.001). However, there was no significant difference between the pirfenidone and nintedanib groups regarding the decline in FVC before and during antifibrotic treatment (p = 0.23).
3.3 Analysis of the Survival Time, Cause of Death, and Risk of Death
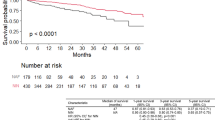
Among 102 patients, 52 deaths were confirmed. Among the 81 patients treated with nintedanib, 38 died. The median survival time of all cases, as determined using the Kaplan–Meier method, was 38.0 (25.9–50.1) months from the start of antifibrotic drug therapy (Fig. 1).

Kaplan–Meier plot of survival probability from the initiation of antifibrotic drug therapy
Among the 102 cases, 13 developed acute exacerbation after initiating antifibrotic therapy, resulting in an annual incidence rate of 5.3%; of these 13 cases, 11 died, and the median survival time from the onset of acute exacerbation was 6.8 (1.3–12.2) months. Among the 81 patients treated with nintedanib, 11 developed acute exacerbation, with an annual incidence rate of 5.4%. Among the 13 cases that developed acute exacerbation, five were classified as “death caused by acute exacerbation” (death occurring within 3 months after the occurrence of acute exacerbation or death during hospitalization due to acute exacerbation), accounting for 9.6% (1.6–17.6%) of all 52 deaths. Additionally, among the 81 patients who received nintedanib, four cases were classified as “death caused by acute exacerbation,” accounting for 10.5% (0.7–20.2%) of all 38 deaths in the nintedanib-treated group.
Four out of 102 cases were newly diagnosed with lung cancer after starting antifibrotic therapy. Among the 81 cases treated with nintedanib, two cases were diagnosed with lung cancer after starting nintedanib. Throughout the entire course of all cases, only one death was due to lung cancer.
Of the 52 deaths, 5 were due to acute exacerbation, 1 was due to lung cancer, 35 were due to worsening of the underlying disease, and 1 each was due to COVID–19, pneumonia, pancreatic cancer, and malignant lymphoma. Moreover, three deaths were due to heart disease, one due to gastrointestinal perforation, two due to cardiac arrest on admission, and one due to unknown causes.
For all 102 cases, we analyzed the predictors of survival after starting pirfenidone or nintedanib antifibrotic therapy using Cox proportional hazard regression models. Independent variables including sex, age at antifibrotic treatment initiation, type of antifibrotic therapy (whether nintedanib was used or not), %FVC, and %DLCO were analyzed. However, none of the variables were found to be statistically significant risk factors for survival (Table 6).
Next, we analyzed 80 cases in which changes in lung function tests were observed during a period of 6–12 months before and after the initiation of antifibrotic agents. We used Cox proportional hazard regression models to identify the predictors of survival time after the initiation of antifibrotic agents, with age at the start of antifibrotic therapy and changes in FVC per year before and during the therapy as independent variables. The results indicated that a decrease in FVC per year during antifibrotic therapy was a risk factor for survival time (Table 7).
3.4 Adverse Drug Reactions and Discontinuation
Nintedanib was discontinued in 55 out of 81 patients, whereas pirfenidone was discontinued in 26 out of 31 patients. Table 8 lists the reasons for treatment discontinuation, with IPF worsening as the most common reason for both drugs.
Table 9 presents the adverse drug reactions (ADRs) for such medications. Nintedanib was discontinued in nine patients because of liver impairment, and eight patients because of diarrhea. Moreover, pirfenidone was discontinued in three patients because of nausea, vomiting, and loss of appetite. No deaths were directly attributed to ADRs.
4 Discussion
Herein, we demonstrated that antifibrotic drugs suppress the decline in FVC in real-world clinical settings in Japan. Among the 52 cases of mortality, 9 cases (9.6%, 1.6–17.6) were due to acute exacerbation and one case was attributed to lung cancer. The median survival time from the initiation of antifibrotic drug therapy was 38.0 months (25.9–50.1), and the decline in FVC during the antifibrotic therapy period was identified as a prognostic factor.
The antifibrotic drugs pirfenidone and nintedanib were approved in Japan on 12 December 2008 and 31 August 2015, respectively.
The Hokkaido study is a study that investigated the prognosis of IPF in the real-world, targeting Japanese patients. This study conducted a survival analysis based on data obtained until 30 September 2011, focusing on patients diagnosed between 1 January 2003 and 31 December 2007. Among the 553 eligible patients, 328 deaths were confirmed, and the median survival time from the time of diagnosis was 35 months [15].
Our study and the Hokkaido study have similar patient backgrounds. Both studies confirmed the diagnosis of IPF by using the CMB through Japan’s healthcare subsidy system. The average age was 71.8 ± 7.5 years in our study and 70.0 ± 9.0 years in the Hokkaido study, with male proportions of 74.5% and 72.7%, respectively. The distribution of severity levels I, II, III, and IV according to the JSC in our study population was 41.1%, 4.9%, 31.4, and 23.5%, whereas that in the Hokkaido study was 43.3%, 12.6%, 22.1%, and 22.1%, respectively [22]. Furthermore, the distribution of stages 1, 2, and 3 according to the GAP model was 52.0%, 35.3%, and 12.7% in our study, and 44.2%, 36.2%, and 19.6% in the Hokkaido study, respectively [22].
The Hokkaido study did not mention the use of antifibrotic drugs, but considering the timeline, it is evident that patients receiving nintedanib were not included. There is a possibility that patients receiving pirfenidone after December 2008 were included, but the impact on the results is limited. Therefore, the primary difference between our study and the Hokkaido study may lie in the use of antifibrotic medications.
Japanese individuals have a higher incidence of diffuse alveolar damage attributed to factors such as medications [23], and acute exacerbation of IPF has been extensively reported in Japan [24, 25]. While the proportion of acute exacerbations as a cause of death in IPF was 18% in the USA [6], it was 40% in the Hokkaido study, and Kondo et al. reported 35.1% [26]. In Japan, a high number of deaths are believed to be associated with acute exacerbation, making acute exacerbation crucial in the clinical practice of IPF management in this country.
In our study, acute exacerbation accounted for 9.6% (1.6–17.6) of all deaths.
The definition of “death due to acute exacerbation” has not been firmly established, making direct comparisons difficult. However, in our study, the proportion of deaths attributed to acute exacerbation among all deaths was lower than that in the Hokkaido study and the report by Kondo et al. determining whether antifibrotic medications can reduce the risk of acute exacerbation and death is a crucial clinical challenge. Further investigation is necessary.
In our study, the antifibrotic therapy suppressed FVC decline, and the rate of mortality caused by acute exacerbation in patients receiving antifibrotic therapy tended to be lower than that of previous reports. However, the median survival duration after initiating antifibrotic therapy was 38.0 (25.9–50.1) months, and we did not observe a substantial difference compared with the median survival duration of 35 months reported in the Hokkaido study.
Collard et al. reported that changes in FVC over a 6–12-month period are more important prognostic factors than the baseline FVC in patients with IPF [27]. Even with equivalent baseline data, a greater decline in FVC over time implies a worse prognosis compared with those with a lesser decline.
In actual clinical practice in Japan, due to the excessive cost of antifibrotic drugs, most patients initiate antifibrotic therapy after receiving CMB under the medical expense assistance program. This program prioritizes the severity level according to the JSC, and for mild cases (JSC I and II), the CMB may be withheld until medical expenses reach a certain level, making it difficult to administer antifibrotic drugs.
Due to such circumstances, many cases with suspected IPF undergo observation, and among them, those showing a progressive trend are more likely to become eligible for enrollment in the medical expense assistance program and antifibrotic drug treatment. In our data, a decline in FVC after treatment initiation was identified as a risk factor for mortality. However, considering the aforementioned circumstances, it is possible that cases with a significant decline in vital capacity are more likely to be selected for antifibrotic drug treatment in real-world clinical practice in Japan.
Among the 80 cases whose changes in vital capacity before and after the initiation of antifibrotic therapy were examined, the decline in FVC during the 6–12 months before treatment initiation was − 484 ± 589 mL/year, and at the time of treatment initiation, a yearly decline in vital capacity of − 17.6% ± 21.4% compared with the pretreatment value was observed. Moreover, the prognostic factor was not the change in vital capacity before treatment initiation but the change in vital capacity after treatment initiation. The tendency in real-world clinical practice in Japan to initiate antifibrotic therapy after waiting for disease progression may result in missed opportunities to improve patients’ prognosis.
Jegal et al. examined the prognostic factors in 179 patients with IPF by performing a multivariate analysis; they found three independent prognostic factors, which included sex, change in vital capacity over time, and diffusing capacity at baseline [28]. In the individuals with impaired diffusion capacity at baseline, initiating antifibrotic therapy without waiting for a decline in vital capacity may contribute to the improved prognosis.
In the Hokkaido study, the third leading cause of death was concomitant lung cancer, accounting for 11% of all deaths. The use of nintedanib combined with docetaxel as a second-line therapy for advanced non-small-cell lung cancer has a significant effect on prolonging the survival time, especially in patients with adenocarcinoma [29]. Otsubo et al. compared the group of patients receiving single-drug chemotherapy with the group of patients receiving chemotherapy plus nintedanib for unresectable non-small-cell lung cancer concomitant with pulmonary fibrosis, and they found that progression-free survival was significantly prolonged in the group receiving chemotherapy plus nintedanib [30]. It is unclear whether nintedanib helps inhibit the onset of lung cancer; however, in the present study, throughout the disease course, six out of 102 patients developed lung cancer, and death due to lung cancer was extremely low, occurring in one of the 102 patients. Excluding cases with concomitant lung cancer at the time of IPF diagnosis may have influenced the low incidence of death due to lung cancer.
In the present study involving 102 patients, the duration from the initiation of antifibrotic therapy to death or censoring was 29.5 ± 21.9 months. However, the mean period of antifibrotic agent administration was 22.9 ± 20.1 months, and antifibrotic agents were discontinued at a mean duration of ~ 7 months before death or censoring. Pirfenidone was discontinued in 4 out of 32 patients because of gastrointestinal symptoms and in 2 patients because of skin symptoms. Furthermore, nintedanib was discontinued in 9 out of 81 patients because of liver impairment and in 10 patients because of gastrointestinal symptoms.
According to Kato et al., in IPF patients receiving nintedanib, the management of side effects of nausea and diarrhea can affect the prognosis [31]. Improving the management of these side effects could contribute to an improvement in prognosis.
Herein, we demonstrated that antifibrotic drugs can attenuate the progressive decline of FVC in real-world clinical settings. Furthermore, the mortality rate due to acute exacerbation, which is the primary cause of death among IPF patients in Japan, was lower in patients receiving antifibrotic therapy in our study than in those in previous reports. However, the survival period after treatment initiation was ~ 3 years and remains low. One of the issues is that in actual clinical practice in Japan, treatment is often not initiated until the disease has progressed. Another issue is that many patients have to discontinue treatment due to its side effects. By selecting appropriate cases for early administration, reducing treatment discontinuation due to side effects through appropriate management, and extending the treatment duration, it is possible to further improve the prognosis of IPF patients.
This study has several limitations. First, it is a retrospective study conducted at a single institution, and the number of cases is limited. There may be biases in case selection; therefore, our results cannot be immediately generalized to the entire Japanese population. Second, as the timing of the tests, criteria for starting the drug administration, and criteria for discontinuation were not established, they were different for each patient, and the amount of missing data was not small, which could have affected the interpretation of the study results. Third, the definition of the diagnostic criteria of IPF and acute exacerbation has changed over time, and the definition of “death due to acute exacerbation” was not established; therefore, it was difficult to compare it with previous studies. We hope that the effect of antifibrotic agents on improving the survival time in patients with IPF will be clearer in the future by conducting IPF registry studies in different countries, including Japan.
5 Conclusions
In real-world clinical practice in Japan, antifibrotic therapy significantly suppressed the decline in FVC, and the decline in FVC after treatment initiation was identified as a risk factor for mortality. Among the total 52 mortality cases, 5 were caused by acute exacerbation, accounting for 9.6% (95% confidence interval: 1.6–17.6). The median survival duration of 102 patients with IPF who received antifibrotic therapy, at 38.0 months (25.9–50.1 months), remained unfavorable.
References
Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–15.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, Selman M, Travis WD, Walsh S, Wilson KC, American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, Latin American Thoracic Society. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44-68.
Schwartz DA, Helmers RA, Galvin JR, Van Fossen DS, Frees KL, Dayton CS, Burmeister LF, Hunninghake GW. Determinants of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149:450–4.
Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199–203.
Mapel DW, Hunt WC, Utton R, Baumgartner KB, Samet JM, Coultas DB. Idiopathic pulmonary fibrosis: survival in population based and hospital-based cohorts. Thorax. 1998;53:469–76.
Fernández Pérez ER, Daniels CE, Schroeder DR, St Sauver J, Hartman TE, Bartholmai BJ, Yi ES, Ryu JH. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137:129–37.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824.
Agarwal R, Jindal SK. Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review. Eur J Intern Med. 2008;19:227–35.
Azuma A, Nukiwa T, Tsuboi E, Suga M, Abe S, Nakata K, Taguchi Y, Nagai S, Itoh H, Ohi M, Sato A, Kudoh S. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171:1040–7.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR, INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Richeldi L, Cottin V, du Bois RM, Selman M, Kimura T, Bailes Z, Schlenker-Herceg R, Stowasser S, Brown KK. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS(®) trials. Respir Med. 2016;113:74–9.
Lancaster L, Crestani B, Hernandez P, Inoue Y, Wachtlin D, Loaiza L, Quaresma M, Stowasser S, Richeldi L. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res. 2019;6: e000397.
Guenther A, Krauss E, Tello S, Wagner J, Paul B, Kuhn S, Maurer O, Heinemann S, Costabel U, Barbero MAN, Müller V, Bonniaud P, Vancheri C, Wells A, Vasakova M, Pesci A, Sofia M, Klepetko W, Seeger W, Drakopanagiotakis F, Crestani B. The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19:141.
Jegal Y, Park JS, Kim SY, Yoo H, Jeong SH, Song JW, Lee JH, Lee HL, Choi SM, Kim YW, Kim YH, Choi HS, Lee J, Uh ST, Kim TH, Kim SH, Lee WY, Kim YH, Lee HK, Lee EJ, Heo EY, Yang SH, Kang HK, Chung MP, Korea ILD Study Group. Clinical features, diagnosis, management, and outcomes of idiopathic pulmonary fibrosis in Korea: analysis of the Korea IPF cohort (KICO) registry. Tuberc Respir Dis (Seoul). 2022;85:185–94.
Natsuizaka M, Chiba H, Kuronuma K, Otsuka M, Kudo K, Mori M, Bando M, Sugiyama Y, Takahashi H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med. 2014;190:773–9.
Collard HR, Richeldi L, Kim DS, Taniguchi H, Tschoepe I, Luisetti M, Roman J, Tino G, Schlenker-Herceg R, Hallmann C, du Bois RM. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur Respir J. 2017;49:1601339.
Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, Kreuter M, Lynch DA, Maher TM, Martinez FJ, Molina-Molina M, Myers JL, Nicholson AG, Ryerson CJ, Strek ME, Troy LK, Wijsenbeek M, Mammen MJ, Hossain T, Bissell BD, Herman DD, Hon SM, Kheir F, Khor YH, Macrea M, Antoniou KM, Bouros D, Buendia-Roldan I, Caro F, Crestani B, Ho L, Morisset J, Olson AL, Podolanczuk A, Poletti V, Selman M, Ewing T, Jones S, Knight SL, Ghazipura M, Wilson KC. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18-47.
Taniguchi H, Kondoh Y. Revised criteria for acute exacerbation of idiopathic pulmonary fibrosis. The annual report by study group of Ministry of Health and Welfare for diffuse lung disease. Diffuse Lung Diseases Research Group from the Ministry of Health, Labor, and Welfare of Japanese Government; 2004. pp. 114–9.
Bestall JC, Paul EA, Garrod R, Garnham R, Jones PW, Wedzicha JA. Usefulness of the Medical Research Council (MRC) dyspnoea scale as a measure of disability in patients with chronic obstructive pulmonary disease. Thorax. 1999;54(7):581–6.
Japanese Respiratory Society’s committee formulating diagnosis and treatment guideline for diffuse lung diseases. Clinical diagnostic and treatment guidelines for idiopathic interstitial pneumonias. Tokyo: Nankodo; 2004. pp. 63–5.
Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE Jr, Collard HR. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–91.
Kondoh S, Chiba H, Nishikori H, Umeda Y, Kuronuma K, Otsuka M, Yamada G, Ohnishi H, Mori M, Kondoh Y, Taniguchi H, Homma S, Takahashi H. Validation of the Japanese disease severity classification and the GAP model in Japanese patients with idiopathic pulmonary fibrosis. Respir Investig. 2016;54:327–33.
Azuma A, Kudoh S. High prevalence of drug-induced pneumonia in Japan. Jpn Med Assoc J. 2007;50:405–11.
Kondoh Y, Taniguchi H, Kawabata Y, Yokoi T, Suzuki K, Takagi K. Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest. 1993;103:1808–12.
Akira M, Hamada H, Sakatani M, Kobayashi C, Nishioka M, Yamamoto S. CT findings during phase of accelerated deterioration in patients with idiopathic pulmonary fibrosis. AJR Am J Roentgenol. 1997;168:79–83.
Kondoh Y, Taniguchi H, Katsuta T, Kataoka K, Kimura T, Nishiyama O, Sakamoto K, Johkoh T, Nishimura M, Ono K, Kitaichi M. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27(2):103–10.
Collard HR, King TE, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168:538–42.
Jegal Y, Kim DS, Shim TS, Lim CM, Do Lee S, Koh Y, Kim WS, Kim WD, Lee JS, Travis WD, Kitaichi M, Colby TV. Physiology is a stronger predictor of survival than pathology in fibrotic interstitial pneumonia. Am J Respir Crit Care Med. 2005;171:639–44.
Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, von Pawel J, Gottfried M, Bondarenko I, Liao M, Gann CN, Barrueco J, Gaschler-Markefski B, Novello S, LUME-Lung 1 Study Group. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomized controlled trial. Lancet Oncol. 2014;15:143–55.
Otsubo K, Kishimoto J, Ando M, Kenmotsu H, Minegishi Y, Horinouchi H, Kato T, Ichihara E, Kondo M, Atagi S, Tamiya M, Ikeda S, Harada T, Takemoto S, Hayashi H, Nakatomi K, Kimura Y, Kondoh Y, Kusumoto M, Ichikado K, Yamamoto N, Nakagawa K, Nakanishi Y, Okamoto I. Nintedanib plus chemotherapy for non-small cell lung cancer with IPF: a randomised phase 3 trial. Eur Respir J. 2022;60:2200380.
Kato M, Sasaki S, Nakamura T, Kurokawa K, Yamada T, Ochi Y, Ihara H, Takahashi F, Takahashi K. Gastrointestinal adverse effects of nintedanib and the associated risk factors in patients with idiopathic pulmonary fibrosis. Sci Rep. 2019;9: 12062.
Acknowledgements
The authors thank Mrs. Kakumi Sasaki for her assistance in data entry and Crimson Interactive Pvt. Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors received no specific funding for this work.
Conflict of interest
Kotaro Iwasaki, Hiroki Wakabayashi, Atsuhito Saiki, Hajime Ueshiba, Yu Murakami, and Yasuo Matsuzawa declare that they have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Ethics approval and consent to participate:
This study and consent procedure were approved by the Ethics Committee of Toho University Sakura Medical Center (approval number: S21084; approval date: 11 October 2022). We utilized an opt-out method to obtain patient consent regarding the use of their personal information, by publishing the research content and our policy on personal information on the hospital website.
Consent for publication
Not applicable.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
Author contributions
Conceptualization: YM and IK; data collection: IK, HW, and YM; formal analysis: YM and IK; supervision: AS and HU; writing—original draft preparation: IK; writing—review and editing: YM; approval of final manuscript: all authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Iwasaki, K., Wakabayashi, H., Saiki, A. et al. Real-World Clinical Efficacy of Antifibrotic Agents for Idiopathic Pulmonary Fibrosis: A Single-Center Retrospective Study in Japan. Drugs - Real World Outcomes 11, 43–52 (2024). https://doi.org/10.1007/s40801-023-00396-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40801-023-00396-w