
Abstract
Due to the high heterogeneity of cancers, it is rather essential to explore driver modules with the help of gene mutation data as well as known interactions between genes/proteins. Unfortunately, latent false positive interactions are inevitable in the Protein-Protein Interaction (PPI) network. Hence in the presented method, a new weight evaluation index, based on the gene-microRNA network as well as somatic mutation profile, is introduced for weighting the PPI network first. Subsequently, the vertices in the weighted PPI network are hierarchically clustered by measuring the Mahalanobis distance of their feature vectors, extracted with the graph embedding method Node2vec. Finally, a heuristic process with dropping and extracting is conducted on the gene clusters to produce a group of gene modules. Numerous experiment results demonstrate that the proposed method exhibits superior performance to four cutting-edge identification methods in most cases regarding the capability of recognizing the acknowledged cancer-related genes, generating modules having relatively high coverage and mutual exclusivity, and are significantly enriched for specific types of cancers. The majority of the genes in the identified modules are involved in cancer-related signaling pathways, or have been reported to be carcinogenic in the literature. Furthermore, many cancer related genes detected by the proposed method are actually omitted by the four comparison methods, which has been verified in the experiments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
cancers, complex diseases with high lethality rates and heterogeneity, are caused by the clonal proliferation of cells, attributing to the selective growth advantage coming from gene mutations [18, 19, 21]. Nevertheless, the majority of mutations are passenger ones and are irrelevant to cancers, they are biologically neutral and do not confer a growth advantage on the cell where they occur. That is to say, only a minority of the mutations, called driver mutations, have been subject to the positive selection and are casually implicated in the clonal proliferation of cells, contributing to the formation and progression of cancers [49]. It will shine a light on cancer pathogenes to differentiate driver genes from passenger ones [38, 40]. Furthermore, studies have demonstrated that driver genes are generally engaged in some critical cellular signaling or regulatory pathways of the human body [24], any one aberrated driver gene is generally enough to disturb the signaling pathway it involves in and leads to the generation of cancer cells. This may illustrate why high heterogeneity exists in cancers. Consequently, it is significant for exploring the heterogeneity to investigate gene mutations in terms of pathway-level instead of gene level [5, 16]. With the rapid development of high-throughput sequencing technology, incredible amounts of cancer omics data have been collected by such cancer genome sequencing projects as the Cancer Genome Atlas (TCGA) [10], and the International Cancer Genome Consortium (ICGC) [30]. It has become realistic to economically detect driver pathways or driver modules (a set of driver genes enriched in cancer-related biological pathways) by using computational methods [14, 25, 26, 64].
A number of studies have been conducted in the identification of driver pathways or driver modules. One kind of approaches, namely de novo identification [52, 63, 65], conduct detection from just genetic data by virtue of the fundamental features of driver pathways or driver modules. The other kinds of ones, namely priori knowledge-based identification approach [1, 34, 42], exploit the known interactions between genes/proteins in addition to genomic data. The study focuses on the latter one.
Among the methods based on prior knowledge, most ones apply the intrinsic topology of biological networks to the identification. The HotNet2 method [42] performs an insulated thermal diffusion process with gene mutation frequencies as well as gene interactions, and constructs a weighted graph for identifying driver modules. The Mutex method [4] hunts for sets of mutually exclusively mutated genes, sharing a common downstream target, from a great gene network. Ahmed et al. [1] regarded that methods which conducts identification employing only mutation frequency may neglect some driver modules with low mutation. Different from the HotNet2 method, their proposed MEXCOWalk method [1] weights the protein-protein interaction (PPI) network in terms of the mutual exclusivity among genes besides the gene mutation frequencies, and conducts an insulated heat diffusion process based on the weights of both vertices and edges. In 2021, Wu et al. [55] pointed out that the amounts of noise contained in biological networks would have unavoidable negative impacts on the performance of identification, and filtered it out by introducing subcellular localization data. Additionally, they devised a parthenogenetic algorithm to solve their proposed recognition model, constructed by introducing hops between genes within a module besides adopting coverage, mutual exclusivity, and network connectivity. The next year, they claimed that attention should be paid to the discrepancy of mutation frequency among different cancers [56]. The HMCEwalk method proposed by them, identifying modules based on a random walk process weights the integrated PPI network by using the harmonic mean of scores concerning coverage as well as mutual exclusivity. In the same year, Wu et al.[54] presented the ECSWalk method based on the method MEXCOWalk, it weights gene interactions in a complex biological network in terms of the similarity of node topological structure besides coverage and mutual exclusivity between mutation genes. There are also some studies attempting to reconstruct or alter the topology of biological networks. The MEMo method [12] builds a graph with edges indicating the functional similarity between a pair of genes, and outputs cliques exhibiting patterns of mutual exclusivity. The MEMCover method [35] uncovers pan-cancer dysregulated pathways from an adjusted functional interaction network in which the interactions fall into the ACROSS\ME level.
Among the above-mentioned identification methods, none of them except IDM-SPS, proposed by Wu et al. [55], has focused on the latent noise such as false positive interactions in biological networks, which may caused by a less precise confidence interval for classification [31, 32]. In this paper, studies are conducted on alleviating the negative effects of noise in virtue of other omics data. We begin with constructing a weighted protein-protein interaction (PPI) network with the aid of the gene-microRNA network as well as the somatic mutation profile, and generate gene feature vectors with the graph embedding method Node2vec. Then a set of gene clusters are produced with DIvisive ANAlysis (DIANA) hierarchical clustering algorithm [48]. Finally, the set of gene clusters are processed based on gene influence to obtain the final set of cancer driver modules. The major contributions are as follows: (1) Introduce a new evaluation index to weight the PPI network. (2) Present the vertices of a PPI network into a low-dimensional vector space with graph embedding methods Node2vec. (3) Devise a heuristic dropping and extracting process on a set of gene clusters, generated from clustering the genes based on their low-dimensional feature vectors. (4) Conduct extensive trials with real pan-cancer datasets, and compare the identification performance with that the other advanced methods Hotnet2, MEXCOwalk, ECSwalk, and HMCEwalk.
Definitions and notations
Given a set of cancer samples S={\(s_i\vert i\)=1, 2, ..., m} as well as a group of mutated genes G={\(g_j\vert j\)=1, 2, ..., n}, let \(A_{m\times n}\) be a binary somatic mutation matrix recording whether gene \(g_j\) mutates in sample \(s_i\) or not, i.e., \(a_{ij}\)=1 (i=1, 2, ..., m, j=1, 2, ..., n) if gene \(g_j\) mutates in sample \(s_i\), and \(a_{ij}\)=0 otherwise. Let PP=(V, E) represents a connected PPI network, where the vertex set V records the proteins expressed from the genes in G (\(n=|V|\)), and edge set E records the undirected interactions between the proteins. For simplifying the later description, the vertex in PP is represented with its corresponding gene \(g_j\) (\(g_j\in G\)). Let PM=(\(V^g\), \(V^m\), \(E^{gm}\), \(W^{gm}\)) denote a gene-microRNA network, where each vertex \(v^g_j\in V^g\) represents a gene (corresponding to the gene \(g_j\in V\)), each vertex \(v^m_k\in V^m\) represents a microRNA, and each edge \(e^{gm}\)(\(v^g_j\), \(v^m_k\))\(\in E^{gm}\) has a weight \(w^{gm}\)(\(v^g_j\), \(v^m_k\))\(\in W^{gm}\), measuring the relationship between gene \(v^g_j\) and microRNA \(v^m_k\). For each \(g_j\in V\), let \(S_j\) record the samples in which gene \(g_j\) is mutated:
Assume that M is a module composed of selected genes. The mutual exclusivity MEX(M) as well as the coverage COV(M) of M are defined in Formulas (2) and (3) [1]:
where MEX(M)=1 means that the genes within M are completely mutually exclusive, i.e., each sample carries at most one mutation coming from the gene of M. COV(M)=1 indicates that each sample carries at least one mutation coming from the gene of M. Let P={\(M_1\), \(M_2\), ..., \(M_r\)} be a group of driver modules, where \(M_i\), \(M_j \subseteq P\), \(M_i \ne M_j\), i, j=1,2, ..., r, \(i \ne j\). The relative size of the module \(M_i\), namely \(RS(M_i)\), is formulated as follows:
Then for the group of driver modules P={\(M_1\), \(M_2\), ..., \(M_r\)}, let MS(P) and CS(P) measure the mutual exclusivity score and the coverage one[1], respectively, defined as follows:
According to the above definitions, an optimization problem for identifying cancer driver modules is depicted as follows: Given a PPI network PP, somatic mutation matrix A, gene-microRNA network PM, the total number of genes Totalg, and the minimum size of a module Mins, identify a group of non-overlapping modules P to maximize Driver Module Set Score DMSS(P), as shown in Formulas (7) to (10).
The ICDM-GEHC method
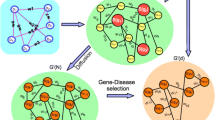
In this section, a method for Identifying Cancer Driver Modules by Graph Embedding and Hierarchical Clustering (ICDM-GEHC) is proposed. The method takes matrix A, PPI network PP, and gene-microRNA network PM as inputs, and produces a set of driver modules P as output. As shown in Fig 1, the method has four main steps, namely assigning weights, extracting features, clustering genes, and constructing driver modules. Each step is depicted detailedly in the four subsequent subsections.

The pipeline of method ICDM-GEHC
Assigning weights
It has been reported that microRNAs (miRNAs) exert critical functions in the progression and development of human cancers through regulating the expression of cancer-related genes [33]. In this paper, the gene-microRNA interaction network is introduced to weight the protein-protein interactions of a PPI network. For the convenience of description, let PP still represent the weighted PPI network, i.e., PP=(V,E,W), where w(\(v_j\), \(v_k\))\(\in W\) denotes the weight of edge (\(v_j\), \(v_k\)).
Given a pair of genes \(g_i\) and \(g_j\) (\(g_i\), \(g_j\in V\)), the confidence between them CF is defined as Formula (11):
where \(NM_{ij}\) records the microRNA neighbors common to genes \(g_i\) and \(g_j\). \(\mu \) is the arithmetic mean of the edge weights in PM (Formula (12)), and \(\lambda \) an adjustable parameter.
Let ME(\(g_i\), \(g_j\)) represent the mutual exclusivity between genes \(g_i\) and \(g_j\) (\(g_i\), \(g_j\in V\)), as calculated in Formula (13):
where Ne(x) records gene x as well as its direct neighbour genes:
Then each edge of the PPI network PP is weighted as Formula (15):
where \(\theta \) is the threshold of mutual exclusivity.
Extracting features
Given an undirected weighted graph PP=(V, E, W), the node embedding algorithm Node2vec [22] is adopted to learn continuous feature representations of the vertices. The feature extraction can be formulated into a maximum likelihood optimization problem:
where f(\(v_x\)) denotes the d-dimensional feature vector representation of vertex \(v_x\) (\(v_x\in V\)) obtained from a process of biased random walking, and \(N_s\)(\(v_x\)) records the network neighbours of vertex \(v_x\) generated with the neighbourhood sampling strategy, i.e., the sequence of vertices in the walking path starting from vertex \(v_x\). Assume that \(v_p\), \(v_c\) and \(v_n\) denote three successive vertices in a walking process, vertex \(v_n\) is chosen with a conditional probability of P(\(v_n|v_c\)):
where Z is a normalization constant, and \(\alpha _{pq}\) is the bias parameter, ascertained as Formula (18):
where parameters p and q indicate whether Deep First Search (DFS) or Breath First Search (BFS) is adopted in the process of random walking.
Clustering genes
In this section, the DIANA hierarchical clustering algorithm [48] is implemented on the weighted PPI network PP to generate a set of gene clusters. Suppose that F={f(\(v_1\)), f(\(v_2\)), ..., f(\(v_n\))} records a set of n feature vectors of d-size, where f(\(v_i\))\(\in F\) represents the feature vector of vertex \(v_i\in PP\). Given f(\(v_i\)), f(\(v_j\))\(\in F\), the Mahalanobis distance [46] \(D_m\)(\(v_i\), \(v_j\)) is adopted to measure the similarity between vertices \(v_i\) and \(v_j\), as shown in Formula (19):
where \(\Sigma \) denotes the covariance matrix between vectors f(\(v_i\)) and f(\(v_j\)). The DIANA-based clustering algorithm is described in Algorithm 1.

Hierarchical Clustering Algorithm.
Constructing driver modules
Based on the set of generated gene clusters P, a dropping and extracting algorithm is designed to construct the driver modules. Suppose that \(v_i\in V\) is a vertex of PPI network PP, let NI(\(v_i\)) measure the node influence of vertex \(v_i\), as defined in Formula (20):
As depicted in Algorithm 2, the algorithm iteratively drops the vertices with the lowest node influence, and extracts connective components in each cluster of P. Specifically, the iteration does not stop until the sum of genes in P is less than or equal to Totalg (Step 2 to Step 23). Each iteration begins with dropping the L vertices with the lowest NI(\(\cdot \)) scores from P and PP, and the edges related to them from PP (Step 2 to Step 11). Then for each cluster \(p_i\) in P, it is substituted with a set of connective components, each of which is extracted from \(p_i\) and with a minimum size of Mins (Step 13 to Step 23). The concrete description is illustrated in Algorithm 2.

Dropping and Extracting Algorithm.
Experiment results and analysis
To test the performance of method ICDM-GEHC, extensive experiments were implemented on real cancer datasets. The TCGA pan-cancer somatic aberration data were acquired from Ahmed et al. [1], consisting of 3110 cancer samples and 11565 genes of 12 cancer types. A widely used H.Sapiens PPI network HINT+HI2012 [13, 42, 60] were adopted, which contained 9859 vertices and 40705 edges. The genes co-existing in somatic mutation data and PPI network were retained, and the processed data are as follows: cancer sample number m=3110, gene number n=6930, edge number |E|=25251. The gene-microRNA network, obtained through feeding the mirDIP database [51] with the 6930 genes, was consisted of 229,135 interactions between 6145 genes and 2734 microRNAs.
We first tested the ICDM-GEHC method under different parameter settings, then compared its performance with four cutting-edge identification methods based on prior knowledge, i.e., Hotnet2 [42], MEXCOwalk [1], HMCEwalk [56], and ECSwalk [54]. All the experiments have been performed on a Workstation with an Intel i7-7700 CPU, 24 GB RAM, a Windows 10 system, and a Python 3.9.12 compiler.
Parameter settings
The settings of parameters adopted in the comparison methods were in consistent with the literatures [1, 42, 54, 56]: the mutual exclusivity threshold \(\theta \)=0.7, the probability \(\beta \)=0.4, and the minimum module size Mins=3. The total number of genes Totalg was set to {100, 200, ..., 2000} for methods Hotnet2, MEXCOwalk and ECSWalk, and {100, 200, ..., 900} for method HMCEwalk. In the ICDM-GEHC method, some parameters were set to the optimal values described in related literatures, i.e., \(\theta \)=0.7, Mins=3, node2vec parameters (p, q)=(4, 1)[1, 22]. Besides, a number of pre-experiments were performed to determine appropriate values for the other parameters required by method ICDM-GEHC.
In the experiments of determining clustering number K, the candidate values of K are calculated as Formula (21):
where function PNum(x) returns the nearest prime number of x, and \(ms\in \){10, 20, ..., 90} denotes the presumptive size of a module. Therefore, the candidate values of \(K\in \){701, 347, 233, 179, 139, 127, 101, 89, 79} corresponding to n=6930. The other parameters are tested as follows: \(\lambda \in \){0.25, 0.5, 0.75, 1}, \(d\in \){16, 32, 48, 64, 80, 96}, \(L\in \){1, 2, 3}, \(wl\in \){20, 40, 60, 80,100}, \(nw\in \){100, 200, 300, 400, 500}(wl and nw are two important parameters used in algorithm Node2vec, where wl represents walk length, and nw denotes the number of walks per node[22]). Figure 2a–f display the DMSS scores under different parameter settings. Based on the pre-experimental results, method ICDM-GEHC has the following parameter settings: \(\lambda \)=0.25, d=48, wl=80, nw=400, K=89, L=1.

The Driver Module Set Score (DMSS) with different parameter settings for ICDM-GEHC
Static evaluation
In this section, static evaluations were conducted in terms of a pair of reference gene sets, such as the COSMIC Cancer Gene Census (CGC) database [17], and the Network of Cancer Genes (NCG) [15]. As previous literature has performed, both Receiver Operating Characteristic Curve (ROC) [7] and Fold Enrichment analysis [2] were adopted to evaluate the capability of detecting known cancer genes, i.e., conducting a comparison between the union of genes in all recognized modules and a cancer reference gene set.
(1) Receiver Operating Characteristic Curve (ROC)
The ROC curve is created by calculating and plotting the True Positive Rate (TPR) against the False Positive Rate (FPR) at various Totalg settings, i.e., each point on the curve indicates a pair of TPR and FPR obtained with a given Totalg. TPR (resp. FPR) is the ratio of the number of identified reference (resp. un-reference) genes to the total number of reference (resp. un-reference) genes, as shown in Formula (22) (resp. Formula (23)). TPR and FPR respectively indicate the sensitivity and the specificity of the method, reflecting the robustness of it [6].
where TP (resp. FP) counts the number of reference genes (resp. un-reference genes) identified as cancer-related genes, and TN (resp. FN) counts the number of un-reference genes (resp. reference genes) that are not identified as cancer-related genes.
(2) Fold Enrichment analysis
Fold Enrichment measures the ratio between the proportion of identified reference genes and the proportion of the identified genes, as calculated as Formula (24):
where Reference counts the number of genes in the reference gene set, Recovered measures how many genes in the reference gene set are identified, All counts the number of genes (vertices) in the HINT+HI2012 network, and Selected denotes the sum of recognized genes. There were 616 and 591 genes contained in the CGC and the NCG databases, respectively, of which 436 and 410 ones were included in the somatic mutation data and the HINT+HI2012 network, respectively. Therefore, Reference was set to 436 for the CGC database, and 410 for the NCG one.
In Fig. 3, the ROC curves were compared among methods Hotnet2, MEXCOwalk, ECSWalk, HMCEwalk, and ICDM-GEHC based on databases CGC and NCG, where Totalg=100, 200, ..., 2000. The vertical dotted lines indicate the ROC values when Totalg=900 and 2000, respectively. The bracketed data in the legends represents the area under the ROC curve (the AUC value ), where \(A_1\) (resp. \(A_2\)) denotes the AUC value of Totalg between 100 and 900 (resp. between 100 and 2000). From this figure, we can observe that the ICDM-GEHC method achieves better identification performance than the other four methods, for it has produced the steepest curve among the five approaches. Take the CGC database for an example, when Totalg ranges from 100 to 900, method ICDM-GEHC acquires the highest AUC value among the five ones. When Totalg ranges from 100 to 2000, the AUC value of method ICDM-GEHC is 0.073, which is still higher than method Hotnet2 (0.055), MEXCOwalk (0.069), and ECSWalk (0.061).

The comparison of ROC curve among different methods
Tables 1 and 2 compare the fold enrichment analysis results based on databases CGC and NCG, respectively. It can be seen from the two tables that the fold enrichment obtained by the ICDM-GEHC Method is higher than or equal to that acquired by the other four methods in most cases.
In addition, the performance of detecting low mutation frequency genes was also evaluated for the five methods, i.e., the above analysis was implemented again under the condition that the reference gene has a mutation rate lower than 1% or 2%. For the CGC database, there are 291 genes with mutation rates lower than 1%, and 374 genes with mutation rates lower than 2%. For the NCG database, there are 266 and 352 genes accordingly.
Tables 3, 4, 5, 6 display the fold enrichments analysis results of various approaches on the two database. From these tables, we can discover that the ICDM-GEHC method exhibit better performance in most cases for reference gene frequency\(\le \)2%, while do not manifest significant advantage when the reference gene frequency is less than or equal to \(\le \)1%.
Table 7 compares the total execution time among the five approaches under the condition that Totalg=100, 200, ..., 900. The experiment results indicate that method ICDM-GEHC takes the longest time among these methods, followed by methods ECSWalk and HMCEwalk, and the least time-consuming methods are Hotnet2 and MEXCOwalk.
Modular evaluation
As referred above, the static evaluation evaluates the performance of identification methods in terms of the union of genes within the detected modules. In this section, modular evaluations were further performed to assess the specific identified modules and their interrelationships based on such two indexes as the Driver Module Set Score (DMSS) and the Cancer Type Specificity Score (CTSS) [1].
The CTSS Score is adopted to estimate the cancer-type specificity of a group of identified modules P={\(M_1\), \(M_2\), ..., \(M_r\)}. Given \(M_i\in P\) and cancer type t, let \(S_{M_i}\) represent the set of samples that have at least one mutated genes belonging to module \(M_i\), i.e., \(S_{M_i}\)=\(\bigcup _{\forall g_j\in M_i}S_j\). Assume that \(S^t\) and \(S_{M_i}^t\) denote the subset of samples in S and \(S_{M_i}\) diagnosed with cancer type t, respectively. The probability \(p_i^t\) is calculated with a Fisher’s exact test from such four values as \(|S_{M_i}^t|\), \(|S^t-S_{M_i}^t|\), \(|S_{M_i}-S_{M_i}^t|\), and |S-\(S^t|-|S_{M_i}-S_{M_i}^t|\). It estimates whether a module \(M_i\) is specific to the cancer type t, and is used to calculate the CTSS score of P, as follows:
where \(p_i^t\) is calculated as follows:
In Fig. 4, the DMSS scores obtained by the five methods are illustrated. From this figure, we can observe that the ICDM-GEHC method obtains the highest DMSS score among the five recognition approaches under different Totalg settings. This demonstrates that the modules identified by method ICDM-GEHC exhibit better coverage and mutual exclusivity than those recognized by the other four methods.

The comparison results of DMSS scores
Figure 5 illustrates the comparisons of the CTSS score obtained by the five approaches. As conducted by Ahmed et al. [1], COLON and RECTAL tumors were grouped together, so that 11 cancer types instead of 12 ones were utilized. From this figure, we can discover that the ICDM-GEHC method still exhibits the best performance in terms of the CTSS score. It can acquire a higher CTSS score than the other methods for each Totalg setting except Totalg=200, 300, 400, suggesting that its output modules are significantly enriched for specific cancer types.

The comparison results of CTSS score
Analysis of ICDM-GEHC modules
Figure 6a illustrates the eight modules detected by method ICDM-GEHC when Totalg=100. The module sizes range between 3 and 25, and the coverage of the modules ranges between 5.24% to 69.99%. Node sizes are proportional with gene mutation frequencies, indicating each gene identified by method ICDM-GEHC has a mutation frequency greater than zero. An edge is colored black if it connects two genes belonging to the same module, and grey otherwise. The thickness of a line is in positive proportion to the edge weight. Color of a module represents the cancer type that has the highest enrichment for mutations in genes of that module. Each module is named after the gene with the highest mutation frequency in that module. Figure 6b exhibits the cancer type specificity, where the rows represent modules, the columns represent cancer types, and the colors of entries indicate the significance of enrichment for cancer types in terms of Fisher’s exact test p-values.

a The modules produced by the ICDM-GEHC method when Totalg=100. The legend for the color codes is displayed on the right. b The results of cancer type specificity
In Fig. 7, the heat maps of gene mutations in three different sizes of modules are displayed. Each column represents a cancer sample (the samples, that do not have mutations in any genes of the three modules, are not exhibited), and each row denotes a gene. The top-left scale indicates the quantity of mutations in a sample. The bottom-left scale as well as the right one denote the proportion and the quantity of samples with mutations on a certain gene, respectively. Distinct colors denote distinct kinds of cancer samples. They mean the same thing in Fig. 8 (see Appendix A). As can be seen from this figure, the quantity of samples covered by a module increases apparently with the increase of module size, i.e., 253 for the CCNE1 module, 523 for the KAT6A module, and 1007 for the EGFR module. Although the three modules exhibit satisfying mutual exclusivity, i.e., most samples mutate in just one gene of the module, it gets worse with the increase in module size. The proportion of samples having more than one gene mutations are 9.52% for the CCNE1 module, 19.69% for the KAT6A module, and 37.91% for the EGFR module. Furthermore, it is discovered the central gene, which has the highest coverage in a module, may not always be the one that has the greatest degree.

The heat maps of gene mutations on three different sizes of modules
As displayed in Fig. 6a, most of the detected modules, i.e., those centered at TP53, CCND1, MYC, PIK3CA, EGFR, and KAT6A, are part of pathways known to be associated with carcinogenesis. In the following analysis, the referred biological pathways are acquired from the KEGG database (https://www.genome.jp/kegg/).
The genes within module TP53 are primarily engaged in such four cancer-related pathways as p53 signaling pathway (PTEN, CDKN2A, TP53, CDK4), Neurotrophin signaling pathway (BRAF, AKT1, TP53), Pathways in cancer (PTEN, BRAF, CDKN2A, TP53, CDK4, AKT1), and PI3K-Akt signaling pathway (PTEN, AKT1, CDK4, TP53). The module involves in eleven cancer types with KIRC (Kidney Clear Cell Carcinoma) being the most specific one, for it possesses the lowest Fisher’s exact test p-value 3.19e\(-\)80. As the central gene of the TP53 module, TP53 exhibits high mutation frequency (41.51%) across the entire samples, while has a comparative low mutation rate (1.91%) in the KIRC samples. This is consistent with the report that gene TP53 mutates at a relatively low frequency in KIRC [53]. In this module, genes TP53 and PTEN share the highest edge weight, and demonstrate moderate strong mutual exclusivity, i.e., they respectively mutate in 1259 and 317 samples, and mutate in 95 ones simultaneously. In addition, several cancer-related genes with low mutation frequencies (<1%), connected directly with gene TP53 or gene PTEN, are recognized in this module, such as WRN, WWOX, HIPK2, BMP1, RNF20 and ZNF384. WRN takes part in the replication, repair, and recombination of DNA, and the Loss-of-function mutation in WRN results in genetic instability and cancer [9]. WWOX has been suggested to be able to exert its tumor suppressive activity, and the suppression of its expression can make cancer cells resistant to death [50]. HIPK2 could represent a significant prognostic marker, and even a therapeutic target [8]. Studies have identified that BMP1 is engaged in the progression of renal cancer as an independent predictor of prognosis in Clear cell renal cell carcinoma (ccRCC) patients [57], and RNF20 overexpression inhibits ccRCC cell proliferation through downregulation of SREBP1c [41]. ZNF384 participates in the genesis and development of tumors as a significant signal molecule [27]. The CCND1 module has the same specific cancer type as the TP53 module. Three cancer related pathways are engaged in as follows: p53 signaling pathway (MDM2, MDM4, CCND1, CDK6), Pathways in cancer (RB1, MDM2, E2F3, CCND1, CDK6), and PI3K-Akt signaling pathway (MDM2, CCND1, CDK6). CTNNB1 shares relatively great edge weight with two cancer genes RB1 and CDK6. The three genes exhibit extremely high mutual exclusivity, they respectively mutate in 89, 117 and 47 samples, while only 5 samples have more than two mutations of them.
Both MYC and SMAD4 module are specific for cancer type CRC (Colorectal Cancer). Besides Colorectal cancer, several genes (MYC, APC, CTNNB1, TCF7L2) in module MYC are also engaged in the following cancer-related pathways: Pathways in cancer, Hippo signaling pathway, Wnt signaling pathway, and Endometrial cancer. In addition, the former three genes exhibit the top three coverage in this module (as shown in Fig. 1d), and have satisfying mutual exclusivity. They mutate in 283, 226 and 89 samples respectively, while just 32 samples have more than two mutations of them. Three genes in module MYC, i.e., NUP153, FAM214A, and MYO6, demonstrate low frequency while covering many kinds of cancers. For example, MYO6 mutate in ten kinds of cancers, while its mutation frequency is only 0.96%. MYO6 has been verified to be an important substance linking miRNA, circRNA, and glucose metabolism in colorectal cancer [29, 44].
The PIK3CA module is most specific for cancer UCEC (Uterine corpus endometrial carcinoma). Four pathways are involved by genes (PIK3CA, PIK3R1, KIT, FGFR1, ARHGEF11) of this module: Rap1 signaling pathway, PI3K-Akt signaling pathway, Pathways in cancer, and Breast cancer. The centering gene PIK3CA has a much coverage in the UCEC samples than in the whole ones, i.e., it mutates in UCEC samples with frequency 50%, while in the whole samples with frequency 19%. It has been verified that the proportion of patients with PIK3CA mutations is very high for cancer UCEC [28]. Furthermore, as a pair of genes having the highest coverage in the module, PIK3CA and PIK3R1 also displays terrific mutual exclusivity. Although they mutate in 596 and 152 samples respectively, the number of samples mutated on both of them is only 18. Genes FGFR1 and ARHGEF11 cover many kinds of cancers while having low mutation frequency, i.e., they mutates in six and eleven types of cancers respectively with about 0.9% mutation frequency.
The EGFR module involves eleven cancer types with GBM (Glioblastoma) being the most specific one, for it possesses the lowest Fisher’s exact test p-value 1.49e\(-\)31. The module contains fourteen genes, eleven of which are engaged in the following seven pathways: FoxO signaling pathway (EGFR, SOS1, IGF1R, IRS1, INSR), MicroRNAs in cancer (EGFR, ERBB2, SOS1, PDGFRB, PDGFRA, IRS1), Rap1 signaling pathway (EGFR, PDGFRB, PDGFRA, IGF1R, INSR, CDH1, TLN1), MAPK signaling pathway (EGFR, ERBB2, PDGFRB, PDGFRA, IGF1R, INSR, SOS1, ERBB4), PI3K-Akt signaling pathway (EGFR, ERBB2, PDGFRB, PDGFRA, IGF1R, INSR, SOS1, ERBB4, IRS1), Pathways in cancer (EGFR, ERBB2, PDGFRB, PDGFRA, IGF1R, SOS1, CDH1), and Prostate cancer (EGFR, ERBB2, SOS1, PDGFRB, IGF1R, PDGFRA). Three of the these pathways have been reported to be closely related to GBM. The alterations in Rap1 signaling pathway are significant in the progression of certain Glioblastoma [23]. MAPK pathway plays an important role in the co-activation of cell proliferation and CREB, which is an essential regulator of cyclin-D1 expression cell in GBM cells [37]. PI3K/AKT signaling has been regarded as one of the most periodically deregulated pathways in glioblastoma, the suppression of it has been acknowledged as a prospective therapeutic target for glioma [43].
The genes in the KAT6A module are primarily engaged in the following two cancer-related pathways: Pathways in cancer (EP300, CREBBP, NFE2L2, NCOA3, RUNX1, KEAP1), Thyroid hormone signaling pathway (EP300, CREBBP, NCOA3). The most specific cancer type of this module is LUSC (Lung Squamous Cell Carcinoma). The centering gene KAT6A exhibits a much higher mutation rate in LUSC samples (about 10.6%) than in the whole samples (about 4.9%). It has been suggested that KAT6A plays an oncogenic role in LUSC [47]. In addition, the EGR1 gene, with the lowest mutation frequency in the module, has been confirmed to play a tumor suppression role for this cancer [62].
The most specific cancer type of the CCNE1 module is OV (Ovarian Cancer). The centering gene CCNE1 shares the highest edge weight with gene FBXW7. They exhibit a high degree of mutual exclusivity, mutating in 145 and 86 patients respectively, and mutating in just 5 patients simultaneously. The amplification of CCNE1 has been demonstrated as a major oncogenic driver in a subset of high-grade serous ovarian cancer [20]. FBXW7 has been identified to inhibit angiogenesis, migration, and invasion of ovarian cancer cells by inhibiting VEGF expression through inactivating \(\beta \)-catenin signaling [39, 66].
The output modules of method ICDM-GEHC were further compared with those produced by the other four methods, where Totalg=100. It is discovered that all of the eight modules produced by the ICDM-GEHC method comprise oncogenes known in COSMIC, while the four comparison approaches generate at least one module that dose not contain any gene in the COSMIC database (8 out of 19 for method Hotnet2, 1 out of 12 for method MEXCOwalk, 2 out of 16 for method ECSWalk, and 1 out of 10 for method HMCEwalk). Methods Hotnet2, MEXCOwalk, ECSWalk, HMCEwalk, and ICDM-GEHC have identified 32, 48, 52, 26, and 52 known oncogenes that were recorded in the COSMIC database, respectively. Among the 100 genes recognized by method ICDM-GEHC, there are 18, 44, 40, and 16 genes that are also identified by methods Hotnet2, MEXCOwalk, ECSWalk, HMCEwalk, respectively. Furthermore, there are 40 genes identified by method ICDM-GEHC being omitted by the other four methods, 14 of these genes have been recorded in the COSMIC database, and 18 of them have been confirmed to be concerned with the development and progression of cancers, or to be engaged in cancer-related pathways (the gene lists are given in Appendix B).
Conclusion
It is both challenging and significant to identify driver modules, for which will contribute to conducting research on cancers. In this study, a novel method ICDM-GEHC was devised. It begins with constructing a weighted PPI network with the help of somatic mutation profiles as well as gene-microRNA networks. The vertices are then manifested with their extracted feature vectors, and are clustered into a set of gene clusters. Eventually, a heuristic process is conducted to produce a group of driver gene modules. Comparison experiments were performed among methods Hotnet2, MEXCOwalk, ECSWalk, HMCEwalk, and ICDM-GEHC by using real biological data. The ICDM-GEHC method exhibits superior performance to the other ones in most cases in terms of the capability of identifying cancer-related genes, producing modules that have relatively high coverage as well as mutual exclusivity, and are significantly involved for specific cancer types. Most genes within the generated modules are engaged in critical cancer-related pathways, or have been verified to be oncogenes or tumor suppressors. Simultaneously, the ICDM-GEHC method actually detected many cancer-related genes that have been omitted by the four comparison methods. The above points of view have been confirmed through a quantity of experiments. Consequently, the ICDM-GEHC method may be regarded as a helpful supplemental tool for recognizing cancer driver modules.
Although the ICDM-GEHC method presents good identification performance by applying advanced machine learning techniques into multi-omics data, it does have some notable limitations. In this method, only somatic mutation data is adopted, other genetic aberrations such as epigenetic changes, copy number variations, translocations, and fusions can be considered in an extended version of the method. In addition, since the PPI networks may vary across different cell types, tissue types, environmental conditions, and time points, the dynamic network should be adopted to replace the static one, so as to increase the flexibility and reliability of the method. In the course of experiments, it is also discovered that a high computational cost is incurred, future efforts should be devoted to further enhance its efficiency through optimizing parameters, simplifying the algorithm, and improving the module refinement.
Data availability
Data supporting the results of this study can be obtained from GitHub at https://github.com/jalexnoel/Retinal-spike-train-decoder.git.The data and the source code that support the findings of this study are openly available at https://github.com/Dengclowninfo/ICDM-GEHC.
References
Ahmed R, Baali I, Erten C et al (2019) Mexcowalk: mutual exclusion and coverage based random walk to identify cancer modules. Bioinformatics 36(3):872–879. https://doi.org/10.1093/bioinformatics/btz655
Amgalan B, Lee H (2014) Wmaxc: a weighted maximum clique method for identifying condition-specific sub-network. PLoS ONE 9(8):e104993. https://doi.org/10.1371/journal.pone.0104993
Anuraga G, Tang WC, Phan NN et al (2021) Comprehensive analysis of prognostic and genetic signatures for general transcription factor iii (gtf3) in clinical colorectal cancer patients using bioinformatics approaches. Curr Issues Mol Biol 43(1):2–20. https://doi.org/10.3390/cimb43010002
Babur Ö, Gönen M, Aksoy BA et al (2015) Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol 16:1–10. https://doi.org/10.1186/s13059-015-0612-6
Boca SM, Kinzler KW, Velculescu VE et al (2010) Patient-oriented gene set analysis for cancer mutation data. Genome Biol 11(11):R112. https://doi.org/10.1186/gb-2010-11-11-r112
Bolboacă SD, Jäntschi L (2014) Sensitivity, specificity, and accuracy of predictive models on phenols toxicity. J Comput Sci 5(3):345–350. https://doi.org/10.1016/j.jocs.2013.10.003
Bradley AP (1997) The use of the area under the roc curve in the evaluation of machine learning algorithms. Pattern Recogn 30(7):1145–1159. https://doi.org/10.1016/S0031-3203(96)00142-2
Calzado MA, Renner F, Roscic A et al (2007) Hipk2, a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle 6(2):139–143. https://doi.org/10.4161/cc.6.2.3788
Cha H, Lee D, Jung H et al (2015) Investigation of werner protein as an early dna damage response in actinic keratosis, bowen disease and squamous cell carcinoma. Clin Exp Dermatol 40(5):564–569. https://doi.org/10.1111/ced.12548
Chang K, Creighton CJ, Davis C et al (2013) The cancer genome atlas pan-cancer analysis project. Nat Genet 45(10):1113–1120. https://doi.org/10.1038/ng.2764
Chen S, Li J, Zhou P et al (2020) Sptbn1 and cancer, which links? J Cell Physiol 235(1):17–25. https://doi.org/10.1002/jcp.28975
Ciriello G, Cerami E, Sander C et al (2012) Mutual exclusivity analysis identifies oncogenic network modules. Genome Res 22(2):398–406. https://doi.org/10.1101/gr.125567.111
Das J, Yu H (2012) Hint: high-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol 6(1):92. https://doi.org/10.1186/1752-0509-6-92
Dimitrakopoulos CM, Beerenwinkel N (2017) Computational approaches for the identification of cancer genes and pathways. Wiley Interdiscip Rev 9(1):e1364. https://doi.org/10.1002/wsbm.1364
Dressler L, Bortolomeazzi M, Keddar MR et al (2022) Comparative assessment of genes driving cancer and somatic evolution in non-cancer tissues: an update of the network of cancer genes (ncg) resource. Genome Biol 23(1):35. https://doi.org/10.1186/s13059-022-02607-z
Efroni S, Ben-Hamo R, Edmonson M et al (2011) Detecting cancer gene networks characterized by recurrent genomic alterations in a population. PLoS ONE 6(1):e14437. https://doi.org/10.1371/journal.pone.0014437
Forbes SA, Beare D, Boutselakis H et al (2016) Cosmic: somatic cancer genetics at high-resolution. Nucleic Acids Res 45(D1):D777–D783. https://doi.org/10.1093/nar/gkw1121
Futreal PA, Kasprzyk A, Birney E et al (2001) Cancer and genomics. Nature 409(6822):850–852. https://doi.org/10.1038/35057046
Futreal PA, Coin L, Marshall M et al (2004) A census of human cancer genes. Nat Rev Cancer 4(3):177–183. https://doi.org/10.1038/nrc1299
Gorski JW, Ueland FR, Kolesar JM (2020) Ccne1 amplification as a predictive biomarker of chemotherapy resistance in epithelial ovarian cancer. Diagnostics 10(5):279. https://doi.org/10.3390/diagnostics10050279
Greenman C, Stephens P, Smith R et al (2007) Patterns of somatic mutation in human cancer genomes. Nature 446(7132):153–158. https://doi.org/10.1038/nature05610
Grover A, Leskovec J (2016) node2vec: scalable feature learning for networks. KDD 2016:855–864. https://doi.org/10.1145/2939672.2939754
Gutmann DH, Saporito-Irwin S, DeClue JE et al (1997) Alterations in the rap1 signaling pathway are common in human gliomas. Oncogene 15(13):1611–1616. https://doi.org/10.1038/sj.onc.1201314
Hahn WC, Weinberg RA (2002) Modelling the molecular circuitry of cancer. Nat Rev Cancer 2(5):331–341. https://doi.org/10.1038/nrc795
Hanahan D, Weinberg R (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70. https://doi.org/10.1016/S0092-8674(00)81683-9
Hirabayashi S, Ohki K, Nakabayashi K et al (2017) Znf384-related fusion genes define a subgroup of childhood b-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 102(1):118. https://doi.org/10.3324/haematol.2016.151035
Huang Q, Zhou Y, Wang B et al (2022) Mutational landscape of pan-cancer patients with pik3ca alterations in Chinese population. BMC Med Genom 15(1):146. https://doi.org/10.1186/s12920-022-01297-7
Huang X, Shen X, Peng L et al (2020) Circcsnk1g1 contributes to the development of colorectal cancer by increasing the expression of myo6 via competitively targeting mir-455-3p. Cancer Manag Res. https://doi.org/10.1186/s12920-022-01297-7
Hudson TJ, Anderson W, Aretz A et al (2010) International network of cancer genome projects. Nature 464(7291):993–998. https://doi.org/10.1038/nature08987
Jäntschi L (2021) Formulas, algorithms and examples for binomial distributed data confidence interval calculation: excess risk, relative risk and odds ratio. Mathematics. https://doi.org/10.3390/math9192506
Jäntschi L (2022) Binomial distributed data confidence interval calculation: formulas, algorithms and examples. Symmetry. https://doi.org/10.3390/sym14061104
Jonas S, Izaurralde E (2015) Towards a molecular understanding of microrna-mediated gene silencing. Nat Rev Genet 16(7):421–433. https://doi.org/10.1038/nrg3965
Kim YA, Cho DY, Dao P et al (2015) Memcover: integrated analysis of mutual exclusivity and functional network reveals dysregulated pathways across multiple cancer types. Bioinformatics 31(12):i284–i292. https://doi.org/10.1093/bioinformatics/btv247
Kim YA, Cho DY, Dao P et al (2015) Memcover: integrated analysis of mutual exclusivity and functional network reveals dysregulated pathways across multiple cancer types. Bioinformatics 31(12):i284–i292. https://doi.org/10.1093/bioinformatics/btv247
Kirana C, Peng L, Miller R et al (2019) Combination of laser microdissection, 2d-dige and maldi-tof ms to identify protein biomarkers to predict colorectal cancer spread. Clin Proteom 16(1):1–13. https://doi.org/10.1186/s12014-019-9223-7
Krishna KV, Dubey SK, Singhvi G et al (2021) Mapk pathway: potential role in glioblastoma multiforme. Interdiscip Neurosurg 23:100901. https://doi.org/10.1016/j.inat.2020.100901
Lawrence MS, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499(7457):214–218. https://doi.org/10.1038/nature12213
Lee I, Kim T, Song H et al (2009) O509 curcumin activates erk and jnk mapk pathways to induce egr1 expression for the inhibition of cell growth in ovarian cancers. Int J Gynecol Obstetr 107:S238–S238. https://doi.org/10.1016/S0020-7292(09)60882-1
Lee JH, Zhao XM, Yoon I et al (2016) Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov 2(1):16025. https://doi.org/10.1038/celldisc.2016.25
Lee JH, Jeon YG, Lee KH et al (2017) Rnf20 suppresses tumorigenesis by inhibiting the srebp1c-pttg1 axis in kidney cancer. Mol Cell Biol 37(22):e00265-17. https://doi.org/10.1128/MCB.00265-17
Leiserson MDM, Vandin F, Wu HT et al (2015) Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 47(2):106–114. https://doi.org/10.1038/ng.3168
Li Q, Liu X, Mao J et al (2023) Rragb-mediated suppression of pi3k/akt exerts anti-cancer role in glioblastoma. Biochem Biophys Res Commun 676:149–157. https://doi.org/10.1016/j.bbrc.2023.07.031
Li W, Lu Y, Ye C et al (2021) The regulatory network of microrna in the metabolism of colorectal cancer. J Cancer 12(24):7454. https://doi.org/10.7150/jca.61618
Ma J, Cui Y, Cao T et al (2019) Pds5b regulates cell proliferation and motility via upregulation of ptch2 in pancreatic cancer cells. Cancer Lett 460:65–74. https://doi.org/10.1016/j.canlet.2019.06.014
McLachlan GJ (1999) Mahalanobis distance. Resonance 4(6):20–26
Partynska A, Piotrowska A, Pawelczyk K et al (2022) The expression of histone acetyltransferase kat6a in non-small cell lung cancer. Anticancer Res 42(12):5731–5741. https://doi.org/10.21873/anticanres.16080
Patnaik AK, Bhuyan PK, Rao KK (2016) Divisive analysis (diana) of hierarchical clustering and gps data for level of service criteria of urban streets. Alex Eng J 55(1):407–418. https://doi.org/10.1016/j.aej.2015.11.003
Stratton MR, Campbell PJ, Futreal PA (2009) The cancer genome. Nature 458(7239):719–724. https://doi.org/10.1038/nature07943
Taouis K, Driouch K, Lidereau R et al (2021) Molecular functions of wwox potentially involved in cancer development. Cells 10(5):1051. https://doi.org/10.3390/cells10051051
Tokar T, Pastrello C, Rossos AEM et al (2017) mirdip 4.1-integrative database of human microrna target predictions. Nucleic Acids Res 46(D1):D360–D370. https://doi.org/10.1093/nar/gkx1144
Vandin F, Upfal E, Raphael BJ (2012) De novo discovery of mutated driver pathways in cancer. Genome Res 22(2):375–385. https://doi.org/10.1101/gr.120477.111
Wang X, Sun Q (2017) Tp53 mutations, expression and interaction networks in human cancers. Oncotarget 8(1):624. https://doi.org/10.18632/oncotarget.13483
Wu H, Chen Z, Wu Y et al (2022) Integrating protein-protein interaction networks and somatic mutation data to detect driver modules in pan-cancer. Interdiscip Sci 14(1):151–167. https://doi.org/10.1007/s12539-021-00475-y
Wu J, Yang J, Li G et al (2021) Idm-sps: identifying driver module with somatic mutation, ppi network and subcellular localization. Eng Appl Artif Intell 106:104482. https://doi.org/10.1016/j.engappai.2021.104482
Wu J, Wu C, Li G (2022) Identifying common driver modules by equilibrating coverage and mutual exclusivity across pan-cancer data. Neurocomputing 492:408–420. https://doi.org/10.1016/j.neucom.2022.04.050
Xiao W, Wang X, Wang T et al (2020) Overexpression of bmp1 reflects poor prognosis in clear cell renal cell carcinoma. Cancer Gene Ther 27(5):330–340. https://doi.org/10.1038/s41417-019-0107-9
Xie Y, Yan J, Cutz JC et al (1822) (2012) Iqgap2, a candidate tumour suppressor of prostate tumorigenesis. Biochimica et Biophysica Acta (BBA) 6:875–884. https://doi.org/10.1016/j.bbadis.2012.02.019
Yang L, Gu Y (2023) Sptbn2 regulates endometroid ovarian cancer cell proliferation, invasion and migration via itgb4-mediated focal adhesion and ecm receptor signalling pathway. Exp Ther Med 25(6):1–11. https://doi.org/10.3892/etm.2023.11977
Yu H, Tardivo L, Tam S et al (2011) Next-generation sequencing to generate interactome datasets. Nat Methods 8(6):478–480. https://doi.org/10.1038/nmeth.1597
Zhang B, Zhao Z, Wang Y et al (2023) Stx5 inhibits hepatocellular carcinoma adhesion and promotes metastasis by regulating the pi3k/mtor pathway. J Clin Transl Hepatol 11(3):572. https://doi.org/10.14218/JCTH.2022.00276
Zhang H, Chen X, Wang J et al (2014) Egr1 decreases the malignancy of human non-small cell lung carcinoma by regulating krt18 expression. Sci Rep 4(1):5416. https://doi.org/10.1038/srep05416
Zhang J, Zhang S (2017) Discovery of cancer common and specific driver gene sets. Nucleic Acids Res 45(10):e86–e86. https://doi.org/10.1093/nar/gkx089
Zhang J, Zhang S (2018) The discovery of mutated driver pathways in cancer: models and algorithms. IEEE/ACM Trans Comput Biol Bioinf 15(3):988–998. https://doi.org/10.1109/TCBB.2016.2640963
Zhang J, Zhang S, Wang Y et al (2013) Identification of mutated core cancer modules by integrating somatic mutation, copy number variation, and gene expression data. BMC Syst Biol 7(2):S4. https://doi.org/10.1186/1752-0509-7-S2-S4
Zhong L, Pan Y, Shen J (2021) Fbxw7 inhibits invasion, migration and angiogenesis in ovarian cancer cells by suppressing vegf expression through inactivation of \(\beta \)-catenin signaling. Exp Ther Med 21(5):1–8. https://doi.org/10.3892/etm.2021.9945
Zhou L, Panté N (2010) The nucleoporin nup153 maintains nuclear envelope architecture and is required for cell migration in tumor cells. FEBS Lett 584(14):3013–3020. https://doi.org/10.1016/j.febslet.2010.05.038
Acknowledgements
This research is supported by the National Natural Science Foundation of China under Grant No. 62366007, Guangxi Natural Science Foundation under Grant No. 2022GXNSFAA035625, the National Natural Science Foundation of China under Grant No. 62302107, Research Fund of Guangxi Key Lab of Multi-source Information Mining & Security (14-A-03-02, 15-A-03-02, 19-A-03-01, 20-A-01-03), “Bagui Scholar” Project Special Funds, Guangxi Collaborative Innovation Center of Multi-source Information Integration and Intelligent Processing.
Author information
Authors and Affiliations
Contributions
SD: Data curation, Software, Writing-original draft. JW: Conceptualization, Methodology, Writing-review and editing. GL: Supervision. JL: Supervision, Validation. YZ: Validation.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix A: The heat maps of gene mutations on five different sizes of modules
See Fig. 8.

The heat maps of gene mutations on five different sizes of modules
Appendix B: The genes identified by method ICDM-GEHC
The 40 genes that are just detected by method ICDM-GEHC [PDS5B, GTF3C1, FGFR1, MYH11, IQGAP2, PIK3C2B, PDGFRB, TCF7L2, SPTBN2, KIT, ADAMTS2, ARID4A, SMAD4, NAV2, PTPRB, MYH10, PDGFRA, STX5, MYO6, NUP98, NUP153, INSR, E2F3, NAV1, CDC27, PTPRM, KEAP1, SOS1, SMC3, ASAP1, EXPH5, SPTBN1, ARHGEF11, FAM214A, BRD4, TRIM33, ERRFI1, NFE2L2, TPR, DOCK3].
14 of these genes are in COSMIC, they are [KIT, FGFR1, TCF7L2, TPR, NUP98, PDGFRB, NFE2L2, KEAP1, TRIM33, SMAD4, PTPRB, PDGFRA, MYH11, BRD4].
18 of these genes have been verified to be carcinogenic, they are [SOS1, INSR, E2F3, PDS5B, GTF3C1, IQGAP2, SPTBN2, ADAMTS2, ARID4A, NAV2, MYH10, STX5, NUP153, CDC27, PTPRM, ASAP1, SPTBN1, ARHGEF11]. Four of which (SOS1, INSR, E2F3, ARHGEF11) are involved in four important pathways. Genes SOS1 and INSR are enriched in PI3K-Akt signaling pathway, MAPK signaling pathway, and Ras signaling pathway.Genes E2F3 and ARHGEF11 are involved in Pathways in cancer. Six of which (ARID4A, NAV2, MYH10, CDC27, PTPRM, ASAP1) have been recorded as being concerned with cancers in GeneCards (https://www.genecards.org/). Eight of which (PDS5B, GTF3C1, IQGAP2, SPTBN2, ADAMTS2, STX5, NUP153, SPTBN1) have been demonstrated to play significant roles in the onset and progression of various cancers. PDS5B is dysregulated in human cancers and concerned with patient prognosis [45]. GTF3C1 belongs to GTF3 family genes, whose expressions are associated with many types of cancers [3]. Loss of IQGAP2 results in the tumorigenesis of prostate tumorigenesis, gastric cancer, and hepatocellular carcinoma [58]. SPTBN2 is concerned with some biological process related to tumorigenesis, and has been demonstrated to be increased in lung adenocarcinoma, colorectal cancer, and endometrial [59]. ADAMTS2 has been reported to be significantly associated with tumor progression, vascular invasion and distant liver metastasis [36]. STX5 can reduce the adhesion between HCC cells and to the extracellular matrix, so as to promote tumor metastasis [61]. The reduction of Nup153 levels in cancer cells can affect their directional migration, which is a elementary feature in the spread of cancer [67]. SPTBN1 has been considered to affect the occurrence, progression, and metastasis of many kinds of cancers [11].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, S., Wu, J., Li, G. et al. ICDM-GEHC: identifying cancer driver module based on graph embedding and hierarchical clustering. Complex Intell. Syst. 10, 3411–3427 (2024). https://doi.org/10.1007/s40747-023-01328-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40747-023-01328-5