
Abstract
Near real-time monitoring of faecal indicator bacteria (FIB) in waters is currently not feasible, and current monitoring methods require field sampling and laboratory testing that inhibits decision-making within a relevant timeframe. While recent studies identified the potential of using specific fluorescence regions for FIB monitoring, sufficient accuracy often requires site-specific calibration due to minor variations in fluorescence peak locations. In this study, a series of lab experiments were completed to address some of the selectivity issues. Specifically, the study explored correlations between wavelength-specific fluorescence signals acquired through fluorescence excitation-emission matrices (EEM) and the amount of E. coli K-12 (E. coli) and E. faecalis (enterococci) in exponential and stationary phase broth cultures. Subsequently, the experiments quantified how the addition of known concentrations of L-tryptophan amplifies an indole pulse, specifically its concentration and the corresponding fluorescence properties. Results show unique peak excitation/emission (λex/λem) wavelengths (± 5 nm) in EEMs for E. coli cell pellet and in M9 broth (~ 280/ ~ 327 nm), enterococci cell pellet (~ 276/ ~ 324 nm), L-tryptophan (~ 278/ ~ 343 nm and ~ 298/ ~ 344 nm), and indole (~ 232/ ~ 321 nm). The findings demonstrate that L-tryptophan concentrations in E. coli broth were reduced. At the same time, the indole content increased throughout the initiation phase to the stationary phase of the bacteria growth curve, with the peak indole pulse occurring approximately at the time of transition from the exponential to stationary phase. Such unique fluorescence signatures for not only FIB but also indole (whose pulse can be triggered by L-tryptophan) provide foundations for developing reliable and near real-time in situ FIB sensors.
Highlights
-
Fluorescence regions specific to common E. coli and enterococci strains, tryptophan and indole are identified.
-
High accuracy between concentrations and fluorescence intensity are observed.
-
E. coli broth with 6 mM L-tryptophan produces 5 mM indole to effectively infer FIB.
Similar content being viewed by others
1 Introduction
Researchers and professionals in the water and sanitary engineering field have long recognised the need for a rapid and effective method of detecting faecal indicator bacteria (FIB). However, traditional tests are time-consuming, and require manual sampling and specially equipped laboratories. Using the fluorescence properties of metabolic/cell components or products, fluorescence sensors could provide near-instantaneous, non-invasive quantification of critical water quality parameters such as E. coli and enterococci if appropriate calibration is performed. Such sensor development has been hindered by several factors, including difficulty in relating its intensity to a concentration for the target FIB, as well as poor specificity and sensitivity (Baker et al. 2015; Walck 2017; Fox et al. 2017; Sorensen et al. 2018, 2021; Bedell et al. 2022; Gunter et al. 2023). A monitoring tool's ability to discriminate between the target organisms and other substances within a detection matrix is known as its specificity. On the other hand, sensitivity is the proportion of target organisms that can be detected within a test matrix (Offenbaume et al. 2020). While there are studies (Fox et al. 2017) that provide a good indication for an E. coli-specific fluorescence excitation/emission (λex/λem) wavelength pair, their results were produced within a model system (autochthonous) with specific growth media, growth, and laboratory conditions. In recent years, nevertheless, researchers have further explored this opportunity. Baker et al. (2015) found a log correlation of r = 0.74 across a 7-log range in E. coli enumeration for sewage-impacted rivers when they tested their proposed fluorescence excitation/emission region (i.e., λex/λem of 280/350 nm). Walck (2017) used an absorption and fluorescence analysis procedure in the laboratory to determine an E. coli fluorescence true excitation peak at 280 nm and a target emission wavelength of 338 nm. However, the study did not provide broth culture and comprehensive laboratory procedure details, a concentration value for E. coli, or address specificity issues, which would make it difficult to reproduce. Fox et al. (2017) performed laboratory research using non-fluorescent minimal media to promote growth within their model system whilst excluding the presence of proteinaceous material. They were able to develop fluorescence and optical density measurements at 600 nm (OD600) data for an E. coli growth curve with time. The peak-T range Fluorescence Intensity (FI) increased due to an increase in E. coli concentration after 360 min. They identified that E. coli had an λex/λem peak in the peak-T region of 280/300 – 380 nm and concluded peak-T fluorescence was ubiquitous within the bacterial cells (either as structural or functional biological molecules) analysed within their study. However, they identified peak-T as representative of intracellular material but also of extracellular fluorescent dissolved organic matter (FDOM) and attributed peak-T to the presence of amino acid (tryptophan). They reported that tryptophan was produced by E. coli as a result of cell multiplication and metabolic processing. However, it was unclear whether the signal in the fluorescence excitation-emission matrix (EEM) was due to E. coli, tryptophan, or both. Because peak-T depended on microbial activity (especially in systems with complex microbial communities), they suggested using peak-T only as a proxy for microbial activity rather than E. coli enumeration (Fox et al. 2017). Bedell et al. (2022) used tryptophan-like fluorescence (TLF), like Fox et al. (2017) and Sorensen et al. (2018), and coupled that with a machine learning (ML) model to predict E. coli from TLF. Bedell et al. (2022) prototype TLF sensor application used ultraviolet light emitting diodes (UV LEDs) to excite tryptophan-like fluorophores at 275 nm and a UV photodiode paired with a bandpass filter centred around 357 nm. Their method was used to predict > 10 colony forming units (CFU)/100 mL E. coli and detected and measured proxies for fouling. However, the integration of the sensor and its data platform into water systems still needed to be considered, as it only triggered an alarm for more extensive traditional testing. In addition, and importantly, the development of ML models typically requires large amounts of data, which will be site-specific, thus requiring field monitoring resources. The Logue et al. (2016) study found a close connection between incubations of various bacterial communities and subsequent changes in fluorescence intensities. Four experimental communities decomposed various components of the Dissolved Organic Matter (DOM) pool. However, the researchers cautioned that the correlation analysis could not be used to unambiguously link a specific bacterial taxon to the degradation and utilisation of a particular DOM. Further research was necessary to examine how specific microbial taxa in complex communities utilise individual organic matter compounds. Consequently, there was no direct link between E. coli or enterococci and the degradation of DOM. Thus, while attempts to quantify E. coli through fluorescence have been performed, several issues, mainly in relation to specificity (Offenbaume et al. 2020), have thus far limited the development of a generalisable fluorescence-based E. coli monitoring approach.
Tryptophan's blanket term TLF is typically used to identify specific compounds of FDOM in the peak-T region of EEMs (Carstea et al. 2016; Sorensen et al. 2018, 2021; Simões and Dong 2018). Tryptophan concentration and its correlation with fluorescence intensity were investigated in a study by Wünsch et al. (2015). They measured tryptophan wavelengths at λex/λem 277/350 nm and quantified molar fluorescence, absorbance, Stokes shift, and quantum yields. Carstea et al. (2016) showed tryptophan has a peak fluorescence at λex/λem 295/353 nm, though a relationship between peak intensity values and concentration was not identified. Simões and Dong (2018) reported tryptophan’s intrinsic fluorescence at λex/λem 280/350 nm, though the concentration(s) was not identified. Given that the same EEM region seems to correlate both to tryptophan and to E. coli based on previous studies, it appears difficult to discern their relative contributions and, in turn, to estimate E. coli cell numbers from this region’s fluorescence alone due to these selectivity issues.
A more generic use of fluorescence signals was proposed in relation to faecal contamination monitoring. Sorensen et al. (2018) determined that fluorescence spectroscopy technology was a viable option for the real-time screening of faecal contamination in drinking waters globally. They found FDOM at TLF λex/λem wavelengths 280/360 nm was a good indicator of faecal contamination in drinking water; however, the correlations were site-specific, implying that the findings did not apply specifically to E. coli but rather to site-specific fluorescence properties during faecal contamination events. Nevertheless, they explain that if tryptophan were readily available in the environment, E. coli would import and hydrolyse tryptophan, almost quantitatively, into indole, which is then excreted. This would enhance any peak-T signal because indole fluoresces at a 33% greater intensity on its own than when it forms part of the tryptophan molecule (Gunter et al. 2023). In addition, peak-T is composed of peak-T1 and peak-T2, where peak-T2 indole has peak λex/λem wavelengths different to the peak-T1 tryptophan. This implies that indole’s fluorescence signal would be much more significant in the peak-T2 region in the presence of tryptophan, given the same amount of E. coli/FIB, thus alleviating E. coli selectivity issues. Despite this suggestion, little quantitative analysis (together at once) of separate E. coli, tryptophan, and indole fluorescence signals against their concentration was performed.
Indole fluorophores in proteins fluoresce strongly in the region common for indole-containing compounds; they are commonly found in wastewaters and are also referred to as tryptophan-like (Eftink and Ghlron 1976; Coble et al. 2014; Carstea et al. 2016). In nature, bacteria are most commonly found in complex communities, and indole production occurs and alters bacterial physiology differently (Lee and Lee 2010; Han et al. 2011; Li and Young 2013; Gaimster et al. 2014; Gaimster and Summers 2015; Kim and Park 2015). It is commonplace to use FIB as a proxy for Total Coliforms (TC) to indicate high-risk faecal contamination in water (Tallon et al. 2005; Bedell et al. 2022; NHMRC and NRMMC 2022), and the measurement of indole production is a traditional method for the identification/differentiation of specific microbial species (Gaimster et al. 2014; Gaimster and Summers 2015; Kim and Park 2015).
Various Gram-positive and Gram-negative bacterial species have been identified in the literature as producing large quantities of indole, including E. coli, E. faecalis (enterococci), and many commensal gut microbes (Gaimster et al. 2014; Han et al. 2011; Lee and Lee 2010; NHMRC and NRMMC 2022). Concerning E. coli, it has been shown that during the bacterial growth transition from exponential to stationary phase, the indole pulse is necessary for long-term stationary phase viability (Gaimster et al. 2014; Gaimster and Summers 2015). Li and Young (2013) and Gaimster and Summers (2015) results suggest that the total amount of indole (up to 5–6 mM) produced by an E. coli broth is limited by the amount of exogenous tryptophan in the growth medium, environmental conditions, and nutrient availability. Concentrations of 0.5 to 1 mM indole have been identified to modulate biofilm formation, virulence, and stress responses, while concentrations of 4 to 5 mM indole added exogenously to an E. coli culture have been shown to inhibit growth and cell division reversibly (Gaimster and Summers 2015). The duration of an indole pulse is somewhat inconsistent due to the complexity of the processes involved, but a pulse can occur in ~ 20 to 30 min (Gaimster et al. 2014). Noting that indole production over time can be different in terms of production rate than the more instantaneous indole pulse near a bacterial broth’s stationary phase. Regarding fluorescence, indole concentration vs intensity has been documented in Wünsch et al. (2015), where they illustrate indole wavelengths being λex/λem 269/340 nm, molar fluorescence, molar absorbance, stokes shift and quantum yield. At the same time, Carstea et al. (2016) show that indole has a fluorescence peak at λex/λem 230/330 – 350 nm, though they did not identify a relationship between intensity and concentration. Sorensen et al. (2018) compared the TLF intensity emitted by dissolved tryptophan and indole. They tested solutions containing 0, 10, 20, 50, and 100 ppb of both compounds, and found that pure indole fluoresced with 33% greater intensity than tryptophan within the TLF region. However, the TLF wavelengths λex/λem pair 280/360 nm were used to collect results for both compounds. A specific λex/λem pair for either tryptophan or indole was not illustrated, and the concentration range was limited to 100 ppb (0.0004897 mM tryptophan and 0.0008536 mM indole).
Given that the indole pulse can be noticed in a different region of an EEM than E. coli and tryptophan, there seems to be potential to concurrently monitor this to better identify/quantify the actual amount of E. coli. Moreover, the same processes with tryptophan and indole are valid for enterococci. Both FIBs demonstrate the ability of bacteria to decompose tryptophan to indole, which accumulates in the medium (Lee and Lee 2010; Gaimster and Summers 2015; Sigma-Aldrich Pty. Ltd. 2021a; Microbiology Info.com 2022).
Research has shown that the E. coli/enterococci growth rate, tryptophan concentration and reduction, and indole production are correlated; however, so far, they have not been thoroughly identified/quantified concurrently in EEMs to determine correlations between their concentrations and fluorescence intensities. Thus, the objectives of this research were to identify and measure varying concentrations of E. coli, enterococci, tryptophan, and indole in laboratory settings with traditional methods as well as with absorbance and fluorescence approaches. To create sufficient evidence that FIB concentrations can be more accurately monitored when using both specific fluorescence wavelength pairs in conjunction with an indole pulse measurement (caused by exogenous tryptophan). Since the TLF region is also a common fluorescence region for other substances/contaminants (e.g., oils), the simultaneous monitoring of indole fluorescence would help rule out these types of non-bacterial contamination. In addition, triggering an indole pulse with exogenous tryptophan could shed light on the magnitude of the faecal contamination event as well as its location since the timing of the pulse (e.g., just before/after the stationary phase) will be related to travel time of FIB from the contamination source. The evidence arising from this study could support the development of more selective and informative in situ faecal contamination warning technologies.
2 Material and Methods
2.1 Bacterial Species
The primary bacterial species cultured for analysis were the E coli K-12 strain and Enterococcus faecalis strain ATCC 19433. E. coli presence in waters can indicate faecal contamination, and their cells can survive outside the body for a limited time (Bogosian et al. 1996; Reisner et al. 2003; Maraccini et al. 2016; UNICEF 2019; NHMRC and NRMMC 2022; NHMRC 2022), making them ideal indicator organisms for environmental samples. Importantlfy, as previously mentioned, E. coli produces fluorescence in the peak-T region of EEMs (Fox et al. 2017). Whereas enterococci form a biofilm, survive harsh conditions and environments, and are the most suitable FIB in many water quality guidelines (Kay et al. 2005; Gilmore et al. 2014; Khalifa et al. 2015; Maraccini et al. 2016; Paul Wood 2020; NHMRC and NRMMC 2022). The E. coli and enterococci were stored and refreshed on agar petri plates (See Supplementary Material SM2).
The laboratory procedures, growth media development, incubation conditions, results production, and analysis were reproducible for either bacterial species. Related studies (Lee and Lee 2010; Gaimster and Summers 2015) identified that indole production was related to tryptophan, growth medium, and growing conditions. However, our preliminary research found that E. coli increased more readily to a higher concentration than enterococci, and thus, we focused our results primarily on E. coli.
2.2 Media
The Escherichia coli K-12 substrate MG1655 Growth Medium: M9 medium with 2% glycerol (See Supplementary Material SM3 and Table SM. 1) was predominately used in this research (EcoCyc 2021). The ingredients for the M9 media listed in Table SM. 1, including the distilled water (dH2O), were sourced from the laboratory. Using a tryptophan assay kit, the initial tryptophan content of the M9 media was found to be ~ 0 µM. Then, different concentrations of L-tryptophan were added to the M9 media to increase the L-tryptophan content as required. The nutrient media CM0001B (Thermo Scientific™ 2021a) and nutrient media No. 2 (Thermo Scientific™ 2021b) were used in a few initial experiments for E. coli and the enterococci cell pellet. A tryptophan assay kit showed the initial tryptophan content in nutrient media No. 2 (N2) to be ~ 170 µM. Similarly, different concentrations of L-tryptophan were calculated and added to the N2 media.
Even though the initial tryptophan content in the broth was estimated, such content was monitored during the experiments to further prove how much was used or produced by E. coli and how that content affected E. coli growth and rate of growth, as well as indole production. This was important because, as Li and Young (2013) and Gaimster and Summers (2015) reported, the amount of indole produced by E. coli can be determined based on the amount of tryptophan in the growth medium, thus making it plausible that indole production slows and then stops as the supply of exogenous tryptophan is exhausted. Because of this assumption, our laboratory work included experiments with extra tryptophan to help predict the indole signal's timing, duration, and magnitude, using the tryptophan content as a guide. Also, the L-tryptophan was added and tested in different amounts to notice differences in bacterial growth with L-tryptophan depletion and, in subsequent indole analyses, to examine the limit to indole production and how this related to the initial L-tryptophan content in isolation.
The L-tryptophan content was added to M9 media using a stock-made 50 mM L-tryptophan solution. Concentrations of 0.5 mM, 1 mM, 2 mM, 5 mM, and 6 mM L-tryptophan were achieved within the M9 media (and the N2 media) to grow E. coli over time, take samples to produce the OD600 bacteria growth curve and for subsequent analyses. Sampling experiments and testing were conducted over time in triplicate samples under the same conditions and time, i.e., multiple M9 media E. coli cultures (and N2 media cultures), with and without additional L-tryptophan. The 1 mL samples for tryptophan and 1 mL samples for indole measurement were pipetted into 1.5 mL centrifuge tubes, centrifuged as described in Sect. 2.3, then frozen at -80 ºC; 3 mL samples were used for OD600 measurements, and 1 mL samples for the EEMs were not centrifuged but directly frozen at -80 ºC for subsequent dilutions. The OD600 readings were recorded as described in Sects. 2.3 and 2.4, the fluorescence EEMs as described in Sects. 2.5 and 2.6, the L-tryptophan concentrations (see Sect. 2.8) were analysed with a tryptophan assay kit and indole (see Sect. 2.9) an indole assay kit.
2.3 Bacterial Growth Curves
To evaluate FIB concentrations, OD600 readings were taken as a measure of bacterial growth for both E. coli and enterococci (Laplace et al. 1997; Turner et al. 2004; Sezonov et al. 2007; Milo et al. 2010; Khalifa et al. 2015; Fox et al. 2017; Yap and Trau 2019). Bacterial growth curves of E. coli (n = ~ 200) and enterococci (n = ~ 12) were produced by inoculating 200 mL of the sterile medium using the stored petri plate cultures. The bacteria in broth were incubated at 37 °C and shaken at 130 rpm while the samples were collected under sterile conditions (See Supplementary Material SM4).
Following OD600 measurement, three 1 mL samples were pipetted into 1.5 mL centrifuge tubes, i.e., one tube for L-tryptophan analysis, one for indole analysis, and one for EEM analysis. The tryptophan’s and indole’s 1 mL samples in 1.5 mL centrifuge tubes were centrifuged at 12,000 rpm at 4 °C for 10 min. The samples for EEM analysis were not centrifuged to capture the entirety of the solution but were placed directly in the freezer at -80 °C. Following the 10 min centrifuge of the tryptophan and indole samples, these were added to the storable containers, placed in the -80 °C freezer, and immediately frozen. All samples were tested, on average, 2–4 weeks later. The frozen samples were subsequently thawed for tryptophan, indole, or EEM analyses. The 1 mL samples for EEMs were diluted in 10 mL sterile centrifuge tubes with Phosphate Buffered Saline (PBS) accordingly.
2.4 Bacterial Growth Curve Analysis
To measure the FIB concentration, first, the OD600 reading was taken, and then their concentration in cell density (Cells/mL) and colony forming units per mL (CFU/mL) was approximated using general equations for (1) E. coli and (2) for enterococci, developed based on correlations resulting from a number of selected samples that were measured for both cell count and absorbance. The calculations were calibrated by taking another series of triplicate samples (at the start of an experiment, with low concentrations and a higher concentration later) (see Supplementary Material SR1 Tables SR. 1 – SR. 4).
For the concentration calculation to be related to the OD600 values, spread plate counts (CFU/mL) were determined for both typical E. coli and enterococci broth dilutions in M9 media, nutrient broth No.2 (N2) and nutrient broth CM0001B (see Supplementary Material SR1 Fig. SR. 1a-b). Based on OD600 and spread plate results shown in Supplementary Material SR1 Tables SR. 1, SR. 2, and SR. 3, this enabled the development of a regression model between these indicators:
where \({E.coli }_{OD600}\) represents the OD600 readings and \({E.coli }_{CFU/mL}\) the respective spread plate counts of the analysed broth dilutions.
Due to lower spread plate counts, the enterococci dilutions were documented for nutrient broth No.2 (N2) samples only. The OD600 and spread plate results data shown in Supplementary Material SR1 Table SR. 4 enabled the enterococci equation below to be developed:
where \({Enterococci }_{OD600}\) represents the OD600 readings and \({Enterococci }_{CFU/mL}\) the respective spread plate counts of the analysed broth dilutions.
The correlation (i.e., regression coefficient) between the two different measurement methods is similar to those previously reported (Laplace et al. 1997; Sezonov et al. 2007; BNID 100985 and BNID 106028 Milo et al. 2010; Khalifa et al. 2015; Yap and Trau 2019). While the accuracy based on mean values is very high (see Supplementary Material SR1 Fig. SR. 1a-b), the large standard deviation reflects the high variability in the results, which is partially caused by the inherent variability of such biological systems and their measurement inconsistency, as well as their growth difference across the different experiments. However, the variation in the relationship between spread plate counts and OD600 may also imply that such measurement methods are not robust and may support the concurrent use of fluorescence-based investigation techniques. To assess the above Eq. (1) and Eq. (2), several triplicate series for E. coli and enterococci OD600 vs spread plates results were verified periodically as described in Sect. 2.3 and Supplementary Material SM4.
2.5 Fluorescence Excitation-Emission Matrices Measurements
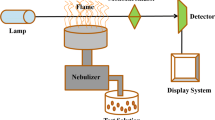
Fluorescence EEM data were amassed using a Duetta spectrofluorometer (Horiba 2020). A micro quartz cuvette (4 mL) with a 10 mm path length was used throughout.
To obtain reliable EEMs for these experiments where samples contain multiple fluorophores, the blank was subtracted from the EEM in subsequent MS Excel data analysis. Additionally, it was deemed appropriate to apply different EEM collection settings for other EEM regions. For example, at short wavelength range EEMs, a large bandpass and integration time were used, while at longer wavelength range EEMs, a smaller bandpass and shorter integration time were applied. Therefore, EEM analysis for the samples in this lab investigation consisted of eighteen individual methods setups with specific code names (see Supplementary Material SM5 and Fig. SM. 1 – SM. 18). The unique method configurations were slightly different in terms of bandwidth, time steps, excitation/emission, and a few other specifics. However, all configurations remained within the same total excitation range within 200–400 nm and emission range within 275–500 nm. Also, the excitation step increment, emission increment, and detection accumulations remained the same for all configurations. The diverse configurations were made to minimise noise and ensure that we could isolate/identify the actual fluorescence signal in different regions of the EEM. Multiple samples were compared/analysed in the same method setup. Then, after the measurements, if required for analysis, we used the peak fluorescence ratio (between the results of sample dilutions or between different time series samples with the same method setup) to align the data and make comparisons.
2.6 Fluorescence Data Analysis
Using the Duetta software, data extraction, and manipulation techniques, the EEM data were saved and investigated. The visual analysis method involved manual interpretation of 2D and 3D contour plots in the Duetta software. Peak-picking techniques, data manipulation, and statistical analysis were deployed. Peak picking was used to visually discriminate real peaks from artifacts and noise (Garrett et al. 1991). This helped identify Rayleigh scattering and noise around the EEM edges. Although there are generalised strategies, such as those described by Tan et al. (2020), for robust Rayleigh scattering correction and spectral resolution improvement of an EEM. However, in our analysis, to remove the diagonal Rayleigh scattering, the Duetta software analysis tool was manually employed (Horiba 2020). See Supplementary Material SM6 and Eq. (SM. 1) for more information and alternative methods using MS Excel. The EEM visualisation was also performed through the development of a dedicated code in R (RStudio 2023.06.01). See Supplementary Material Sections: SR_MS (Duetta EzSpec Software compilation of main manuscript EEMs figures) and SR_SMA (Duetta EzSpec Software compilation of Supplementary Material EEMs figures) for better visualisations of the fluorescence intensity in the lower or higher ranges (Horiba 2020).
Peak-picked data was analysed to provide a visual representation of the fluorescence development over time. Using the EEM fluorescence and OD600, the relationship between fluorescence at identified wavelength pairs and E. coli concentration was estimated.
2.7 E. coli and Enterococci Cell Pellet Measurements
To gather the E. coli and enterococci cell pellet, stationary phase cultures were centrifuged in 50 mL centrifuge containers at 5000 × g at 4 °C for 5 min. The supernatant was pipetted off and filtered using a 0.22 μm PVDF membrane filter to guarantee all cells were removed and immediately frozen. The cell pellet was resuspended and washed five times in 5 mL PBS pH 7.4 at 25 °C to ensure that any supernatant or media was no longer present. The final cell pellet solution had the OD600 value recorded, and further dilutions in PBS were expressed against that value. Finally, the fluorescence EEM of the cell pellet was recorded for those dilutions and measured against the OD600 values.
Control measures were placed on the E. coli and enterococci cell pellet to provide confidence that the resultant pellet was pure. Gram staining of diluted E. coli and enterococci cell pellet were monitored under a microscope for samples at regular bacteria growth phase intervals. See Supplementary Material SR2 Fig. SR. 2 and Fig. SR. 3, which represent the gram staining of stationary phase (a) E. coli and (b) E. faecalis (enterococci) cell pellet samples. Additionally, OD600, 16-streak plate counts, and spread plate counts were produced for the bacteria broth in triplicate at regular intervals, noted in laboratory notebooks, and for future reference photographed.
Before fluorescence EEM analysis, the chosen E. coli and enterococci cell pellet and supernatant samples were diluted tenfold, 100-fold, and 1000-fold in PBS. Then, the cell pellet and supernatant samples produced fluorescence EEMs as described in Sects. 2.5 and 2.6 and concentrations expressed with the general equations for (1) E. coli and (2) enterococci, as described in Sect. 2.4.
2.8 Tryptophan Measurements
The L-tryptophan (LTP) reagent grade 25 g was sourced through Sigma-Aldrich Pty. Ltd. (2021b) and processed in the laboratory in dH2O to achieve calculated concentrations. An initial solution was developed using ~ 2.3 g of the weighed powder in 225 mL dH2O in a glass bottle with a lid, then sterilised by autoclaving at 121 °C for 20 min. This provided a 50 mM L-tryptophan solution further diluted in dH2O to 10 mM / 5 mM / 2 mM / 1 mM / 0.5 mM / 0.2 mM / 0.1 mM / 0.05 mM / 0.02 mM / 0.01 mM / 0.005 mM. Additionally, an L-tryptophan standard 1000 uM (from the tryptophan assay kit) was diluted tenfold, 100-fold, 1000-fold, and 10000-fold for analysis. A tryptophan assay kit, described later, was subsequently used to confirm the accuracy of the dilutions. Then, multiple EEMs were acquired for those diluted solution samples using the Duetta analysis methods described in Sects. 2.5 and 2.6.
The L-tryptophan content was determined using the Bridge-It® L-Tryptophan Fluorescence Assay kit protocol and fluorescence microplate reader (Mediomics 2021). Following the assay protocol and methods from Sect. 2.3 of this report, the frozen E. coli broth samples (-80 °C) were thawed for L-tryptophan analyses in the PC2 laboratory.
2.9 Indole Measurements
Indole for synthesis was sourced through Sigma-Aldrich Pty. Ltd. (2021c) and processed in the laboratory in Ethanol to achieve calculated concentrations. An initial indole solution was developed using the Indole bottle contents (25 g) and Ethanol (500 mL). This provided a ~ 426.8 mM indole solution subsequently diluted in ethanol to 100 mM / 60 mM / 30 mM / 17 mM / 10 mM / 8.5 mM / 6 mM / 5 mM / 4 mM / 3 mM / 2 mM / 1 mM / 0.5 mM / 0.2 mM / 0.1 mM / 0.005 mM solutions, in 10 mL centrifuge tubes wrapped in aluminium foil and masking tape stored without light. Additionally, the 10 mM indole standard from the indole assay kit was diluted in Ethanol to 1 mM / 0.1 mM / 0.01 mM / 0.001 mM / 0.0001 mM for analysis. Excitation-emission matrices were then acquired for those diluted solution samples using the methods described in Sects. 2.5 and 2.6. Relationships between fluorescence at identified wavelength pairs and indole concentration were used to inform the following research tasks.
To understand how much E. coli grows over time and when to expect and maximise indole signalling, testing was conducted with different M9 media E. coli culture samples, with and without L-tryptophan addition, with and without additional M9 medium and compared over time (See Sects. 2.2 and 2.10). We examined how the initial L-tryptophan concentration in an E. coli broth relates to the indole magnitude over time (Gaimster et al. 2014; Gaimster and Summers 2015) and whether to expect the concentration of indole, the timing and duration of the shift from exponential to stationary phase to differ depending on initial L-tryptophan concentration in media. According to Gaimster and Summers (2015), there are two distinct modes of indole signalling, i.e., pulse signalling with high transient levels of indole and persistent signalling due to long-lasting levels of indole. They reported that the cell-associated indole, which peaks and falls rapidly, is much higher than the free indole. Therefore, to provide a broad assessment of the levels of indole, our sampling for free indole started every ~ 4 h before, during, and after the exponential phase until E. coli went into its stationary phase; from that point, the sampling frequency was once every 24 h until the death phase. The 1 mL samples for indole measurement were pipetted into 1.5 mL centrifuge tubes, centrifuged as described in Sect. 2.3, and then frozen at -80 ºC. The OD600 readings were collected as identified in Sects. 2.3, and the EEMs produced as identified in Sects. 2.5 and 2.6.
Another experiment tested whether a 5 mM indole signal was produced after adding three 2 mM L-tryptophan supplements to an E. coli broth over time. This was to test whether (i) we could get more than 5 mM of indole and (ii) whether adding L-tryptophan in smaller amounts over time provides different results than altogether at the start. Adapted from Gaimster and Summers (2015), a fresh E. coli culture was inoculated into a fresh M9 medium. Subsequently, two different triplicate broths were incubated at 37 °C with shaking at 130 rpm for 24 h. The supernatant indole concentration in the stationary phase broths was assayed. One sub-culture triplicate was incubated at 37 °C with shaking for a further 192 h. The other sub-culture triplicate was also incubated with shaking at 37 °C, but three 2 mM supplements of L-tryptophan were added at 52, 76 and 100 h (for a total of 6 mM added L-tryptophan). Samples were taken from each broth every 24 h for 72 h, and the supernatant was assayed for indole at 76 / 100 / 124 and 194 h.
Two indole assay kits were used (Cell Biolabs Inc. 2021; Sigma-Aldrich Pty. Ltd. 2021a) for measuring free indole in samples in a bacterial growth medium. Both assay kits made use of typical laboratory procedures and a fluorescence microplate reader. The kit from Cell Biolabs Inc. (2021) was a colorimetric assay that measured the total amount of free indole present in bacterial samples. The sample indole concentrations were determined by comparison with a known indole standard, and the kit had a detection sensitivity limit of 31 µM indole. The second kit from Sigma-Aldrich Pty. Ltd. (2021a) was based on a version of Ehrlich’s and Kophanovac’s reagents. The intensity of the produced coloured compound was directly proportional to the indole in the sample, and this indole assay kit has a linear detection range of 3 to 100 µM indole. Following the related assay protocols (Cell Biolabs Inc. 2021; Sigma-Aldrich Pty. Ltd. 2021a) and Sect. 2.3 of this report, the frozen 1 mL centrifuged E. coli broth samples (-80 °C) were thawed for indole analyses. The assay method protocols were followed and conducted in the PC2 laboratory. The fluorescence microplate reader was set up according to each indole assay kit protocol. Subsequently, the indole concentration data was saved and investigated using the fluorescence microplate reader software and data extraction and manipulation techniques.
2.10 E. coli and Enterococci Bacterial Broth Measurements
Before fluorescence EEMs vs concentration analysis, the chosen E. coli and enterococci broth samples (stationary phase samples and time series samples) were not filtered to capture the entirety of the solution, i.e., the bacterial cell pellet in the broth with biochemicals produced over time. These samples were thawed, vortexed and diluted tenfold, 100-fold, and 1000-fold in PBS. Then, these samples produced fluorescence EEMs, as described in Sects. 2.5 and 2.6, and concentrations expressed with the general equations for (1) E. coli and (2) enterococci, as described in Sect. 2.4.
2.11 Exploratory Lab Representation of Field Monitoring Potential
As E. coli and enterococci in the environment are not typically found in their exponential growth phase but in the stationary phase, additional laboratory experimentation was conducted to further understand the relationship between E. coli, tryptophan, and indole. We specifically assessed whether a predetermined indole pulse produced from a stationary phase E. coli broth with additional L-tryptophan supplementation could be triggered. The justification was that if such an indole pulse could be achieved, there would be potential for a fluorescence-based field sensor to be developed, which applies L-tryptophan in the field to confirm E. coli presence through E. coli and indole fluorescence EEM regions. These samples were analysed and developed using methods identified in previous Sects. 2.1 – 2.10.
3 Results and Discussion
3.1 Excitation-Emission Matrices vs Concentration
3.1.1 E. coli and Enterococci Cell Pellet
E. coli and enterococci cell pellets contain material/biochemicals that fluoresce in the peak-T region of EEMs. Our analysis suggests E. coli have a peak λex/λem pair of ~ 280/ ~ 327 nm (± 5 nm) while enterococci have a peak λex/λem pair of ~ 276/ ~ 324 nm (± 5 nm) (see Supplementary Material SR2 Tables SR. 5 and SR. 6). Their EEM peak λex/λem values and regions are almost identical. However, our results suggest that in typical broths, E. coli predictably concentrated higher (both in terms of CFU/mL and OD600) than enterococci. Their bacteria profile could be the direct cause (see Supplementary Material SR2 Fig. SR. 2, gram staining): E. coli are rod-shaped, form long strands, chain-like structures, and clumps, whereas enterococci (see Supplementary Material SR2 Fig. SR. 3, gram staining) are circular/oval shaped and as seen under the microscope gram staining, they did not form long clumps, chains, or strands. Still, when moving away from broths and developing both cell pellet solutions, the concentrations became highly comparable (See Fig. 1a, b).

The Method M34 EEMs of (a) E. coli cell pellet (tenfold dilution OD600 = 0.18) and (b) enterococci cell pellet (tenfold dilution OD600 = 0.19). (See SR_MS Fig. SR_MS. 1)
The E. coli and enterococci cell pellets were diluted tenfold, 100-fold, and 1000-fold; EEMs were analysed for all method setups. For both E. coli OD600 = 1.8 (Fig. 2a-c) and enterococci OD600 = 1.9 (Fig. 3a-c) cell pellet, the EEMs (method M34) provide a visual representation of the typical mean fluorescence reduction resulting from a dilution series, as well as the cell pellet peak λex/λem value and region similarities.

The Method M34 EEMs of E. coli cell pellet (a) tenfold, (b) 100-fold and (c) 1000-fold dilutions

The Method M34 EEMs of enterococci cell pellet (a) tenfold, (b) 100-fold and (c) 1000-fold dilutions
The reader can refer to Supplementary Material SR_MS Fig. SR_MS. 2a-b-c and Fig. SR_MS. 3a-b-c for better visualisation of the fluorescence intensity in the low range for dilutions 100-fold and 1000-fold.
The mean results of the dilution series (tenfold, 100-fold, and 1000-fold) were used to determine the mean peak λex/λem pair for E. coli presence. For mean OD600 values of 0.178, 0.018, and 0.0020. Subsequently, the mean peak fluorescence for E. coli cell pellet and Eq. (1) (see Sect. 2.4) were used to develop the FI vs concentration (Conc.) relationship (R2 = 0.99). This is presented with the standard deviation of the method M4 mean results for E. coli cell pellet (Fig. 4a). Similarly, the enterococci cell pellet was analysed using mean values derived from multiple dilution series (tenfold, 100-fold, and 1000-fold) for mean OD600 values of 0.131, 0.013, and 0.001. This allowed the identification of the mean peak λex/λem pair for enterococci presence. The mean peak fluorescence for enterococci cell pellet and Eq. (2) (see Sect. 2.4) were used to develop the FI vs Conc. relationship (R2 = 0.99) and is presented with the standard deviation of the method M4 mean results for enterococci cell pellet (Fig. 4b). As the sample became more diluted, there was typically less fluorescence. However, our analysis found that the presence of E. coli and enterococci in the EEMs remained. Although the peak fluorescence reduced, it did so relative to its surroundings.
Additionally, when combined, the E. coli and enterococci cell pellet mean peak λex/λem pair was ~ 278/ ~ 326 nm (± 5 nm), and their concentration(s) was determinable using OD600 and Eq. (1) or Eq. (2).

Method M4 peak FI vs concentration results (with standard deviation) for the (a) E. coli cell pellet dilution series (tenfold, 100-fold, and 1000-fold) and the (b) enterococci cell pellet dilution series (tenfold, 100-fold, and 1000-fold)
The cell pellet development(s) were a combination of broths centrifuged and washed together. The FI and CFU/mL were higher or larger depending on the cell pellet concentration. The significant standard deviation for the high concentration results in Fig. 4a, b is due to a combination of multiple samples with different OD600 values (E. coli between ~ 1.0 and ~ 2.0). Similarly, the enterococci cell pellet saw one of the triplicate samples with a higher OD600 value (between ~ 1.1 and ~ 1.9) and, therefore, concentration. However, both curves follow a similar trend (See Fig. 4a, b).
3.1.2 L-tryptophan
The mean results for the method M4 set up (Fig. 5a, b) found the mean λex/λem wavelength pair to predict concentrations between 0.001 to 0.2 mM L-tryptophan was ~ 280/ ~ 343 nm (± 5 nm) with R2 = 0.91. Similarly, the mean λex/λem wavelength pair of method M200400 results (Fig. 6a, b) between 0.001 to 0.2 mM L-tryptophan was ~ 275/ ~ 344 nm (± 5 nm) with an accuracy of R2 = 0.90. The mean λex/λem of both methods, M4 and M200400 (see Supplementary Material SR3 Table SR. 7) was ~ 278/ ~ 343 nm (± 5 nm) for concentrations between 0.001 to 0.2 mM L-tryptophan. A smaller peak (see Fig. 6a and Supplementary Material SR3 Fig. SR. 4a) occurred at lower, indole-like excitation wavelengths in the peak-T2 region, though with proportionally much lower fluorescence intensity.

The (a) Method M4 EEM of 0.1 mM L-tryptophan, and (b) the peak FI vs concentration trend for the peak λex/λem ~ 280/ ~ 343 nm (± 5 nm) pair and L-tryptophan concentrations between 0.001 to 0.2 mM with standard deviation. (See SR_MS Fig. SR_MS. 4)

The (a) Method M200400 EEM of 0.1 mM L-tryptophan, and (b) the peak FI vs concentration trend for the peak λex/λem ~ 275/ ~ 344 nm (± 5 nm) pair and L-tryptophan concentrations between 0.001 to 0.2 mM with standard deviation. (See SR_MS Fig. SR_MS. 5)
As the concentration between 0.001 and 0.2 mM L-tryptophan increased, so did the fluorescence intensity (Fig. 5 and Fig 6). For higher concentrations (0.5 to 6 mM, see Supplementary Material SR3 Table SR. 8, Fig. SR. 4a-b, SR. 5a-b and SR. 6d-f), the peak mean λex/λem wavelength pair shifted slightly (~ 298/ ~ 344 nm (± 5 nm)), and fluorescence intensity decreased. We surmise that this is due to the inner filter effect and fluorescence quenching, including collisional quenching or even apparent quenching of the sample (Eftink and Ghlron 1976; Knappskog and Haavik 1995; Lakowicz 2006; Wang et al. 2017; Zu et al. 2017; Chen et al. 2018; Kumar Panigrahi and Kumar Mishra 2019) not automatically accounted for in the EEMs collection phase, and it occurred for concentrations high enough not to be expected in freshwater environments (Sorensen et al. 2018). According to the literature (Eftink and Ghlron 1976; Joshi et al. 1990; Knappskog and Haavik 1995; Lakowicz 2006), various substances and molecular interactions act as fluorescence quenchers. A decreasing trend is common for many organic dyes and carbon dots; the fluorescence decreases when the concentration increases above certain thresholds (Zu et al. 2017). Molecular oxygen is one of the best-known collisional quenchers (which quenches almost all known fluorophores), and there is also apparent quenching caused by the optical properties of a sample (Lakowicz 2006).
Tryptophan falls within a specific TLF excitation and emission range contained within peak T (T1 and T2), and our identified λex/λem peak wavelength pair evolution is seen in the literature (Baker et al. 2015; Bridgeman et al. 2015; Wünsch et al. 2015; Carstea et al. 2016; Fox et al. 2017; Yin et al. 2020). Baker et al. (2015) identified that fluorescence at 280 nm excitation and 350 nm emission wavelengths was attributed to TLF. Bridgeman et al. (2015) determined that TLF fluorescence was emitted at 340–370 nm under excitation at both 220–240 nm and 270–280 nm. Wünsch et al. (2015) illustrated that the wavelengths for tryptophan were λex/λem 277/350 nm. Carstea et al. (2016) presented tryptophan’s λex/λem being 295/353 nm, with potential sources in wastewater being proteins, peptides, and livestock wastewater. Fox et al. (2017) discussed FDOM protein-like fluorescence within the λex/λem 230–280/330–360 nm range. Referred to as peak T, protein-like FDOM of microbial origin λex/λem 275/340 nm was tryptophan-like and 230/305 nm tyrosine-like. At the same time, Fox et al. (2017) identified the fluorescence peak for tryptophan presence as being λex/λem 280/300–380 nm. Yin et al. (2020) identified two typical fluorescence peaks in freshwater aquatic samples: peak T1-tryptophan-like λex/λem 275/340–350 nm and peak T2-tryptophan-like λex/λem 225–237/340–381 nm. Peak-T1 materials were related to/associated with soluble microbial products and peak-T2 materials proteins of aromatic structures. Our results in terms of novel λex/λem fluorescence intensity peaks align with previous research findings; identifying EEM peak features with varying concentrations is a novel output of our work.
Table 1 illustrates the mean primary and secondary (Method M200400) fluorescence peak percentage differences between 0.1 and 6 mM L-tryptophan. Results show that for 0.1 to 0.2 mM L-tryptophan, the fluorescence peaks had a difference of approximately half, for 0.5 mM L-tryptophan one-third and for 6 mM L-tryptophan four-fifths, respectively. At concentrations ≥ 0.5 mM, the difference is partly due to/affected by fluorescence quenching and the inner filter effect.
In summary, L-tryptophan peak excitation wavelength(s) differ depending on concentration, but the peak emission remains at ~ 345 nm. There is also a secondary peak excitation at ~ 230 nm, but this has a significantly lower intensity than that for an equivalent concentration of indole, which has a similar excitation wavelength peak.
3.1.3 Indole
For six methods setups and indole concentrations between 0.05 to 5 mM (see Supplementary Material SR4 Table SR. 9), the mean λex/λem pair was ~ 232/ ~ 321 nm (± 5 nm), also showing good consistency in the EEM area of detection at higher concentrations. A smaller peak (Supplementary Material SR4 Fig. SR. 9a and Fig. SR. 10a) occurred at higher, TLF-like excitation wavelengths, though with proportionally much lower fluorescence intensity.
As the indole concentration increased from 0.1 to 0.5 mM, so did the fluorescence (Fig. 7b) for Method MR1 with R2 = 0.90 (for concentrations between 0.1 to 1 mM, see Supplementary Material SR4 Fig. SR. 7b).

The (a) Method MR1 EEM of 0.5 mM indole, and (b) the peak FI vs concentration trend for the peak λex/λem ~ 228/ ~ 320 nm (± 5 nm) pair and indole concentrations between 0.1 to 0.5 mM with standard deviation. (See SR_MS Fig. SR_MS. 6)
For Method M200400, as indole concentrations increased from 0.05 to 0.5 mM. so did the fluorescence (see Supplementary Material SR4 Fig. SR. 9b). The results for three method setups (MR1, M200400 and IND3) showed that fluorescence decreased steadily for indole concentrations beyond 0.5 mM, up to 5 mM (see Supplementary Material SR4 Table SR. 10, Fig. SR. 8b, Fig. SR. 10b, Fig. SR. 11b and Fig. SR. 12a-f) due to the inner filter effect and fluorescence quenching of the material (not automatically accounted for in the EEMs collection phase) (Eftink and Ghlron 1976; Joshi et al. 1990; Lakowicz 2006; Wang et al. 2017; Zu et al. 2017; Chen et al. 2018; Kumar Panigrahi and Kumar Mishra 2019).
Interestingly, (see Supplementary Material SR4 Fig. SR. 9a-b, Fig. SR. 10a-b and Fig. SR 12 a-f) between 0.05 to ≤ 3 mM with increased indole concentration, the primary fluorescence peak λex/λem remained at ~ 230/ ~ 320 nm, while a secondary (lower fluorescence) peak at λex/λem ~ 270/ ~ 323 nm shifted to λex/λem ~ 296/ ~ 322 nm (between ~ 0.5 to ≤ 6 mM). From 0.2 to 0.5 mM, the primary peaks’ (λex/λem ~ 230/ ~ 320 nm) fluorescence increased. Above concentrations of 0.5 mM, it decreased but remained the most dominant fluorescence up to concentrations of 3 mM, when the fluorescence fell sharply, in comparison to the secondary peak levelling off. If such result was consistent even after accounting for the inner filter effect and fluorescence quenching, the indole peak could be considered concentration-dependent. This result could be a way to identify indole in solution and to determine its concentration in correlation with the fluorescence intensity maximum. While the focus of this work was analysing the same relationship for E. coli (concentration vs fluorescence), these results seem to be promising and set the foundation for further work in this regard, though fieldwork should complement lab work to ensure that potential selectivity and sensitivity issues are identified and addressed.
Table 2 illustrates an assessment of the (Method M200400) λex 229 nm peak mean fluorescence differences between 0.1 to 5 mM indole and L-tryptophan.
At 0.1 mM, the two peaks differed by ~ 70%; at 0.5 mM, they differed by ~ 60%; at 5 mM, they differed by ~ 80%. It is important to note that this difference is in part due to/affected by fluorescence quenching and the inner filter effect at high concentrations (≥ 0.5 mM). Even though the indole fluorescence intensity was much stronger, we could also distinguish the tryptophan peak due to its λem maximum being located in a different spot. It was discovered that the λem peak for L-tryptophan was ~ 341 nm rather than ~ 321 nm, and therefore, the difference lies not only in the magnitude of fluorescence but also in the wavelength at which the peak occurs.
The study by Wünsch et al. (2015) illustrated the optical properties of DL-tryptophan and indole. Molar fluorescence was provided for the absorbance maximum, and the quantum yield was (stated as) the percentage of the estimated quantum yield. The quantum yield result for DL-tryptophan was 0.15 (± 7%), while for indole, the result was 0.28 (± 10%). These results were higher than those of Sorensen et al. (2018), who illustrated that the intensity of indole on its own was 33% greater than when it formed part of the tryptophan molecule. They used a general TLF λex/λem pair of 280/360 nm to collect fluorescence results for both compounds, and the concentration range was limited to 100 ppb (0 to 0.0004897 mM tryptophan and 0 to 0.0008536 mM indole). In comparison, in terms of peak fluorescence intensity difference (%), Wünsch et al. (2015) results were more consistent with ours (see Tables 2 and 3, Method M200400 results). Between 0.05 to ≤ 0.5 mM, examining the L-tryptophan and indole peaks, indole had a mean ~ 59% (standard deviation ~ 13%) higher FI, given the same concentration. Noting that our results were drawn from the peak fluorescence intensity in EEMs without an absorbance maximum correlation. The inner filter effect and fluorescence quenching were mentioned to contribute at higher concentrations (≥ 0.5 mM) but were not further investigated.
However, the likely range of values for FIB-related indole in the environment depends upon indole production from tryptophan. As identified by Sorensen et al. (2018), Tryptophan-like thresholds for individual risk categories for drinking water studies are low 1.3 ppb (0.0064 µM), medium 2.4 ppb (0.0118 µM), high 6.9 ppb (0.0338 µM) and very high 27.1 ppb (0.1327 µM). Nowicki et al. (2019) identified TLF as a measure of microbial contamination risk in groundwater by risk class (correlated to E. coli risk class) from low to very high using the thresholds of 1 ppb (0.0049 µM) and 3.9 ppb (0.0191 µM). These thresholds were well below our tryptophan values. Therefore, indole values > 0.5 mM are not expected in waters like those above.
In summary, indoles’ prominent peak λex slightly differs (~ 228/ ~ 232 nm) depending on concentration, but the peak λem remains consistently ~ 321 nm. Indole also showed a second peak λex/λem at ~ 280 to 295/ ~ 320 nm (± 5 nm), but the fluorescence intensity was much lower than the equivalent L-tryptophan concentrations’ FI when tested by the same method setup. Moreover, the peak-T2 range (λex/λem ~ 232/ ~ 321 nm) is of most interest because this region offers a point of difference to E. coli/enterococci and tryptophan.
3.1.4 E. coli and Enterococci in Media
E. coli grown in minimal media M9 is represented in the method M24 EEM (Fig. 8b). Results show that when M9 media (Fig. 8a) diluted tenfold is compared to (Fig. 8b) E. coli in M9 media (OD600 ~ 0.7) diluted tenfold there is a significant difference in region intensity. This implies that it can be stated that the region-specific fluorescence is due to E. coli rather than the media. Similarly, enterococci broth in N2 media was distinguishable when manually subtracted from the N2 media alone, and the analysed EEM peak λex/λem pair and intensity were comparable to enterococci cell pellet alone (see Supplementary Material SR5 Fig. SR. 13a-b).

The (a) Method M24 EEM of the M9 media solution (tenfold dilution) compared against the (b) Method M24 EEM of the stationary phase E. coli M9 media broth (tenfold dilution). (See SR_MS Fig. SR_MS. 7)
3.2 Excitation-Emission Matrices vs Concentration Evolution over Time
The method M4 EEMs for E. coli in M9 media broths were used to analyse the E. coli growth region over time (see Fig. 9 and Supplementary Material SR6 Fig. SR. 14a-f and Fig. SR. 15a-f results for all samples, taken at 6 h, 24 h, 30 h, 54 h, 76 h and 102 h). The accuracy (R2 = 0.81) of the method M4 mean results was determined from the mean E. coli M9 media broth FI vs Eq. (1) E. coli concentration, and is presented with the standard deviation of both (Fig. 9). The FI increases with E. coli concentration, demonstrating the effectiveness of the determined mean E. coli λex/λem pair, and the magnitude of the relationship is in line with the values shown for the pellet alone (Fig. 4a).

The trend seen in the Method M4 E. coli M9 media broth FI vs E. coli concentration with standard deviation (sampled over time) results using the peak λex/λem pair ~ 280/ ~ 327 nm (± 5 nm) and Eq. (1)
Nutrient media type affects E. coli growth. This is evident from the different growth curves observed for E. coli grown in M9 and N2 media broths under the same incubation conditions (see Fig. 10 and Supplementary Material SR6 Fig. SR. 16a-b for breakdown of media type results). The mean E. coli OD600 value in the N2 media broth peaks at OD600 ~ 1.5, while the M9 media broth peaks at OD600 ~ 0.8. In addition, the average difference in mean OD600 values for M9 media and N2 media E. coli broths was ~ 51% over time. However, the exponential and stationary phases occur similarly. Higher concentrations of L-tryptophan in media are also reflected in their respective E. coli growth OD600 vs time charts (Fig. 10). Findings show that if additional L-tryptophan was initially present within either medium M9 or N2, the growth of E. coli over time was lower than that for the broth developed in that media alone. The average percent difference in OD600 values between the straight M9 media broth and (i) the M9 media + 0.5 mM L-tryptophan broth was 27%; (ii) the M9 media + 1 mM L-tryptophan broth was 19%; (iii) the M9 media + 2 mM L-tryptophan broth was 22%; (iv) the M9 media + 5 mM L-tryptophan broth was 18%; (v) the M9 media + 6 mM L-tryptophan broth was 19%. The average percent difference between the straight N2 media broth and the N2 media + 6 mM L-tryptophan broth was 49%. Therefore, these results suggest that the addition of L-tryptophan consistently reduced the E. coli concentration in the growth medium. Otherwise, as previously identified, indole production contributed to this OD600 reduction. Concentrations of 4 to 5 mM indole added exogenously to an E. coli culture were shown to reversibly inhibit growth and cell division (Gaimster and Summers 2015). This remained true; however, in our experiments, we extended this to include lower indole concentrations between 0.5 and 5 mM.

E. coli broth OD600 growth curves with standard deviation for straight M9 media, M9 + 0.5 mM, M9 + 1 mM, M9 + 2 mM, M9 + 5 mM, and M9 + 6 mM L-tryptophan; straight N2 media and N2 + 6 mM L-tryptophan
Throughout these experiments, indole production was detected; we found specific regions in EEMs to monitor both indole and E coli. Our results showed that the indole pulse concentration was maximised at the end of the exponential phase/onset of the stationary phase. Further, the indole pulse concentration in FIB broths was related to the initial exogenous tryptophan content in media, obtainability of growth media and conditions for growth. Specifically, E. coli converted, almost quantitatively, exogenous tryptophan to indole for exogenous tryptophan concentrations up to 5 mM. A comparison of OD600 vs concentration results for E. coli, L-tryptophan, and indole in E. coli (a) M9 broths and E. coli (b) M9 + 6 mM L-tryptophan broths illustrates this conversion (see Supplementary Material SR6 Fig. SR. 17 for a breakdown of results). Mean OD600 values for E. coli and mean concentrations for indole in M9 media broths are shown in Fig. 11 and in N2 media broths Fig. 12 throughout time (also see Supplementary Material SR7 Fig. SR. 22). While a clear peak is not noticeable, indole consistently increased and reached its maximum levels at the start of the stationary phase.

E. coli broth OD600 growth curves with standard deviation for (a) straight M9 media, (b) M9 + 2 mM L-tryptophan and (c) M9 + 6 mM L-tryptophan, compared against indole concentrations with standard deviation

E. coli broth OD600 growth curves with standard deviation for (a) straight nutrient media No.2 (N2) and (b) N2 + 6 mM L-tryptophan, compared against indole concentrations with standard deviation
Furthermore, concentrations of E. coli and indole in media are reflected in their respective FI vs concentration charts (see Figs. 13 and 14 and Supplementary Material SR6 Fig. SR. 18). The Indole FI vs concentration downward trend seen in both Figs. 13i and 14f is due to the inner filter effect and fluorescence quenching (Eftink and Ghlron 1976; Joshi et al. 1990) for concentrations over 500 µM (0.5 mM) indole. It can also be noticed that, with the addition of L-tryptophan, the magnitude of the relationship between FI and E. coli concentration and between FI and indole concentration changes substantially (e.g., Fig. 13b, c vs e, f vs h, i, Supplementary Material SR6 Fig. SR. 18b-c, and Fig. 14b-c vs e-f).

E. coli (a-c) M9 media, (d-f) M9 + 2 mM L-tryptophan, and (g-i) M9 + 6 mM L-tryptophan broth (a, d, g) OD600 growth curves compared against indole concentrations with standard deviation, (b, e, h) the trends seen in the Method M4 E. coli λex/λem ~ 280/ ~ 327 nm (± 5 nm) pair peak FI vs E. coli concentrations with standard deviation, and (c, f, i) the trends seen in the Method MR1 indole λex/λem ~ 232/ ~ 321 nm (± 5 nm) pair peak FI vs indole concentrations with standard deviation

E. coli (a-c) nutrient media No.2 (N2) and (d-f) N2 + 6 mM L-tryptophan broth (a, d) OD600 growth curves compared against indole concentrations with standard deviation; (b, e) the trends seen in the Method M4 E. coli λex/λem ~ 280/ ~ 327 nm (± 5 nm) pair peak FI vs E. coli concentrations with standard deviation, and (c, f) the trends seen in the Method MR1 indole λex/λem ~ 232/ ~ 321 nm (± 5 nm) pair peak FI vs indole concentrations with standard deviation
This is likely due to a combination of the following opposite effects, i.e., overlapping fluorescence in the same region due to L-tryptophan itself (and indole) and quenching for high concentrations/intensities. Interestingly, however, such an FI and concentration relationship is instead more consistent for those experiments with the same exogenous L-tryptophan, regardless of the media (e.g., no L-tryptophan addition: Figs. 13b and14b; 6 mM L-tryptophan addition: Figs. 13h and 14e), confirming the above hypothesis stating that L-tryptophan is a cause of changes in the E. coli FI-concentration relationship due to its fluorescence contribution. However, there was also the possibility that more L-tryptophan was already converted to indole in the 2 to 6 mM L-tryptophan concentrations range; the FI magnitude increased with L-tryptophan concentration (and indole concentration) and then after a certain concentration, the self-quenching mechanisms of L-tryptophan and indole reduced the FI magnitude, after that point.
Some examples in the literature show that E. coli broth-derived indole is produced rapidly. Gaimster and Summers (2015) saw peak results between 120–180 min and 2–8 h while identifying that the peak indole production rate occurred during stationary phase entry and could last only 10–15 min. However, due to our sampling program, indole production typically peaked between 24 and 48 h (onset of stationary phase). There were a couple of reasons for this: (i) the media—the higher nutrient media N2, produced larger OD600 values and saw the completion of the growth curve phases slightly more rapidly than the M9 minimal medium, which provided a bare-bones complement of nutrients (EcoCyc 2021); (ii) broth volume—our broths were originally 200 mL because the sampling program required 6 mL samples per time step, 1 mL for the indole assay, 1 mL for the tryptophan assay, 1 mL for E.coli fluorescence EEMs and 3 mL for OD600. Incubating and shaking the Erlenmeyer flasks with lower volumes produced growth more rapidly. Initial experiments with ~ 25 mL M9 and N2 media saw E. coli broths reaching the stationary phase within ~ < 8 h. However, this was not a viable option due to our sampling requirements.
3.3 Exploratory Lab Representation of Field Monitoring Potential
Supplementary Material SR7 provides the resulting chart (Fig. SR. 19) for a triplicate stationary phase E. coli broth with 2 mM, 2 mM, and 2 mM L-tryptophan sequentially added at 52 h, 76 h and 100 h from the start of the experiment. Our results show that additional supplementation of 2 mM L-tryptophan alone without additional media up to 6 mM L-tryptophan did not produce an increase in mean OD600. There was, however, a smaller mean indole pulse, with a maximum of ~ 469 µM at 76 h, ~ 542 µM at 100 h, ~ 622 µM at 124 h and ~ 590 µM at 194 h. Therefore, following additional L-tryptophan supplementation to the stationary phase E. coli broth there was a potentially measurable indole production. We suggest that the smaller than expected indole amount can be because in the E. coli broth, being in stationary phase, individual cells had stopped growing/multiplying and were moving towards their death phase, or additionally, the growth media was exhausted before L-tryptophan supplementation (and not enough nutrients/growth media was left for the remaining live bacteria).
Consequently, we then tested whether E coli regrowth could be reproduced in a stationary phase broth with additional sterile M9 media and L-tryptophan supplementation and whether, in turn, an indole pulse could be triggered. The E. coli M9 broth grown to stationary phase (Fig. SR. 20), with additional M9 media and 6 mM L-tryptophan (Fig. SR. 21a-b-c) and with additional M9 media and 5 mM L-tryptophan (Fig. SR. 21d) are presented in Supplementary Material SR7. The different tests show that although the volume of the fresh media was not exceedingly high in comparison to the original volume, the most significant media addition of 20 mL had similar OD600 reproduction but slightly different indole than the 10 mL and 15 mL fresh media results. The most successful setup was the addition of ~ 6 mM L-tryptophan with 10 mL fresh M9 media. This produced an increasing amount of indole from ~ 147 μM at 2 h to ~ 594 μM after 48 h; thus, it seems that in the field, the addition of the right amount of tryptophan could trigger a small but detectable indole pulse even if E. coli is already in stationary phase.
Regardless of the potential of the in situ addition of tryptophan to trigger an indole pulse, it may be that indole was already produced (and therefore detectable) by the E. coli cells in the water source, depending on the amount of tryptophan. As previously summarised, tryptophan concentrations in freshwater sources are relatively low, with thresholds for the highest microbial contamination risks identified as 0.1327 µM (27.1 ppb) for drinking water by Sorensen et al. (2018) and 0.0191 µM (3.9 ppb) for groundwater as reported by Nowicki et al. (2019). These thresholds are below our tested tryptophan values for waters. This makes a high-selectivity (i.e., confirming that E. coli, acting as the FIB of the total population of bacteria, is present by simultaneously detecting indole) real-time fluorescence monitoring of E. coli potentially possible since tryptophan fluorescence would be marginal and not substantially affecting a fluorescence-based E. coli concentration estimation, as instead, it occurred in some of our experiments (e.g., Figs. 13 and 14). In addition, since E. coli cells typically need several hours in a broth with tryptophan to use it and produce indole, a direct field detection of E. coli and indole would likely imply a distant source of contamination and may contribute to microbial source tracking endeavours. Despite similar, partially overlapping fluorescence peaks, which could cause a false detection of E coli due to the in-field tryptophan addition, future work could further quantify the fluorescence of tryptophan at varying suggested concentrations. The results could then be "discounted" from the collected fluorescence of the in situ sensor to ensure that only E. coli contribution is estimated.
An important issue with a potential in situ E. coli monitoring device of this kind is that it will detect TLF and indole fluorescence also in the case of no faecal contamination (i.e., background fluorescence) due to the microbiological community ordinarily present in the water course. While this can be perceived as a strong limitation, a suitable monitoring approach could focus on the relative difference between ambient and contamination event conditions. This is because if there were indeed a faecal contamination event, then TLF and indole fluorescence would suddenly and dramatically increase, as can be expected (Lee and Lee 2010; Han et al. 2011; Gaimster et al. 2014; NHMRC and NRMMC 2022; NHMRC 2022) to detect much more bacteria (especially FIB) in case of contamination. This would allow us to provide early warning of faecal contamination and, based on the timing of the triggered indole pulse, a rough estimation of the distance of the contamination source. While not directly quantifying E. coli concentration, this could be done by site-specific calibration, i.e., by collecting a number of TLF and indole peak fluorescence intensities during contamination events and correlating that with corresponding laboratory E. coli results: we would expect the number of site-specific field data required for calibration to be much smaller than the dataset needed for a site-specific machine learning prediction model. Our lab results presented herein, correlating E. coli, exogenous tryptophan, and indole concentrations, could also be used to shorten the site-specific calibration process. Alternatively, another sensing technology such as Enzyme-Linked Immunosorbent Assay (ELISA), Biosensors, or Microfluidic Devices (Offenbaume et al. 2020) could be used and adapted (i.e., miniaturised, automated) for remote use, jointly with the TLF and indole fluorescence sensors. Importantly, by having an indole fluorescence sensor, non-bacterial contamination types (e.g., oil) can be excluded, thus reducing selectivity issues, and saving the application of potential reagents to only very specific events (i.e., deemed highly likely contamination events by the fluorescence sensors). As such, the need for reagent refills, and in turn, for either bigger sensors or more regular field visits, would be kept to a minimum, making the adaptation of other sensing technologies to remote use more feasible. While the findings are promising, we caution readers that at this stage, they should only be used for framing future laboratory and field experiments before definitive conclusions can be made about the development of any multi-spectral fluorescence sensing device.
4 Conclusions
Laboratory experiments compared growth, concentrations, and related fluorescence signatures of E. coli, enterococci and indole produced under different media and L-tryptophan addition scenarios. Key results include:
-
We identify an λex/λem peak fluorescence pair for E. coli presence being ~ 280/ ~ 327 (± 5 nm). An λex/λem peak fluorescence pair for enterococci presence is ~ 276/ ~ 324 (± 5 nm). An L-tryptophan peak excitation wavelength depends on concentration (between ~ 280 and ~ 298 nm), but the peak emission remains ~ 344 nm. While for indole, the primary λex/λem peak pair for concentrations between 0.05 to 6 mM is ~ 232/ ~ 321 nm (± 5 nm). These λex/λem peak pair combinations offer a clear point of difference between E. coli/enterococci, tryptophan, and indole.
-
Fluorescence spectrophotometer results show fluorescence quenching of L-tryptophan and indole above 0.5 mM for all method setups under laboratory conditions.
-
High accuracy is achieved when correlating wavelength-specific fluorescence with concentrations of E. coli and indole.
-
E. coli converts exogenous tryptophan into a similar amount of indole. Indole production by E. coli depends directly on the amount of growth media and tryptophan in the external growth medium. Higher concentrations of indole are seen at the end of the FIB exponential/stationary phase.
These results are promising in relation to the development of a remote, in situ FIB monitoring tool. While monitoring only the E. coli fluorescence region would offer poor selectivity, concurrently monitoring the fluorescence region for indole would exclude non-bacterial contamination events. At the same time, higher selectivity in relation to faecal contamination could be achieved by combining simple site-specific assessments and calibration, additional sensors, and/or fluorescence signal variation analysis.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author upon reasonable request.
References
Baker A, Cumberland SA, Bradley C et al (2015) To what extent can portable fluorescence spectroscopy be used in the real-time assessment of microbial water quality? Sci Total Environ 532:14–19. https://doi.org/10.1016/j.scitotenv.2015.05.114
Bedell E, Harmon O, Fankhauser K et al (2022) A continuous, in-situ, near-time fluorescence sensor coupled with a machine learning model for detection of fecal contamination risk in drinking water: Design, characterization and field validation. Water Res 220:118644. https://doi.org/10.1016/j.watres.2022.118644
Bogosian G, Sammons LE, Morris PJL et al (1996) Death of the Escherichia coli K-12 strain W3110 in soil and water. Appl Environ Microbiol 62:4114–4120. https://doi.org/10.1128/aem.62.11.4114-4120.1996
Bridgeman J, Baker A, Brown D, Boxall JB (2015) Portable LED fluorescence instrumentation for the rapid assessment of potable water quality. Sci Total Environ 524–525:338–346. https://doi.org/10.1016/j.scitotenv.2015.04.050
Carstea EM, Bridgeman J, Baker A, Reynolds DM (2016) Fluorescence spectroscopy for wastewater monitoring: A review. Water Res 95:205–219. https://doi.org/10.1016/j.watres.2016.03.021
Chen S, Yu YL, Wang JH (2018) Inner filter effect-based fluorescent sensing systems: A review. Anal Chim Acta 999:13–26. https://doi.org/10.1016/j.aca.2017.10.026
Coble PG, Lead J, Baker A, et al (2014) Aquatic organic matter fluorescence. In: Cambridge University Press. Cambridge Environmental Chemistry Series. https://www.cambridge.org/core/books/abs/aquatic-organic-matterfluorescence/aquatic-organic-matter-fluorescence/0F72C6DF1C5D9231CE32F32B96714985. Accessed 22 Nov 2022
EcoCyc (2021) Escherichia coli K-12 substr. MG1655 growth medium: M9 medium with 2% glycerol. In: Curr. Database Escherichia coli K-12 substr. MG1655 Ref. genome. https://biocyc.org/ECOLI/NEW-IMAGE?type=Growth-Media&object=MIX0-59. Accessed 1 Dec 2019
Eftink MR, Ghlron CA (1976) Fluorescence quenching of indole and model micelle systems. J Phys Chem 80:486–493. https://doi.org/10.1021/j100546a014
Fox BG, Thorn RMS, Anesio AM, Reynolds DM (2017) The in situ bacterial production of fluorescent organic matter; an investigation at a species level. Water Res 125:350–359. https://doi.org/10.1016/j.watres.2017.08.040
Gaimster H, Summers D (2015) Regulation of indole signalling during the transition of E. coli from exponential to stationary phase. PLoS One 10:4–5. https://doi.org/10.1371/journal.pone.0136691
Gaimster H, Cama J, Hernández-Ainsa S et al (2014) The indole pulse: A new perspective on indole signalling in Escherichia coli. PLoS One 9. https://doi.org/10.1371/journal.pone.0093168
Garrett DS, Powers R, Gronenborn AM, Clore GM (1991) A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson 95:214–220. https://doi.org/10.1016/j.jmr.2011.09.007
Gilmore MS, Clewell DB, Ike Y, Shankar N (2014) Enterococci: From commensals to leading causes of drug resistant infection. Natl. Libr. Med. https://www.ncbi.nlm.nih.gov/books/NBK190424/pdf/Bookshelf_NBK190424.pdf. Accessed 2 Sep 2021
Gunter H, Bradley C, Hannah DM et al (2023) Advances in quantifying microbial contamination in potable water: Potential of fluorescence-based sensor technology. Wiley Interdiscip Rev Water 10:1–19. https://doi.org/10.1002/wat2.1622
Han TH, Lee JH, Cho MH et al (2011) Environmental factors affecting indole production in Escherichia coli. Res Microbiol 162:108–116. https://doi.org/10.1016/j.resmic.2010.11.005
Horiba (2020) Duetta fluorescence and absorbance spectrometer. Horiba Co. https://www.horiba.com/int/products/detail/action/show/Product/duetta-1621/
Cell Biolabs Inc. (2021) Indole assay kit. Product manual. Catalog Number MET-5122. https://www.cellbiolabs.com/indole-assay-kit. Accessed 14 Jan 2022
Joshi GC, Bhatnagar R, Doraiswamy S, Periasamy N (1990) Diffusion-controlled reactions: Transient effects in the fluorescence quenching of indole and N-acetyltryptophanamide in water. J Phys Chem 94:2908–2914. https://doi.org/10.1021/j100370a033
Kay D, Stapleton CM, Wyer MD et al (2005) Decay of intestinal enterococci concentrations in high-energy estuarine and coastal waters: Towards real-time T90values for modelling faecal indicators in recreational waters. Water Res 39:655–667. https://doi.org/10.1016/j.watres.2004.11.014
Khalifa L, Brosh Y, Gelman D et al (2015) Targeting enterococcus faecalis biofilms with phage therapy. Appl Environ Microbiol 81:2696–2705. https://doi.org/10.1128/AEM.00096-15
Kim J, Park W (2015) Indole: a signaling molecule or a mere metabolic byproduct that alters bacterial physiology at a high concentration? J Microbiol 53:421–428. https://doi.org/10.1007/s12275-015-5273-3
Knappskog PM, Haavik J (1995) Tryptophan fluorescence of human phenylalanine hydroxylase produced in escherichia coli. Biochemistry 34:11790–11799. https://doi.org/10.1021/bi00037a017
Kumar Panigrahi S, Kumar Mishra A (2019) Inner filter effect in fluorescence spectroscopy: As a problem and as a solution. J Photochem Photobiol C Photochem Rev 41:100318. https://doi.org/10.1016/j.jphotochemrev.2019.100318
Lakowicz JR (2006) Quenching of fluorescence. Springer, Boston, MA, Principles of fluorescence spectroscopy. https://doi.org/10.1007/978-0-387-46312-4_8. Accessed 17 Nov 2022
Laplace JM, Thuault M, Hartke A et al (1997) Sodium hypochlorite stress in Enterococcus faecalis: Influence of antecedent growth conditions and induced proteins. Curr Microbiol 34:284–289. https://doi.org/10.1007/s002849900183
Lee JH, Lee J (2010) Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev 34:426–444. https://doi.org/10.1111/j.1574-6976.2009.00204.x
Li G, Young KD (2013) Indole production by the tryptophanase TnaA in escherichia coli is determined by the amount of exogenous tryptophan. Microbiol (United Kingdom) 159:402–410. https://doi.org/10.1099/mic.0.064139-0
Logue JB, Stedmon CA, Kellerman AM et al (2016) Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J 10:533–545. https://doi.org/10.1038/ismej.2015.131
Maraccini PA, Mattioli MCM, Sassoubre LM et al (2016) Solar inactivation of enterococci and escherichia coli in natural waters: effects of water absorbance and depth. Environ Sci Technol 50:5068–5076. https://doi.org/10.1021/acs.est.6b00505
Mediomics (2021) Mediomics assay protocol: Bridge-It® L-tryptophan fluorescence assay. www.mediomics.com. Accessed 3 Feb 2021
Microbiology Info.com (2022) Indole test- principle, reagents, procedure, result interpretation and limitations. MicrobiologyInfo.com. https://microbiologyinfo.com/indole-test-principle-reagents-procedure-result-interpretationand-limitations/. Accessed 24 Apr 2022
Milo R, Jorgensen P, Moran U et al (2010) BioNumbers the database of key numbers in molecular and cell biology. Nucleic Acids Res 38:750–753. https://doi.org/10.1093/nar/gkp889
NHMRC (2022) Guidelines for managing risks in recreational water. National Health and Medical Research Council, Commonwealth of Australia Canberra. https://www.nhmrc.gov.au/about-us/publications/guidelines-managing-risks-recreational-water. Accessed 10 Oct 2022
NHMRC and NRMMC (2022) Australian drinking water guidelines paper 6 national water quality management strategy. National Health and Medical Research Council, Natural Resource Management Ministerial Council, Commonwealth of Australia, Canberra. https://www.nhmrc.gov.au/about-us/publications/australian-drinking-water-guidelines. Accessed 10 Oct 2022
Nowicki S, Lapworth DJ, Ward JST et al (2019) Tryptophan-like fluorescence as a measure of microbial contamination risk in groundwater. Sci Total Environ 646:782–791. https://doi.org/10.1016/j.scitotenv.2018.07.274
Offenbaume KL, Bertone E, Stewart RA (2020) Monitoring approaches for faecal indicator bacteria in water: Visioning a remote real-time sensor for e. coli and enterococci. Water (Switzerland):12. https://doi.org/10.3390/w12092591
Paul Wood PE (2020) Treating for enterococci. Wastewater Dig. https://www.wwdmag.com/microorganismsbacteria-viruses/treating-enterococci. Accessed 25 Apr 2022
Reisner A, Haagensen JAJ, Schembri MA et al (2003) Development and maturation of Escherichia coli K-12 biofilms. Mol Microbiol 48:933–946. https://doi.org/10.1046/j.1365-2958.2003.03490.x
Sezonov G, Joseleau-Petit D, D’Ari R (2007) Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189:8746–8749. https://doi.org/10.1128/JB.01368-07
Sigma-Aldrich Pty. Ltd (2021a) Indole assay kit catalog number MAK326. Sigma-Aldrich. https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Bulletin/2/mak326bul.pdf. Accessed 3 Feb 2021
Sigma-Aldrich Pty. Ltd (2021b) L-tryptophan, reagent grade, >=98% (HPLC manufacturer part ID: T0254). Merck KGaA https://www.sigmaaldrich.com/AU/en/product/sial/t0254. Accessed 4 Jun 2021
Sigma-Aldrich Pty. Ltd (2021c) Indole for synthesis. Merck KGaA https://www.sigmaaldrich.com/AU/en/product/mm/822281?cm_sp=Insite-_-rvRecBlock_recentlyViewed_userHistory-_-recentlyViewed5-3. Accessed 20 Jun 2021
Simões J, Dong T (2018) Continuous and real-time detection of drinking-water pathogens with a low cost fluorescent optofluidic sensor. Sensors (Switzerland) 18:1–14. https://doi.org/10.3390/s18072210
Sorensen JPR, Baker A, Cumberland SA et al (2018) Real-time detection of faecally contaminated drinking water with tryptophan-like fluorescence: defining threshold values. Sci Total Environ 622–623:1250–1257. https://doi.org/10.1016/j.scitotenv.2017.11.162
Sorensen JPR, Nayebare J, Carr AF et al (2021) In-situ fluorescence spectroscopy is a more rapid and resilient indicator of faecal contamination risk in drinking water than faecal indicator organisms. Water Res 206:117734. https://doi.org/10.1016/j.watres.2021.117734
Tallon P, Magajna B, Lofranco C, Leung KT (2005) Microbial indicators of faecal contamination in water: a current perspective. Water Air Soil Pollut 166:139–166
Tan L, Du W, Zhang Y et al (2020) Rayleigh scattering correction for fluorescence spectroscopy analysis. Chemom Intell Lab Syst 203:104028. https://doi.org/10.1016/j.chemolab.2020.104028
Thermo ScientificTM (2021a) Nutrient broth (Dehydrated). Catalog number: CM0001B. https://www.thermofisher.com/order/catalog/product/CM0001B?SID=srch-hj-CM0001B. Accessed 3 Feb 2021
Thermo ScientificTM (2021b) Nutrient broth No. 2 (Dehydrated). Catalog number: CM0067B. https://www.thermofisher.com/order/catalog/product/CM0067B. Accessed 3 Feb 2021
Turner SR, Love RM, Lyons KM (2004) An in-vitro investigation of the antibacterial effect of nisin in root canals and canal wall radicular dentine. Int Endod J 37:664–671. https://doi.org/10.1111/j.1365-2591.2004.00846.x
UNICEF (2019) UNICEF target product profile rapid E. coli detection tests. UNICEF. https://www.unicef.org/supply/media/2511/file/Rapid-coli-detection-TPP-2019.pdf. Accessed 1 Jun 2020
United States Environmental Protection Agency (EPA) (2021) Indicators: enterococci. Natl Aquat Resour Surv. https://www.epa.gov/national-aquatic-resource-surveys/indicators-enterococci
Walck MA (2017) Prompt and in situ diagnosis of live/dead bacteria. In: Undergraduate research scholars program. Texas A&M University College Station. https://hdl.handle.net/1969.1/157700. Accessed 29 May 2018
Wang T, Zeng LH, Li DL (2017) A review on the methods for correcting the fluorescence inner-filter effect of fluorescence spectrum. Appl Spectrosc Rev 52:883–908. https://doi.org/10.1080/05704928.2017.1345758
Wünsch UJ, Murphy KR, Stedmon CA (2015) fluorescence quantum yields of natural organic matter and organic compounds: implications for the fluorescence-based interpretation of organic matter composition. Front Mar Sci 2:1–15. https://doi.org/10.3389/fmars.2015.00098
Yap PY, Trau D (2019) Direct E.coli cell count at OD600. Tip Biosyst. https://tipbiosystems.com/wpcontent/uploads/2020/05/AN102-E.coli-Cell-Count_2019_04_25.pdf. Accessed 27 Jan 2021
Yin H, Wang Y, Yang Y et al (2020) Tryptophan-like fluorescence as a fingerprint of dry-weather misconnections into storm drainage system. Environ Sci Eur 32. https://doi.org/10.1186/s12302-020-00336-3
Zu F, Yan F, Bai Z et al (2017) The quenching of the fluorescence of carbon dots: A review on mechanisms and applications. Microchim Acta 184:1899–1914. https://doi.org/10.1007/s00604-017-2318-9
Acknowledgements
The Authors would like to thank the Griffith University School of Engineering and Built Environment, Queensland Micro- and Nanotechnology Centre and School of Environment and Science – Bioscience staff for providing support when needed.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported by the Griffith University Higher Degree Research (HDR) department and the Australian Government Research Training Program (RTP) Scholarship.
Author information
Authors and Affiliations
Contributions
Kane L. Offenbaume: conceptualisation, methodology, investigation, data collection and curation, validation, formal analysis, modelling, simulation, writing, review, and editing.
Edoardo Bertone: project administration, conceptualisation, validation, resources, supervision, review, and editing.
Rodney A. Stewart: project administration, conceptualisation, validation, resources, supervision, review, and editing.
Dechao Chen: validation, resources, supervision, review, and editing.
Qin Li: validation, resources, supervision, review, and editing.
Helen Stratton: validation, resources, supervision, review, and editing.
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Offenbaume, K.L., Bertone, E., Chen, D. et al. Multi-wavelength Fluorescence Monitoring of Faecal Contamination in Waters: A Laboratory-based Quantification. Environ. Process. 11, 21 (2024). https://doi.org/10.1007/s40710-024-00696-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40710-024-00696-5