
Abstract
Purpose of Review
This review focuses on the opposing effects on the immune system of radiotherapy (RT) and the consequences for combined cancer treatment strategies of RT with immunotherapies, including hyperthermia (HT). How RT and HT might affect cancer stem cell populations is also briefly outlined in this context.
Recent Findings
RT is one of the crucial standard cancer therapies. Most patients with solid tumors receive RT for curative and palliative purposes in the course of their disease. RT achieves a local tumor control by inducing DNA damage which can lead to tumor cell death. In recent years, it has become evident that RT does not only have local effects, but also systemic effects which involves induction of anti-tumor immunity and possible alteration of the immunosuppressive properties of the tumor microenvironment. Though, often RT alone is not able to induce potent anti-tumor immune responses since the effects of RT on the immune system can be both immunostimulatory and immunosuppressive.
Summary
RT with additional therapies such as HT and immune checkpoint inhibitors (ICI) are promising approaches to induce anti-tumor immunity effectively. HT is not only a potent sensitizer for RT, but it might also improve the efficacy of RT and certain chemotherapeutic agents (CT) by additionally sensitizing resistant cancer stem cells (CSCs).
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer management is still a huge challenge to modern medicine. There were 19.3 million new cases of cancer diagnosed in 2020 and almost 10 million deaths were caused by cancer worldwide [1]. Radiotherapy (RT) is one of the most crucial cancer treatments following surgery and chemotherapy. Approximately 60% of patients with solid tumors receive RT in the course of their disease and the need for this classical tumor therapy tends to increase in the next years [2]. For cancer therapy, ionizing radiation is used to achieve local tumor control by inducing cancer cell death and inhibition of the cancer cell proliferation. DNA damage, such as single- and double-strand breaks, induced by ionizing radiation is either caused directly or indirectly by high oxidative stress caused by ionization of other molecules [3]. If clusters of DNA damage accumulate beyond the cells repair capabilities, it causes cancer cell death or senescence [4]. The severity of DNA damage from RT is also known to be influenced by the state of the tumor microenvironment (TME), specifically if it is hypoxic or not [4, 5]. Unfortunately, normal cells are affected by ionizing radiation as well. However, since as tumor cells have an impaired DNA repair machinery, damage to normal tissue can be minimized by applying RT in fractions of smaller doses. Furthermore, the implementation of advanced modulated beam therapies such as intensity-modulated RT and spatially fractionated RT improve the efficacy of RT at the same time with decreased damage to normal tissue [6].
Additionally, RT has also some non-targeted, systemic effects. For a long time, RT was known for its immunosuppressive properties thus has been used to condition the immune system before hematopoietic stem cell transplantation. In recent years, however, the immunostimulatory effects of RT are being increasingly recognized as an effective way to induce anti-tumor immune responses. In patients with advanced cancer, local RT has the ability to induce systemic anti-tumor responses [7]. Due to its immunostimulatory properties, RT is now being investigated in combination with other therapies, specifically immunotherapy (IT), in several clinical trials, and it already showed improvement of local and systemic tumor control in e.g. head and neck cancer as well as lung cancer [7, 8]. The idea to exploit the patient’s own immune system to fight the cancer by using immunotherapies has always been in interest. In recent decades immunotherapies have been developed and applied to induce anti-tumor immune responses in different settings. However, the response rate of the immunotherapies is so far relatively low. Thus, applying RT with further immune modulation, e.g., by hyperthermia (HT) treatment is an ideal option. In this review, we will briefly discuss how RT affects the anti-tumor immune response and how it modulates the tumor microenvironment. Possibilities to combine RT with IT and with further immunomodulation by HT are outlined. Lastly, the effects of RT and HT on cancer stem cells (CSCs) will be addressed.
Immune Modulatory Effects of RT
RT has been long time known only for its local effects. However, there are also non-targeted effects (NTE) [4, 9] of RT that are caused by the release of danger signals and cytokines from the RT-treated area into the surrounding tissue [10, 11]. There are two key types of NTE, a) “bystander effects” which determine the tumor regression of non-irradiated area surrounding tumor sites after local RT and b) “abscopal effects” which is tumor regression in distant tumor sites [12]. The latter are known from clinical observations of tumor regression outside the irradiation field in tumor masses elsewhere in the body. This was the very first evidence that RT stimulates the immune system. These findings were later supported by preclinical studies and proof-of-principle clinical studies [13,14,15] which confirmed that these effects are immune-mediated, strongly via the induction of immunogenic tumor cell death (ICD) [16, 17]. Unfortunately, these NTE occur rarely after RT alone, thus indicating that RT as single therapy is not enough to induce an anti-tumor immune response [18]. Recently, immunotherapies (IT) in combination with RT are currently tested in clinical trials to improve the response rate of IT and improve the immune activating effects of RT and in the ideal case the overall patient survival [19].
Moreover, after RT, the expression of MHC molecules, stress ligands, and death receptors increase on the surface of tumor cells [20, 21••]. Ionizing radiation also modifies the tumor immune phenotype and its microenvironment [22,23,24]. When tumor cells escape from host anti-tumor immune response, they develop their own microenvironment to support the tumor cell growth and to protect them from the host’s immune system by harnessing immune cells and avoid cancer immune surveillance [25]. This self-sustaining TME is a very complex system with different cellular components that each have their distinct role. The TME includes supporting cells, such as fibroblasts and endothelial cells, and cells of the adaptive and innate immune system [26]. Furthermore, it is enriched with immunosuppressive chemokines and cytokines, which together with the tumor-associated immune cells, resulting in further tumor progression and immune suppression, including a reduced function of cytotoxic T-lymphocytes (CTL) and natural killer (NK) cells [27]. Furthermore, tumor cells express immune inhibitory checkpoint molecules (ICMs) on their surface (e.g., programmed death ligand 1 and 2 (PD-L1, PD-L2), and herpesvirus entry mediator (HVEM)) to inhibit T cell responses against tumor cells. These inhibitory ICMs are normally used by antigen-presenting cells (APCs) physiologically to downregulate the T cell response after a pathogen is eliminated to prevent T cells to attack their own body cells [28]. Thus, tumor cells mask themselves with these inhibitory ICMs to successfully escape from immune surveillance.
However, RT can alter this immunosuppressive TME by inducing immunogenic tumor cell death (ICD). ICDs is cell death forms that induce the release of damage-associated molecular pattern (DAMPs) which happens typically when cells lose their integrity after necrosis and necroptosis [29]. DAMPs are normally present only inside of the cells, but when they are released outside of the cell, they attract and activate cells of the innate and adaptive immune system. The well-known DAMPs that are induced by RT includes chromatin–binding protein high mobility group box 1 (HMGB1), calreticulin, adenosine triphosphate (ATP), and heat shock proteins (HSPs) [18, 30•, 31]. Each of these DAMPs have different effects, for example, HMGB1 facilitates tumor antigen presentation and type I interferon–mediated dendritic cell (DC) maturation [32]. Type I interferons (IFN α and IFNβ) stimulate DCs to cross-present the antigen to T cells and further facilitate T cell responses [33]. HSPs (preferably HSP70, HSP90) and calreticulin act as an “eat me” signal to APCs leading to the release of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and IL-6, which further lead to the activation of other immune cells [31, 34]. Additionally, HSPs activate NK cells [35] and facilitate the APC’s cross presentation of antigen to CD8+ T cells, ideally leading to their activation and subsequent T cell–mediated eradication of tumor cells [36, 37] (Fig. 1). ATP attracts DCs into the tumor and promotes phagocytic clearance of tumor cells [38]. Moreover, RT modulates the immunogenicity of the tumor cells by increasing the expression of stimulatory molecules, such as cluster of differentiation 80 (CD80), and major histocompatibility complex class 1 (MHC I) [39,40,41].

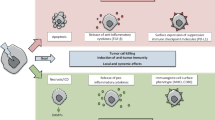
The effects of ionizing radiation on the immune system. Radiotherapy (RT) has both immune stimulatory and immune inhibitory effects. After RT, tumor cells also die of necrosis, resulting in the release of damage associated molecular patterns (DAMPs). DAMPs including HSPs and HGMB1 activate dendritic cells. HSPs bound to tumor antigen facilitate antigen presentation by e.g. DCs and initiate cytotoxic T cell responses against tumor cells. However, on the other hand when tumor cells are treated with RT, it can also lead to immunosuppression. When tumor cells die of apoptosis, DAMPs are not released, instead apoptotic cells are taken up by phagocytes which release anti-inflammatory signals, such as TGF-β and CCL-2. Latter induces differentiation of tumor-associated macrophages (TAM) which promotes tumor progression. Also, tumor cells inhibit T cell activation by upregulating the expression of immune inhibitory checkpoint molecules, thus escaping from the T cell–mediated tumor clearance. DAMPs, damage-associated molecular patterns; DC, dendritic cell; HSP, heat shock protein; HMGB1, high mobility group box protein 1; ATP, adenosine triphosphate; MHC I, major histocompatibility complex class 1; TGF-β, tumor growth factor β; TAM, tumor-associated macrophages; CCL2, monocyte chemoattractant protein-1; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T-lymphocyte–associated protein 4
Besides its immunostimulatory effects, RT is also known for its immunosuppressive properties [42]. Unlike necrosis, apoptosis is a non- or even anti-inflammatory cell death form (Fig. 1), as the cell membrane remains intact, and thus danger signals are not released [43,44,45]. Furthermore, when cells undergo apoptosis, they expose phosphatidylserine on the outer membrane leaflet, which serves as phagocytic signal for particularly macrophages. The latter thereby release anti-inflammatory cytokines, such as TGF- β [44, 46]. Additionally, they increase the expression of immune inhibitory checkpoint molecules like PD-L1 to make the tumor microenvironment immune suppressive and these effects can even be amplified by RT [47,48,49]. Furthermore, RT induces the production of chemokines that attract immune cells into the tumor to sustain the immune suppressive TME [50]. CCL2 is one of those chemokines attracting monocytes into the tumor, which then differentiate into tumor-associated macrophages (TAMs) and further promote tumor growth [51].
Based on these opposing effects of RT with immunosuppression counteracting the stimulation by ICD induction, RT alone often does not induce anti-tumor responses effectively. Fortunately, recently preclinical and clinical studies have shown that by complementing RT with additional immunotherapies, RT-induced anti-tumor immune responses can be augmented and the immune suppressive effects of RT can be reduced [30•, 52, 53].
The immunological effects of RT are highly influenced by radiation dose and fractionation. However, no “perfect” immune-stimulating radiation dose and fractionation scheme has been identified to date, as it also depends on the size and stage of the tumor and numerous other factors. But generally speaking, in most tumor entities, RT of 1.8–2.0 Gray (Gy) per day, 5 times a week, for 5–6 weeks is used for solid tumors. Hypofractionation with a single dose higher than 2.0 Gy per day was reported to have a better outcome and modulates the immune response by causing more immunogenic cell death compared to lower dose radiation [12, 54, 55]. In preclinical studies, hypo-fractionated RT of 8 Gy ×3 was more effective to induce abscopal effects compared to a single fractionation of 20 Gy when it is combined with ICIs [56, 57], but 8 Gy ×3 is also less immunogenic as 8 Gy ×2 when being combined with an autologous tumor cell–ased vaccine [58]. It has already also clinically been proven that locally delivered RT induces systemic modulation of the immune system in tumor patients, but different RT approaches appear to specifically affect distinct immune components [59].
Combination of Radiotherapy and Immunotherapies
Cancer immunotherapy is increasingly becoming one of the cornerstones of cancer treatments. The ability of the immune system to recognize and to eliminate tumors was discovered more than a century ago and ever since broadly studied and clinically applied in the form of immunotherapy successfully in recent years. Consequently, (re-)inducing anti-tumor immune responses by inactivating inhibitory immune receptors (a.k.a., immune checkpoint molecules (ICMs)) has been developed and initially used as a monotherapy and showed great success [60,61,62]. Immune checkpoint inhibitors (ICIs) directed against anti-cytotoxic T-lymphocyte–associated protein a (CTLA-4) and the PD-1/PD-L1 axis successfully (re)activates the immune response and improves the survival of advanced stage cancer patients [60, 62, 63]. ICIs are clinically approved for many tumor entities but despite the promising results the response rate of ICIs is still lower than 15% [64]. It has become clear that when RT is used in combination with IT, local tumor control is improved and even systemic anti-tumor immune response can be induced. ITs that are used in combination with RT include cytokines (e.g., IL-12, IFN), growth factors (e.g., Flt3-L), and immune checkpoint inhibitors (ICIs) (anti-CTLA-4, anti-PD1, anti-HVEM). IT improves the clinical outcome and local tumor control [65,66,67]. The efficacy of RT is more prominent when it is combined with IT. There are several factors that affect the effectiveness of RT and IT combinations such as tumor burden, immunotolerance, and scheduling of RT and IT [56, 61]. Nevertheless, there has been extensive studies tried to explore the responsible mechanisms and predictive markers and combination treatment options to increase the efficacy of ICIs. One major predictive marker of the response of PD-L1 inhibitor is the amount of cytotoxic CD8+ effector T cells infiltrating the tumor site [68•, 69]. Therefore, it has been suggested that therapies which increase the infiltration of CTLs into tumor sites like RT would increase response to IT.
It has been further observed that the expression of PD-L1 increases on tumor cells following irradiation [70, 71], thus suppressing antigen presentation of APCs and dampen CTLs in effector phase [63]. Thus, using RT in combination with anti-PD-1/anti-PD-L1 will improve the number of responders and also at the same time effectiveness of both therapies [21••, 30•]. Recently, the number of clinical trials that study the efficacy and the safety of the ICIs are increasing and even some clinical trials reported that RT does not increase the ICI-related side effects [72, 73]. There are also other novel strategies of RT and IT combination being developed, such as the combination of RT with agonists of immune costimulatory molecules like OX-40L and CD40 [65, 74,75,76]. However, combining these treatments is a complex matter and several factors should be considered when planning the treatments. The timing of the RT and ICIs is one of the most important factors to consider when these treatments are combined. For example, it has been reported that giving CTLA-4 inhibitors 7 days prior to irradiation is much more effective than giving it 1 or 7 days after the treatment in a CT26 murine model [77]. PD-1 inhibitors showed best results when they are given simultaneously with RT compared to 7 days after the irradiation [68•, 78]. In contrast, the agonistic OX40 antibody is better used right after RT [77]. Each ICI has different functions and thus RT should be matched with the individual property of each ICI.
Another factor to consider is the tumor burden; high tumor burden is negatively correlated with the efficacy of ICI [79]. In preclinical data, a smaller tumor volume is correlated with better response of PD-1 inhibitors in non-small cell lung cancer (NSCLC) [80], and vice versa large and advanced stage tumors are resistant to PD-1 inhibitors [81]. This negative correlation was also observed in patients with metastatic melanoma [79, 82]. Higher tumor volume was predictive of poor response to PD-1 inhibitors [79, 82]. NSCLC patients with high metabolic tumor volume had poor response to PD-1 inhibitors and overall survival [83]. However, the exact threshold to describe low and high tumor burden is not determined yet, thus other measures which are more dynamic than the tumor size, for example, Huang et.al suggested the ratio of exhausted T cell subpopulation to pretreatment tumor burden is more suitable to predict which patients will benefit more from ICIs [84, 85]. Also, high tumor mutation burden was an effective predictor of anti-PD-1 response [86]. Taken together, there is increasing evidence in preclinical and clinical settings that a combination of RT and IT improves the immunogenicity of the tumor and increases the efficacy of both treatments. To conclude, several factors have to be considered to achieve the most synergistic effects and high response rates. The efficacy of RT can further be optimized, especially regarding ways to reduce radioresistance. Tumor hypoxia is one of the main reasons of radiotherapy resistance. This can be addressed by additionally applying hyperthermia treatment.
Hyperthermia as an Additional Immunomodulator
HT is an anti-cancer treatment which locally heats the tumor to supraphysiological temperatures (39–45 °C), and is a potent sensitizer for radio- and chemotherapies (CT). HT is commonly used in clinical settings as a combination with RT and CT in various cancer entities and has shown to significantly improve local tumor control and survival of the patients without inducing severe side effects [87,88,89,90,91,92].
Regarding the key modes of actions, HT can improve the delivery of oxygen into predisposed hypoxic tumor tissue [93]. However, one should keep in mind that due to chaotic tumor vasculature the relative increase in blood flow is only about two-folds in tumor tissue and exhibits pronounced heterogeneity compared to normal tissues [94]. Furthermore, heating tissues aggravates the protein aggregation and DNA damage which makes the tumor more sensitive to RT and CT [95, 96]. HT has direct killing effects on tumor cells, starting with temperatures of about 43 °C [97] and has been proven to modulate the innate and adaptive immune system by inducing anti-tumor immune responses in direct and indirect ways [95, 98] (Fig. 2). The local effects on tumor cells are mediated by following key mechanisms: cause of membrane dysfunction, protein denaturation and aggregation, inhibition of DNA repair mechanisms, and induction of cancer cell death by necrosis and apoptosis [99]. By increasing the perfusion, HT can increase the immune cell infiltration into the tumor tissue [100]. HT-induced cancer cell death causes release of DAMPs, such as HMGB1, HSPs, and DNA as well as RNA fragments of cancer cells which are highly immunogenic [101, 102] (Fig. 2). Once these DAMPs are taken up by dendritic cells and macrophages, it further triggers the anti-tumor immune response [95, 98, 103]. Specifically, HSPs released by HT promotes antigen presentation and activates NK and CTLs, thus providing a potent anti-tumor immune response [104,105,106]. Also, when HT is combined with RT, it significantly induces more necrosis which leads to increased release of DAMPs, further inducing the anti-tumor immune response more effectively [107].

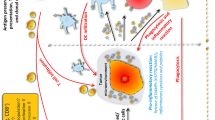
The effect of radiotherapy (RT) in combination with immune checkpoint inhibitors and hyperthermia treatment on the immune system. a After RT certain inhibitory immune checkpoint molecules (ICMs) are upregulated on tumor cells. ICIs (anti-PD-L1, anti-CTLA-4) that inhibit these inhibitory ICMs will diminish the effects of these inhibitory ICMs on T cell activation and further activate anti-tumor immune response. DC’s cross presentation of tumor antigen with MHCI molecule leads to activation of CTLs which eliminate tumor cells. b HT sensitizes the tumor for RT. It boosts the effect of RT by increasing tumor cell death and release of DAMPs. DAMPs that are released from tumor cells (HSPs, HMGB1, DNA, RNA fragments, and other antigens) are taken up by a DC which further activates CTLs leading to the activation of adaptive anti-tumor immune responses. HSPs also activate NK cells. HT increases tumor blood supply and immune cell infiltration into the tumor site. All these effects are leading to the activation of innate and adaptive anti-tumor immune responses. DAMPs, damage-associated molecular patterns; HSP, heat shock protein; HMGB1, high mobility group box protein 1; MHC I, major histocompatibility complex class 1; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T-lymphocyte–associated protein 4; TLR, toll-like receptor; OX40, CD134; OX40-L, OX40 ligand
Besides the positive effects of adding HT to RT, CT also benefits from additional HT. HT improves the perfusion and membrane permeability of the tumor cells, and thus augments the effects of certain chemotherapeutic agents (summarized in [108•]). Due to these beneficial properties, HT is implemented in multimodal treatment settings. The initial clinical trials, such as HYCAN trial (ClinicalTrials.gov Identifier: NCT02369939) of anal carcinoma reports that when HT is added to radiochemotherapy the cells of the innate immune system recover faster in peripheral blood [21••].
The Effects of Radiotherapy and Hyperthermia on Cancer Stem Cells
Cancer stem cells (CSCs) are a small population of tumor cells that has the ability to regenerate and differentiate into tumor cells when transplanted into another animal host. It is believed that tumor cells originated from these CSCs, and CSCs are responsible for tumor metastasis [99]. CSCs have important clinical relevance because CSCs are highly resistant to standard RT and CT, and they are an important target to anti-cancer therapies [109]. CSCs are also thought to be responsible for the drug resistance of tumors [110]. CSCs activate the DNA damage checkpoints in response to irradiation making the DNA repair system of tumor cells more precise [111]. Furthermore, they upregulate the expression of ROS scavenger proteins to decrease the amount of DNA damage caused by RT [99]. Also the environment where CSCs reside plays an important role in its resistance to RT and CT [112]. They reside in hypoxic areas without rich blood vessels, thus exposure to chemotherapeutic agents is decreased. The low level of ROS in these hypoxic areas also results in decreased DNA damage caused by free radicals after RT. Additionally, CSCs can upregulate BCL2 family proteins to protect themselves from the cytotoxic effects of CT [113]. Likewise, CSCs have the ability to protect themselves from the effects of RT and CT in many ways, and the CSCs that remain after the treatments are believed to cause cancer recurrence [114].
The origin of the CSCs is still controversial. Whether cancer cells gained the abilities of stem cells or distinct CSCs exist from the beginning is still not clear, and there is currently no specific biomarker to successfully distinguish CSCs from normal stem cells [115]. This lack of specific biomarkers is a current obstacle in CSC research and the development for treatments that target CSCs [30•, 115]. CSCs evade from immune system in various ways, such as inhibiting the antigen presentation and T cell activation [44] and by actively suppressing the immune system. Because of all these functions of CSCs, they are highly resistant to single cancer therapy, thus in order to eradicate cancer cells it is necessary to combine RT and CT with additional treatments, such as HT and IT [114].
HT has the potential to eliminate CSCs and sensitize them for RT and CT [99, 116]. It can improve the efficacy of RT and CT in many mechanisms. First, HT can specifically affect the hypoxic tumor areas with poor blood supply by increasing the perfusion, thus increasing the oxygen supply and ROS in that area. Second, HT can inhibit multiple DNA repair pathways that CSCs upregulate to protect themselves. Third, HT can change the immunosuppressive microenvironment of the tumor and enhance anti-tumor immune response to eliminate CSCs. Finally, HT can improve CSC’s low immunogenicity by increasing cancer cell death, revealing CSC’s antigen to APCs. However, these effects of HT can vary depending on whether it is used in combination with RT and CT, and also from the tumor entity itself [117]. In some tumor entities including breast cancer [117] and glioblastoma [118], HT alone did not have significant effect on CSCs, though when combined with RT it effectively reduced CSCs regeneration and proliferation. Based on all these facts, targeting CSCs with HT in combination with other treatment strategies shows huge promise and may lead to the eradication of CSCs, preventing the recurrence of the tumor.
Conclusions
RT does not only have local cell killing effects on tumor cells, but it also has a systemic effect which is caused by the activation of anti-tumor immune responses. Recently, it has been shown that RT can elicit anti-tumor immunity by inducing ICD and reshaping the tumor microenvironment. However, RT has both immune suppressive and stimulatory effects, and thus RT alone often is not able to induce anti-tumor immunity effectively. This is partly due to the fact that after RT tumor cells suppress the immune system by upregulating immune suppressive ICMs. Thus, it is beneficial to use RT with additional ITs, which further boost RT’s effects and enhance the anti-tumor immune response. Furthermore, when using RT in combinational settings, RT planning should be strongly considered. Additionally, to promote RT’s effects, HT can be combined with RT to sensitize cancer cells to RT and counteract the radioresistance of cancer cells. HT is not only a potent sensitizer for RT and CT, but it can also effectively contribute to induce anti-tumor immune responses, increasing the efficacy of RT and CT. Furthermore, multimodal treatments of radiochemotherapy together with HT can target CSCs, which are resistant to RT and CT alone and further prevent tumor metastases and recurrence.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ferlay J, et al. Cancer statistics for the year 2020: An overview. Int J Cancer. 2021;149(4):778–89.
Borras JM, et al. How many new cancer patients in Europe will require radiotherapy by 2025? An ESTRO-HERO analysis. Radiother Oncol. 2016;119(1):5–11.
Jeggo P, Löbrich M. Radiation-induced DNA damage responses. Radiat Prot Dosimetry. 2006;122(1–4):124–7.
Lomax ME, Folkes LK, O’Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin Oncol. 2013;25(10):578–85.
Saitoh T, Oda T. DNA damage response in multiple myeloma: the role of the tumor microenvironment. Cancers. 2021;13(3):504.
Asur R, et al. High dose bystander effects in spatially fractionated radiation therapy. Cancer Lett. 2015;356(1):52–7.
D’Andrea MA, Reddy GK. Systemic immunostimulatory effects of radiation therapy improves the outcomes of patients with advanced NSCLC receiving immunotherapy. Am J Clin Oncol. 2020;43(3):218–28.
Daly ME, Monjazeb AM, Kelly K. Clinical trials integrating immunotherapy and radiation for non–small-cell lung cancer. J Thorac Oncol. 2015;10(12):1685–93.
Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3.
Nikitaki Z, et al. Systemic mechanisms and effects of ionizing radiation: A new ‘old’ paradigm of how the bystanders and distant can become the players. Semin Cancer Biol. 2016;37–38:77–95.
Kadhim MA, Hill MA. Non-targeted effects of radiation exposure: recent advances and implications: Figure 1. Radiat Prot Dosimetry. 2015;166(1–4):118–24.
Wang Y. Advances in hypofractionated irradiation-induced immunosuppression of tumor microenvironment. Front Immunol. 2021;11:612072.
Demaria S, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–70.
Rodriguez-Ruiz ME, et al. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Can Res. 2016;76(20):5994–6005.
Golden EB, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015;16(7):795–803.
Zhu M, et al. Immunogenic cell death induction by ionizing radiation. Front Immunol. 2021;12:705361.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11–7.
Walle T, et al. Radiation effects on antitumor immune responses: current perspectives and challenges. Ther Adv Med Oncol. 2018;10:175883401774257.
Johnson CB, Jagsi R. The promise of the abscopal effect and the future of trials combining immunotherapy and radiation therapy. Int J Radiat Oncol Biol Phys. 2016;95(4):1254–6.
Frey B, et al. Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol Immunother. 2014;63(1):29–36.
•• Hader M, et al. Immune biological rationales for the design of combined radio- and immunotherapies. Cancer Immunol Immunother. 2020;69(2):293–306. Key aspects are reviewed that suggest the use of hyperthermia treatment as a adjuvant treatment for radiotherapy and immunotherapies based on several direct and indirect effects of these treatments on the immune system.
Ngwa W, et al. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18(5):313–22.
Jarosz-Biej M, et al. Tumor microenvironment as a “game changer” in cancer radiotherapy. Int J Mol Sci. 2019;20(13):3212.
Colton M, et al. Reprogramming the tumour microenvironment by radiotherapy: implications for radiotherapy and immunotherapy combinations. Radiat Oncol. 2020;15(1):254.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22(1):329–60.
Giraldo NA, et al. The clinical role of the TME in solid cancer. Br J Cancer. 2018;120(1):45–53.
Yu YR, Ho PC. Sculpting tumor microenvironment with immune system: from immunometabolism to immunoediting. Clin Exp Immunol. 2019;197(2):153–60.
Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47(5):765–79.
Krysko O, et al. Necroptotic cell death in anti-cancer therapy. Immunol Rev. 2017;280(1):207–19.
• Rückert M, et al. Radiotherapy and the immune system: more than just immune suppression. Stem Cells. 2021;39(9):1155–65. In this review, both the stimulating and suppressing effects of radiotherapy are explained in detail, and the use of radiotherapy in combination with distintc immune therapies is discussed.
Keam S, et al. Enhancing the efficacy of immunotherapy using radiotherapy. Clin Transl Immunol. 2020;9(9):e1169.
Apetoh L, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9.
Burnette BC, et al. The efficacy of radiotherapy relies upon induction of type I interferon–dependent innate and adaptive immunity. Can Res. 2011;71(7):2488–96.
Galluzzi L, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2016;17(2):97–111.
Schmid TE, Multhoff G. Radiation-induced stress proteins - the role of heat shock proteins (HSP) in anti- tumor responses. Curr Med Chem. 2012;19(12):1765–70.
Skitzki JJ, Repasky EA, Evans SS. Hyperthermia as an immunotherapy strategy for cancer. Curr Opin Investig Drugs. 2009;10(6):550–8.
Werthmöller N, et al. Combination of ionising radiation with hyperthermia increases the immunogenic potential of B16–F10 melanoma cells in vitro and in vivo. Int J Hyperth. 2016;32(1):23–30.
Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–6.
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–65.
Reits EA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Kaur P, Asea A. Radiation-induced effects and the immune system in cancer. Front Oncol. 2012;2:191.
Carvalho HA, Villar RC. Radiotherapy and immune response: the systemic effects of a local treatment. Clinics (Sao Paulo). 2018;73(suppl 1):e557s.
Voll RE, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390(6658):350–1.
Zhang W-J, Zheng S-S. In vitro study of immunosuppressive effect of apoptotic cells. J Zhejiang Univ Sci. 2005;6B(9):919–25.
Willems JJLP, Arnold BP, Gregory CD. Sinister self-sacrifice: the contribution of apoptosis to malignancy. Front Immunol. 2014;5:299.
Chen W, et al. TGF-β released by apoptotic T Cells contributes to an immunosuppressive milieu. Immunity. 2001;14(6):715–25.
Wu C-T, et al. The role of PD-L1 in the radiation response and clinical outcome for bladder cancer. Sci Rep. 2016;6(1):19740.
Sato H, Okonogi N, Nakano T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int J Clin Oncol. 2020;25(5):801–9.
Derer A, et al. Chemoradiation increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. 2016;7:610.
Wennerberg E, et al. Barriers to radiation-induced in situ tumor vaccination. Front Immunol. 2017;8:229.
Mondini M, et al. CCR2-dependent recruitment of tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol Res. 2019;7(3):376–87.
Cushman TR, et al. Combining radiation plus immunotherapy to improve systemic immune response. J Thorac Dis. 2018;10(S3):S468–79.
Arina A, Gutiontov SI, Weichselbaum RR. Radiotherapy and Immunotherapy for cancer: from “systemic” to “multisite.” Clin Cancer Res. 2020;26(12):2777–82.
Chajon E, et al. The synergistic effect of radiotherapy and immunotherapy: a promising but not simple partnership. Crit Rev Oncol Hematol. 2017;111:124–32.
Kim M-S, et al. Radiobiological mechanisms of stereotactic body radiation therapy and stereotactic radiation surgery. Radiat Oncol J. 2015;33(4):265.
Demaria S, et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J Immunother Cancer. 2021;9(4):e002038.
Dewan MZ, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti–CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Rückert M, et al. Combinations of radiotherapy with vaccination and immune checkpoint inhibition differently affect primary and abscopal tumor growth and the tumor microenvironment. Cancers. 2021;13(4):714.
Frey B, et al. Systemic modulation of stress and immune parameters in patients treated for prostate adenocarcinoma by intensity-modulated radiation therapy or stereotactic ablative body radiotherapy. Strahlenther Onkol. 2020;196(11):1018–33.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018;33(4):581–98.
Okazaki T, et al. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–8.
Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86.
Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019;2(5):e192535.
Mondini M, et al. Radiotherapy–immunotherapy combinations – perspectives and challenges. Mol Oncol. 2020;14(7):1529–37.
Twyman-Saint Victor C, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520(7547):373–7.
Antonia SJ, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N Engl J Med. 2017;377(20):1919–29.
• Kabiljo J, et al. Radiotherapy as a backbone for novel concepts in cancer immunotherapy. Cancers. 2019;12(1):79. Evidence is provided that when radiotherapy is combined with immunotherapy, it locally induces immunogenic cell death and reconditions the tumor microenvironment. The preclinical insights of the combined treatments are explained and how these can be translated into clinical studies.
Uryvaev A, et al. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med Oncol. 2018;35(3):25.
Narits J, Tamm H, Jaal J. PD-L1 induction in tumor tissue after hypofractionated thoracic radiotherapy for non-small cell lung cancer. Clin Transl Radiat Oncol. 2020;22:83–7.
Kordbacheh T, et al. Radiotherapy and anti-PD-1/PD-L1 combinations in lung cancer: building better translational research platforms. Ann Oncol. 2018;29(2):301–10.
Hecht M, et al. Safety and efficacy of single cycle induction treatment with cisplatin/docetaxel/ durvalumab/tremelimumab in locally advanced HNSCC: first results of CheckRad-CD8. J Immunother Cancer. 2020;8(2):e001378.
Xing D, Siva S, Hanna GG. The abscopal effect of stereotactic radiotherapy and immunotherapy: fool’s gold or El Dorado? Clin Oncol. 2019;31(7):432–43.
Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
Yokouchi H, et al. Anti-OX40 monoclonal antibody therapy in combination with radiotherapy results in therapeutic antitumor immunity to murine lung cancer. Cancer Sci. 2008;99(2):361–7.
Gough MJ, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33(8):798–809.
Mattei F, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. Plos One. 2016;11(6):e0157164.
Dovedi SJ, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Can Res. 2014;74(19):5458–68.
Kim SI, Cassella CR, Byrne KT. Tumor burden and immunotherapy: impact on immune infiltration and therapeutic outcomes. Front Immunol. 2021;11:629722.
Guisier F, et al. A rationale for surgical debulking to improve anti-PD1 therapy outcome in non small cell lung cancer. Sci Rep. 2019;9(1):16902.
Duraiswamy J, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors–response. Can Res. 2014;74(2):633–4.
Joseph RW, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res. 2018;24(20):4960–7.
Chardin D, et al. Baseline metabolic tumor volume as a strong predictive and prognostic biomarker in patients with non-small cell lung cancer treated with PD1 inhibitors: a prospective study. J Immunother Cancer. 2020;8(2):e000645.
Huang AC, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–5.
Hossain MA, et al. Reinvigorating exhausted CD8+cytotoxic T lymphocytes in the tumor microenvironment and current strategies in cancer immunotherapy. Med Res Rev. 2020;41(1):156–201.
Zheng Y, Yao M, Yang Y. Higher tumor mutation burden was a predictor for better outcome for NSCLC patients treated with PD-1 antibodies: a systematic review and meta-analysis. SLAS Technol. 2021;26(6):605–14.
International Collaborative Hyperthermia Group, et al. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: results from five randomized controlled trials. Int J Radiat Oncol Biol Phys. 1996;35(4):731–44.
Overgaard J, et al. Randomised trial of hyperthermia as adjuvant to radiotherapy for recurrent or metastatic malignant melanoma. Lancet. 1995;345(8949):540–3.
van der Zee J, et al. Comparison of radiotherapy alone with radiotherapy plus hyperthermia in locally advanced pelvic tumours: a prospective, randomised, multicentre trial. Lancet. 2000;355(9210):1119–25.
Ott OJ, et al. Chemoradiotherapy with and without deep regional hyperthermia for squamous cell carcinoma of the anus. Strahlenther Onkol. 2018;195(7):607–14.
Zee JVD, González DG. The Dutch deep hyperthermia trial: results in cervical cancer. Int J Hyperthermia. 2009;18(1):1–12.
Willner A, et al. Neoadjuvant concurrent chemoradiotherapy with and without hyperthermia in retroperitoneal sarcomas: feasibility, efficacy, toxicity, and long-term outcome. Strahlenther Onkol. 2021;197(12):1063–71.
Song CW, et al. Tumour oxygenation is increased by hyperthermia at mild temperatures. Int J Hyperth. 2009;12(3):367–73.
Vaupel PW, Kelleher DK. Pathophysiological and vascular characteristics of tumours and their importance for hyperthermia: heterogeneity is the key issue. Int J Hyperthermia. 2010;26(3):211–23.
Frey B, et al. Old and new facts about hyperthermia-induced modulations of the immune system. Int J Hyperth. 2012;28(6):528–42.
Horsman MR, Overgaard J. Hyperthermia: a potent enhancer of radiotherapy. Clin Oncol. 2007;19(6):418–26.
Elming PB, et al. Hyperthermia: the optimal treatment to overcome radiation resistant hypoxia. Cancers. 2019;11(1):60.
Li Z, et al. Hyperthermia targeting the tumor microenvironment facilitates immune checkpoint inhibitors. Front Immunol. 2020;11:595207.
Huang H, et al. It’s getting hot in here: targeting cancer stem-like cells with hyperthermia. J Stem Cell Transplant Biol. 2017;2(2):113.
Lee S, et al. Immunogenic effect of hyperthermia on enhancing radiotherapeutic efficacy. Int J Mol Sci. 2018;19(9):2795.
van der Zee J, de Bruijne M, van Rhoon GC. Thermal medicine, heat shock proteins and cancer. Int J Hyperthermia. 2006;22(5):433–7.
Schildkopf P, et al. Application of hyperthermia in addition to ionizing irradiation fosters necrotic cell death and HMGB1 release of colorectal tumor cells. Biochem Biophys Res Commun. 2010;391(1):1014–20.
Chen T, et al. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via tLR4 pathway. J Immunol. 2009;182(3):1449–59.
Schildkopf P, et al. Radiation combined with hyperthermia induces HSP70-dependent maturation of dendritic cells and release of pro-inflammatory cytokines by dendritic cells and macrophages. Radiother Oncol. 2011;101(1):109–15.
Milani V, et al. Heat shock protein 70: role in antigen presentation and immune stimulation. Int J Hyperth. 2009;18(6):563–75.
Multhoff G, Hightower LE. Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones. 1996;1(3):167.
Hader M, et al. Differences of the immune phenotype of breast cancer cells after ex vivo hyperthermia by warm-water or microwave radiation in a closed-loop system alone or in combination with radiotherapy. Cancers. 2020;12(5):1082.
• Datta NR, et al. Local hyperthermia combined with radiotherapy and-/or chemotherapy: recent advances and promises for the future. Cancer Treat Rev. 2015. 41(9):742–53. The key radio- and chemosensitzing effects of hyperthermia are summarized and the need of randomized trials with hyperthermia in multimodal settings is stressed.
Yu Z, et al. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51.
Phi LTH, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells International. 2018;2018:1–16.
Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60.
Peitzsch C, et al. Cancer stem cells in radiation response: current views and future perspectives in radiation oncology. Int J Radiat Biol. 2019;95(7):900–11.
Konopleva M, et al. The anti-apoptotic genes Bcl-XLand Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+cells. Br J Haematol. 2002;118(2):521–34.
Arnold CR, et al. The role of cancer stem cells in radiation resistance. Front Oncol. 2020;10:164.
Nie D, Bartram I, Jeschke JM. Do cancer stem cells exist? A pilot study combining a systematic review with the hierarchy-of-hypotheses approach. Plos One. 2019;14(12):e0225898.
Oei AL, et al. Targeting therapy-resistant cancer stem cells by hyperthermia. Int J Hyperth. 2017;33(4):419–27.
Burke AR, et al. The resistance of breast cancer stem cells to conventional hyperthermia and their sensitivity to nanoparticle-mediated photothermal therapy. Biomaterials. 2012;33(10):2961–70.
Man J, et al. Hyperthermia sensitizes glioma stem-like cells to radiation by inhibiting AKT signaling. Can Res. 2015;75(8):1760–9.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research has been funded by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 955625, Hyperboost, and by the Bavarian Research Foundation (MikroHyperTumImmun, AZ-1495–20, Bayerische Forschungsstiftung).
Author information
Authors and Affiliations
Contributions
The structure and the content of the manuscript was conceptualized by USG, MR, MH, and AS. The drafts of the manuscripts were written by AS, MR, USG, BF. The final manuscript was written by AS, MR, USG, BF, MH, RF, OJO. All authors reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Udo S. Gaipl and Michael Rückert contributed equally as senior authors.
This article is part of the Topical Collection on Radiation Biology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sengedorj, A., Hader, M., Frey, B. et al. Interaction of Radiotherapy and Hyperthermia with the Immune System: a Brief Current Overview. Curr Stem Cell Rep 8, 129–138 (2022). https://doi.org/10.1007/s40778-022-00215-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-022-00215-y