
Abstract
Purpose of Review
Obstructive sleep apnoea (OSA) is increasingly found to have an impact on neurodegeneration. In this review, we summarise recent findings on the association between OSA and brain morphology, cognition, and processes related to Alzheimer’s dementia (AD) and Parkinson’s disease (PD).
Recent Findings
Associations between OSA and alterations in grey and white matter, brain diffusivity, and deficits in memory, attention, and executive control were reported. Furthermore, OSA was correlated with higher risks of developing AD and PD and associated pathophysiology. Treatment was found to alleviate but not reverse some of the damage.
Summary
There are strong indications that OSA plays a major role in neurodegenerative processes. The broad picture however remains elusive, likely due to insufficient sample sizes, heterogeneous outcomes, and OSA definitions failing to quantify the disorder’s sub-processes. While studies resolving these issues are required, the available evidence shows OSA to be a promising target to slow neurodegeneration and delay the onset of related disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
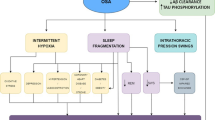
Obstructive sleep apnoea (OSA) is a common form of sleep-disordered breathing affecting around one-seventh of the world’s population [1]. The disorder is characterised by recurrent obstruction of the upper airway, resulting in periods of reduced or absent breathing (intermittent hypoxia) and sleep fragmentation. While often asymptomatic, symptoms can include among others excessive daytime sleepiness, loud snoring, and mood changes such as depression or irritability and morning headaches [2]. The gold standard in diagnosing OSA is through overnight polysomnography performed either in a sleep lab or at home, with the primary metric being the apnoea-hypopnea index (AHI), which quantifies multiple characteristics such as absence or reductions in airflow, oxygen desaturations, or arousal [2,3,4]. Treatments typically include lifestyle changes to counteract risk factors such as obesity, alcohol intake, lack of exercise or smoking, and continuous positive airway pressure (CPAP) during the night, which keeps the airways open [2]. Alternatively, oral devices or, in extreme cases, surgical procedures are available [2]. A growing body of evidence has shown the impact of OSA on reduced cognition [5,6,7,8,9], brain morphology [3, 10,11,12,13], and neurodegenerative pathophysiology [11, 14,15,16,17,18]. Furthermore, it has been shown that the treatment of OSA mitigates some of its negative consequences [13, 16, 19,20,21], suggesting that, with readily available treatment options, OSA is a promising target to delay the onset of neurodegenerative disorders such as dementia, Alzheimer’s disease (AD), or Parkinson’s disease (PD). The present review summarises findings based on adult populations published between 2018 and 2021 (see Tables 1 and 2) regarding the effect of obstructive sleep apnoea on brain morphology, cognition, and the two most common neurodegenerative disorders: Alzheimer’s dementia and Parkinson’s disease.
Sleep Apnoea and Brain Structure
Obstructive sleep apnoea is marked by sleep fragmentation and intermittent hypoxia, which have both been associated with alterations in brain structures. However, recent studies analysing grey matter (GM) provided inconsistent results. While some studies found that the presence and severity of OSA are associated with reduced GM volume in cortical (e.g. frontal and parietal cortex and cingulate/paracingulate gyrus) and subcortical cerebral regions (e.g. hippocampus, amygdala, basal ganglia, and thalamus) and the cerebellum [10, 22], others have found OSA to be associated with increased GM volume. André et al. (2020) (N = 127) found OSA to be associated with increased GM volume, perfusion, and metabolism, mainly in the posterior cingulate, cuneus, and precuneus [11], as did Kim et al. (2021) (N = 2560), who identified increased total, frontal, parietal, and temporal GM volumes in men, and increased total, frontal, and parietal GM volumes in women [12]. Taylor et al. (2018) (N = 41) found mild-severe OSA to be associated with both increased and decreased GM, with an association with increased volume in the bilateral thalamic regions using a voxel-based morphometry analysis (VBM) and increased cortical thicknesses in the left-mid cingulate and decreased thicknesses in the left dorsal posterior insular cortex [23]. Macey et al. (2018) (N = 1045, 65 with clinically diagnosed OSA) reported OSA to be associated with increased hippocampal volume, reflected as surface displacement from the mean, in the bilateral CA1, subiculum and uncus, and decreased volumes in the right CA3/dentate, with some gender-specific variation [20]. Analysing the hypoxia and sleep fragmentation separately, Cross et al. (2018) (N = 83) found that oxygen desaturations were associated with decreased cortical thicknesses in the temporal lobe, while increased sleep fragmentation was associated with decreased cortical thicknesses in the right frontal, central, and occipital regions but increased volume in the left hippocampus and amygdala [24]. While it is possible that some of these inconsistencies may at least, in part, be attributable to the small sample sizes or methodological differences such as different OSA definitions, age ranges, or uncontrolled confounders, these results might not be as contradictory as such. Rodents exposed to intermittent hypoxia have been shown to have increased brain water content, while sleep fragmentation and breathing pattern changes associated with obstructions have been shown to be independently associated with blood pressure fluctuations in humans, and increased GM was found to be co-localised with greater amyloid burden [11, 21]. Furthermore, a recent study performed by Baril et al. (2020) (N = 65) found that mild OSA was associated with widespread areas of lower diffusivity along the skeleton in the centre of white matter (WM) in projection, association, and commissural fibres but not the brainstem, as well as lower free-water fraction and no changes in fractional anisotropy (FA) or WM hyperintensity volume, while subjects with moderate to severe OSA showed lower axonal diffusivity in the corpus callosum (CC) [13]. A similar result has also been reported by Zhang et al. (2019) (N = 44), where subjects with moderate-severe OSA exhibited significant lower FA and higher mean and radial diffusivity in the anterior CC [25]. Furthermore, Koo et al. (2020) (N = 79, male only) found OSA to be associated with lower FA in the bilateral anterior thalamic radiations and the right uncinated fasciculus [26]. Low FA is considered to be an indication of poor WM integrity, while low diffusivity has been observed in acute pathological processes associated with restricted water movement in cells, such as reactive gliosis, axonal damage, or cytotoxic oedema [13]. Together, it was hypothesised that increased GM may represent pre-symptomatic stages of OSA-caused brain degeneration characterised by cerebral oedema, increased amyloid deposition, and reactive gliosis, which could eventually lead to reduced GM and WM integrity as the disease progresses [11, 21]. Indeed, signs of OSA-related brain degeneration were detected by Weihs et al. (2021) (N = 690), who found that OSA severity, defined by both AHI and ODI, is associated with age-related local brain atrophy [3]. While no studies regarding treatment were published recently, previous studies found indications that treatment of OSA was able to alleviate OSA-associated damage to the brain [13, 20, 26].
Sleep Apnoea and Cognition
Sleep apnoea has been associated with cognitive dysfunction. In a meta-analysis based on 19,940 subjects, those with OSA were 2.44 times more likely to develop mild cognitive impairment (MCI), with women being at a higher risk (RR = 2.06) than men (RR = 1.18) [27]. Similarly, Beaudin et al. (2020) (N = 1084) found OSA presence and nocturnal hypoxia to be associated with higher cognitive impairment and the presence of moderate-severe OSA with higher odds of having MCI [5]. Interestingly, according to a study performed on 101 subjects by Gagnon et al. (2019), subjects with OSA and MCI seem to be less aware of their cognitive deficits than subjects without OSA [28]. There are two prevalent schools of thought in which OSA is believed to impact cognition, which likely act simultaneously. In the short term, cognitive impairment can be a cause of OSA-induced sleep fragmentation and daytime sleepiness. Non-rapid eye movement (NREM) sleep for example plays an important role in memory processing and consolidation [29]. In an experiment performed by Djonlagic et al. (2020) (N = 53), subjects were asked to perform a motor sequence test in the evening and again in the morning to assess motor memory consolidation [6]. Subjects suffering from OSA during rapid eye movement (REM) and NREM sleep showed significantly lower improvements in the morning tests compared to subjects with no OSA or OSA exclusively during REM sleep [6]. In the long term, the impact of OSA could be the result of hypoxia and sleep fragmentation-induced brain changes (see above), resulting in cognitive dysfunction. This area was investigated by most of the recent studies, but due to the wide variety of different cognitive tests, comparing the results is complicated. An attempt to resolve this was proposed by D’Rozario et al. (2018), who developed a brief 30-min assessment which evaluates neurobehavioural function [7]. Overall, associations were found between OSA and decreased attention [5, 7, 8, 30, 31], memory [5, 26, 32], and executive function [7, 8], which are generally in line with previous findings. The same can be seen in the results of analyses studying OSA-associated severity markers such as AHI or ODI, where the results often fail to replicate the associations between the cognitive markers and the presence of OSA [5, 7, 30, 31]. Specifically, André et al. (2020) found no significant correlations between OSA-associated parameters and cognition (global cognitive function, processing speed, attention, working memory, executive function, and episodic memory) [11]. These discrepancies might in part not only be due to low sample sizes and differences in study populations and methodologies but also be due to the presence of OSA-associated comorbidities, which might influence cognition or the impact of the length between the beginning of the disorder and diagnosis.
Short-term CPAP treatment has been shown to improve, but not reverse some cognitive deficits. Bhat et al. (2018) (N = 182) found significant improvements in objective vigilance in subjects with severe OSA after at least 1 month of CPAP treatment [33]; Jackson et al. (2018) (N = 141) found that 3 months of CPAP resulted in significant improvements, but not reversal to normal neuropsychological function (verbal fluency, psychomotor performance, complex cognitive function, memory, set shifting, mood, quality of life, but not working memory) in subjects with mild-moderate OSA [8]; and Pecotic et al. (2019) (N = 48) reported slight significant improvements in convergent thinking, perception, and psychomotor performance after 1 year of CPAP treatment [34]. Furthermore, in a meta-analysis based on 1926 subjects, M.L. Wang et al. (2020) reported that CPAP treatment (average treatment length: 6 weeks) had a (borderline) significant effect on attention and information processing speed in subjects with severe OSA, with no effects being identified for attention and speed of information processing, executive function, or memory [9]. After the onset of MCI, Richards et al. (2019) (N = 54) and Y. Wang et al. (2020) (N = 17) found that subjects with MCI and mild OSA showed improved psychomotor/cognitive processing speed after 1 year of CPAP treatment [35, 36]. One reason for the lack of strong effects is due to poor CPAP treatment compliance. It is however also likely that the improvements do not represent long-term permanent changes but are rather related to reduced sleepiness and sleep fragmentation as a result of the CPAP treatment. Indeed, all but M.L. Wang et al. (2020) and Y. Wang (2020) reported a significant decrease in daytime sleepiness after treatment [8, 9, 33, 34, 36], although Bhat et al. (2018) did not find changes in sleepiness to be predictive of improved vigilance [33]. However, older studies based on limited data have found that cognitive function did not improve after CPAP in subjects who did not experience subjective daytime sleepiness, irrespective of OSA severity [9, 37].
Sleep Apnoea and Alzheimer’s Dementia
Alzheimer’s dementia (AD) is an irreversible and deadly neurodegenerative disorder characterised by deteriorating cognitive abilities. While its cause is still poorly understood, progression of the disorder is largely associated with amyloid plaques, neurofibrillary tangle consisting of tau protein, and loss of neuronal connections in the brain [18]. Regarding sleep apnoea, there is a complex relationship between OSA and Alzheimer’s dementia. While none is responsible for the other, both influence each other’s pathological processes resulting in a possible bidirectional relationship [38]. In the one direction, AD-related changes in the brain result in sleep dysregulation and, as a consequence, high prevalence of sleep disorders such as OSA in Alzheimer’s disease patients [38]. In the other direction, OSA has been proposed as a risk factor for AD as it promotes or enhances AD-related subclinical pathological processes. In fact, multiple recent studies based on large cohorts have shown that subjects with OSA are, depending on the study, between 1.49 and 2.21 times more likely to develop AD than individuals not suffering from OSA [16, 17, 39,40,41]. Furthermore, Bubu et al. (2021) showed that individuals with OSA have shorter progression times between cognitively normal (CN) to mild cognitive impairment (MCI) or MCI to AD [17]. One proposed mechanism through which OSA could have an effect on AD pathology is via a dysregulation of the Aβ metabolism caused by intermittent hypoxia and reduced clearance from interstitial to cerebrospinal fluid (CSF) caused by sleep fragmentation, resulting in decreased CSF Aβ 40 and 42 levels and increased Aβ plaque formation. Recent studies support this, with Liguori et al. (2019) finding that CSF Aβ40 and 42 levels were lower in OSA patients than those in control subjects but higher than those in AD subjects [14]; Jackson et al. (2020) (N = 46) finding that OSA severity, specifically during NREM sleep, was associated with increased brain Aβ burden [42]; and André et al. (2020) identifying a significant association between increased florbetapir, a marker for amyloid plaques, uptake, and OSA presence [11]. Longitudinally, in a 2-year follow-up study on 208 CN subjects, Sharma et al. (2018) identified a significant association between the annual rate of change of Aβ 42 and OSA severity, which was stronger than the change predicted by ApoE4, currently the strongest risk factor known for AD [15]. This was also observed by a study from Bubu et al. (2019) on 1639 CN and MCI subjects (mean follow-up period: 2.52 ± 0.51 years), who additionally observed that subjects with OSA experienced a greater annual rate of change in florbetapir uptake, indicating a greater buildup of amyloid plaques and providing further validity to this mechanism [18]. Similar results were found in autopsied hippocampi and brainstems of 34 subjects with OSA [43]. While not identifying significant correlations in the brainstem, the authors found hypoxia severity to be a significant predictor of Aβ plaque burden in the hippocampus [43]. Concerning tau, the relationship between the protein and sleep apnoea is even less understood. While some studies found no association between OSA and CSF total, phosphorylated tau, or neurofibrillary tangles [14, 15, 43], others did, although it remains to be seen if these are caused by OSA itself or if they are age-related early manifestations of AD-related pathological processes [18, 44, 45]. With no AD treatment being available, prevention through treatment of risk factors is currently the only way to delay the onset of AD, with OSA being a viable target. Indeed, greater CPAP-induced OSA improvement was associated with decreased CSF Aβ and Tau levels in 18 OSA subjects, who underwent 1–4 months of CPAP treatment, and OSA subjects receiving CPAP were found to have a lower risk of developing AD than subjects without CPAP treatment [16, 19].
Sleep Apnoea and Parkinson’s Disease
Parkinson’s disease (PD) is a progressive and, currently, untreatable neurodegenerative disorder primarily affecting the motor system. OSA often coincides with PD, although reported prevalence varies widely between 20 and 70.1% [46]. There are indications that OSA may act as a risk factor before the onset of PD. In a recent meta-analysis performed by Sun et al. (2020), subjects with OSA were 1.56 times more likely to develop PD than controls [47]. The exact mechanisms at play are still not fully understood, but, similar to AD, OSA, although not causing the disorder, likely plays a role in promoting or enhancing PD-associated pre-clinical pathological processes. Concurrent with this, Sun et al. (2019) (N = 88) reported that both OSA severity and hypoxia markers were associated with increased levels of plasma α-synuclein, a key protein involved in PD pathology, in healthy adults [48]. With the onset of PD, the relationship between OSA and PD becomes more complex. While there is no evidence that the incidence of OSA is higher in the PD than that in the non-PD population, OSA has an impact on the disorder when present [47]. A meta-analysis performed by Elfi et al. (2020) found that subjects with PD and OSA showed greater cognitive and motor deficits than subjects with PD but without OSA [49]. Similar results were also observed by Meng et al. (2020) and Kaminska et al. (2018) (same sample, N = 67), who additionally found that 12-month CPAP treatment resulted in improved PD-associated non-motor symptoms and a stabilisation of motor function [50, 51]. While this indicates that OSA has a detrimental effect on PD-associated cognitive and motor functions, there are also findings that PD has an effect on OSA severity. In the early stages of PD, the disorder has protective effects due to PD-induced weight loss, one of the biggest risk factors for OSA, while PD-related factors such as impaired ventilation control and upper airway motor instability might increase OSA severity as the disorder progresses [52]. Support for the latter was published by Bahia et al. (2019) (N = 48), where PD subjects with a laryngopharyngeal motor dysfunction were three times more likely to have OSA than those without the dysfunction [53].
Conclusion
There is a complex relationship between OSA and neurodegeneration, with both influencing each other and different aspects of the disorder having different effects. In this review, we have summarised recent findings on the association between OSA and brain structure, cognition, and the two most common neurodegenerative disorders, namely Alzheimer’s dementia and Parkinson’s disease. Overall, recent studies reported associations between OSA and grey and white matter alterations [3, 10,11,12, 20, 22,23,24], and changes in brain diffusion [13, 25, 26], as well as impaired cognition, specifically regarding memory [5, 6, 26, 32], attention [5, 7, 8, 30, 31], and executive control [7, 8]. Furthermore, subjects with OSA were found to have a higher risk of developing mild cognitive impairment (MCI) [5, 27], Alzheimer’s dementia [16, 17, 39,40,41], and Parkinson’s disease [47], and show shorter progression times between cognitively normal and MCI or MCI and Alzheimer’s dementia [17]. But while these studies have added further insights, there are some discrepancies in their results and large gaps remain to get a comprehensive overview of the exact mechanism at play here. Next to the problem of generally small sample sizes and the presence of a complex and dynamic system influenced by a variety of factors, the lack of conclusive effects might be due to the way OSA itself is defined. A large majority of studies considered in this review have defined OSA as a categorical variable based on various AHI cutoffs, medical diagnoses, or self-reported symptoms. Next to the difficulty of comparing such results between different studies, there is also the question of what such an association represents, as such a broad phenotype makes it close to impossible to distinguish between effects caused by OSA and the ones caused by OSA-associated comorbidities such as obesity, hypertension, diabetes, or depression [54]. Using the continuous AHI instead could be a viable solution, although this does not resolve all issues either. While this index, in combination with other symptoms, is enough to diagnose OSA in a clinical setting, it might not be valid to investigate specific OSA-related pathways [4]. Firstly, the index combines both hypoxic and sleep fragmentation-related events, which individually influence neurodegenerative processes, but not necessarily in an additive fashion. Furthermore, the index also only assesses the frequency, while completely ignoring the length of the individual events. A subject with few but very long events would therefore be considered “healthier” than a subject with numerous but short events, especially if cutoffs are used. Alternative scores such as the arousal index and oxygen desaturation index, or to incorporate length, metrics such as percentage/time of sleep spent below a certain oxygen saturation threshold, could prove to be much more informative.
In conclusion, while not being the cause, there are strong indications that OSA is a major risk factor for neurodegeneration and neurodegenerative disorders. OSA treatment was shown to alleviate some of the damage and improve cognitive deficits. The underlying mechanisms, however, are yet to be fully understood, highlighting the need for large, preferably longitudinal studies based on standardised metrics, and more importantly, assessing OSA-related hypoxia and sleep fragmentation separately. However, with no viable cure available for most neurodegenerative disorders, OSA shows to be a promising target to delay their onset.
References
Benjafield AV, Ayas NT, Eastwood PR, Heinzer R, Ip MSM, Morrell MJ, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019;7:687–98.
Lee JJ, Sundar KM. Evaluation and management Of adults with obstructive sleep apnea syndrome. Lung. 2021;199:87–101.
Weihs A, Frenzel S, Wittfeld K, et al. Associations between sleep apnea and advanced brain aging in a large-scale population study. Sleep. 2021;44:1–15.
Kapur VK, Auckley DH, Chowdhuri S, Kuhlmann DC, Mehra R, Ramar K, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an american academy of sleep medicine clinical practice guideline. J Clin Sleep Med. 2017;13:479–504.
Beaudin AE, Raneri JK, Ayas NT, et al (2020) Cognitive function in a sleep clinic cohort of patients with obstructive sleep apnea. Ann Am Thorac Soc AnnalsATS.202004-313OC
Djonlagic I, Guo M, Igue M, Malhotra A, Stickgold R. REM-related obstructive sleep apnea: when does it matter? Effect on motor memory consolidation versus emotional health. J Clin Sleep Med. 2020;16:377–84.
D’Rozario AL, Field CJ, Hoyos CM, Naismith SL, Dungan GC, Wong KKH, et al. Impaired neurobehavioural performance in untreated obstructive sleep apnea patients using a novel standardised test battery. Front Surg. 2018;5:1–8.
Jackson ML, McEvoy RD, Banks S, Barnes M. Neurobehavioral impairment and cpap treatment response in mild-moderate obstructive sleep apnea. J Clin Sleep Med. 2018;14:47–56.
Wang M-L, Wang C, Tuo M, Yu Y, Wang L, Yu J-T, et al. Cognitive effects of treating obstructive sleep apnea: a meta-analysis of randomized controlled trials. J Alzheimer’s Dis. 2020;75:705–15.
Marchi NA, Ramponi C, Hirotsu C, Haba-Rubio J, Lutti A, Preisig M, et al. Mean oxygen saturation during sleep is related to specific brain atrophy pattern. Ann Neurol. 2020;87:921–30.
André C, Rehel S, Kuhn E, Landeau B, Moulinet I, Touron E, et al. Association of sleep-disordered breathing with Alzheimer disease biomarkers in community-dwelling older adults. JAMA Neurol. 2020;77:716–24.
Kim REY, Abbott RD, Kim S, Thomas RJ, Yun C-H, Kim H, et al. Sleep duration, sleep apnea, and gray matter volume. J Geriatr Psychiatry Neurol. 2021:089198872098891. https://doi.org/10.1177/0891988720988918.
Baril A, Gagnon K, Descoteaux M, et al. Cerebral white matter diffusion properties and free-water with obstructive sleep apnea severity in older adults. Hum Brain Mapp. 2020;41:2686–701.
Liguori C, Mercuri NB, Nuccetelli M, Izzi F, Cordella A, Bernardini S, et al. Obstructive sleep apnea may induce orexinergic system and cerebral β-amyloid metabolism dysregulation: is it a further proof for Alzheimer’s disease risk? Sleep Med. 2019;56:171–6.
Sharma RA, Varga AW, Bubu OM, Pirraglia E, Kam K, Parekh A, et al. Obstructive sleep apnea severity affects amyloid burden in cognitively normal elderly. A longitudinal study. Am J Respir Crit Care Med. 2018;197:933–43.
Tsai MS, Li HY, Huang CG, Wang RYL, Chuang LP, Chen NH, et al. Risk of Alzheimer’s disease in obstructive sleep apnea patients with or without treatment: real-world evidence. Laryngoscope. 2020;130:2292–8.
Bubu OM, Umasabor-Bubu OQ, Turner AD, Parekh A, Mullins AE, Kam K, et al. Self-reported obstructive sleep apnea, amyloid and tau burden, and Alzheimer’s disease time-dependent progression. Alzheimer’s Dement. 2021;17:226–45.
Bubu OM, Pirraglia E, Andrade AG, et al. Obstructive sleep apnea and longitudinal Alzheimer’s disease biomarker changes. Sleep. 2019;42:1–13.
Ju Y-ES, Zangrilli MA, Finn MB, Fagan AM, Holtzman DM. Obstructive sleep apnea treatment, slow wave activity, and amyloid-β. Ann Neurol. 2019;85:291–5.
Macey PM, Prasad JP, Ogren JA, Moiyadi AS, Aysola RS, Kumar R, et al. Sex-specific hippocampus volume changes in obstructive sleep apnea. NeuroImage Clin. 2018;20:305–17.
Baril A-A, Gagnon K, Brayet P, Montplaisir J, De Beaumont L, Carrier J, et al. Gray matter hypertrophy and thickening with obstructive sleep apnea in middle-aged and older adults. Am J Respir Crit Care Med. 2017;195:1509–18.
Huang X, Tang S, Lyu X, Yang C, Chen X. Structural and functional brain alterations in obstructive sleep apnea: a multimodal meta-analysis. Sleep Med. 2019;54:195–204.
Taylor KS, Millar PJ, Murai H, Haruki N, Kimmerly DS, Bradley TD, et al. Cortical autonomic network gray matter and sympathetic nerve activity in obstructive sleep apnea. Sleep. 2018;41:1–10.
Cross NE, Memarian N, Duffy SL, Paquola C, LaMonica H, D’Rozario A, et al. Structural brain correlates of obstructive sleep apnoea in older adults at risk for dementia. Eur Respir J. 2018;52:1800740.
Zhang B, Zhu D, Zhao W, Zhang Y, Yang Y, Zhang C, et al. Selective microstructural integrity impairments of the anterior corpus callosum are associated with cognitive deficits in obstructive sleep apnea. Brain Behav. 2019;9:1–9.
Koo DL, Kim HR, Kim H, Seong J-K, Joo EY. White matter tract-specific alterations in male patients with untreated obstructive sleep apnea are associated with worse cognitive function. Sleep. 2020;43:1–10.
Zhu X, Zhao Y. Sleep-disordered breathing and the risk of cognitive decline: a meta-analysis of 19,940 participants. Sleep Breath. 2018;22:165–73.
Gagnon K, Baril A-A, Montplaisir J, Carrier J, de Beaumont L, D ' Aragon C, et al. Disconnection between self-reported and objective cognitive impairment in obstructive sleep apnea. J Clin Sleep Med. 2019;15:409–15.
Rauchs G, Desgranges B, Foret J, Eustache F. The relationships between memory systems and sleep stages. J Sleep Res. 2005;14:123–40.
Alomri RM, Kennedy GA, Wali SO, Ahejaili F, Robinson SR. Differential associations of hypoxia, sleep fragmentation, and depressive symptoms with cognitive dysfunction in obstructive sleep apnea. Sleep. 2020:1–9.
Simões EN, Padilla CS, Bezerra MS, Schmidt SL. Analysis of attention subdomains in obstructive sleep apnea patients. Front Psychiatry. 2018;9:435.
Delhikar N, Sommers L, Rayner G, Schembri R, Robinson SR, Wilson S, et al. Autobiographical memory from different life stages in individuals with obstructive sleep apnea. J Int Neuropsychol Soc. 2019;25:266–74.
Bhat S, Gupta D, Akel O, Polos PG, DeBari VA, Akhtar S, et al. The relationships between improvements in daytime sleepiness, fatigue and depression and psychomotor vigilance task testing with CPAP use in patients with obstructive sleep apnea. Sleep Med. 2018;49:81–9.
Pecotic R, Dodig IP, Valic M, Galic T, Kalcina LL, Ivkovic N, et al. Effects of CPAP therapy on cognitive and psychomotor performances in patients with severe obstructive sleep apnea: a prospective 1-year study. Sleep Breath. 2019;23:41–8.
Richards KC, Gooneratne N, Dicicco B, Hanlon A, Moelter S, Onen F, et al. CPAP adherence may slow 1-year cognitive decline in older adults with mild cognitive impairment and apnea. J Am Geriatr Soc. 2019;67:558–64.
Wang Y, Cheng C, Moelter S, Fuentecilla JL, Kincheloe K, Lozano AJ, et al. One year of continuous positive airway pressure adherence improves cognition in older adults with mild apnea and mild cognitive impairment. Nurs Res. 2020;69:157–64.
Steiropoulos P, Galbiati A, Ferini-Strambi L. Detection of mild cognitive impairment in middle-aged and older adults with obstructive sleep apnoea: does excessive daytime sleepiness play a role? Eur Respir J. 2019;53:1801917.
Liguori C, Maestri M, Spanetta M, Placidi F, Bonanni E, Mercuri NB, et al. Sleep-disordered breathing and the risk of Alzheimer’s disease. Sleep Med Rev. 2021;55:101375.
Lee JE, Yang SW, Ju YJ, Ki SK, Chun KH. Sleep-disordered breathing and Alzheimer’s disease: a nationwide cohort study. Psychiatry Res. 2019;273:624–30.
Lutsey PL, Misialek JR, Mosley TH, Gottesman RF, Punjabi NM, Shahar E, et al. Sleep characteristics and risk of dementia and Alzheimer’s disease: the atherosclerosis risk in communities study. Alzheimer’s Dement. 2018;14:157–66.
Shi L, Chen S-J, Ma M-Y, Bao Y-P, Han Y, Wang Y-M, et al. Sleep disturbances increase the risk of dementia: a systematic review and meta-analysis. Sleep Med Rev. 2018;40:4–16.
Jackson ML, Cavuoto M, Schembri R, Doré V, Villemagne VL, Barnes M, et al. Severe obstructive sleep apnea is associated with higher brain amyloid burden: a preliminary pet imaging study. J Alzheimer’s Dis. 2020;78:611–7.
Owen JE, Benediktsdottir B, Cook E, Olafsson I, Gislason T, Robinson SR. Alzheimer’s disease neuropathology in the hippocampus and brainstem of people with obstructive sleep apnea. Sleep. 2021;44:1–10.
Motamedi V, Kanefsky R, Matsangas P, Mithani S, Jeromin A, Brock MS, et al. Elevated tau and interleukin-6 concentrations in adults with obstructive sleep apnea. Sleep Med. 2018;43:71–6.
Díaz-Román M, Pulopulos MM, Baquero M, Salvador A, Cuevas A, Ferrer I, et al. Obstructive sleep apnea and Alzheimer’s disease-related cerebrospinal fluid biomarkers in mild cognitive impairment. Sleep. 2021;44:1–8.
Shen Y, Shen Y, Dong Z-F, Pan P-L, Shi H, Liu C-F. Obstructive sleep apnea in Parkinson’s disease: a study in 239 Chinese patients. Sleep Med. 2020;67:237–43.
Sun A-P, Liu N, Zhang Y-S, Zhao H-Y, Liu X-L. The relationship between obstructive sleep apnea and Parkinson’s disease: a systematic review and meta-analysis. Neurol Sci. 2020;41:1153–62.
Sun H, Sun B, Chen D, Chen Y, Li W, Xu M, et al. Plasma α -synuclein levels are increased in patients with obstructive sleep apnea syndrome. Ann Clin Transl Neurol. 2019;6:788–94.
Elfil M, Bahbah EI, Attia MM, Eldokmak M, Koo BB. Impact of obstructive sleep apnea on cognitive and motor functions in Parkinson’s disease. Mov Disord. 2021;36:570–80.
Meng L, Benedetti A, Lafontaine A-L, Mery V, Robinson AR, Kimoff J, et al. Obstructive sleep apnea, CPAP therapy and Parkinson’s disease motor function: a longitudinal study. Parkinsonism Relat Disord. 2020;70:45–50.
Kaminska M, Mery VP, Lafontaine A-L, Robinson A, Benedetti A, Gros P, et al. Change in cognition and other non-motor symptoms with obstructive sleep apnea treatment in Parkinson disease. J Clin Sleep Med. 2018;14:819–28.
Zeng J, Wei M, Li T, Chen W, Feng Y, Shi R, et al. Risk of obstructive sleep apnea in Parkinson’s disease: a meta-analysis. PLoS One. 2013;8:e82091.
Bahia CMCS, Pereira JS, Lopes AJ. Laryngopharyngeal motor dysfunction and obstructive sleep apnea in Parkinson’s disease. Sleep Breath. 2019;23:543–50.
Bonsignore MR, Baiamonte P, Mazzuca E, Castrogiovanni A, Marrone O. Obstructive sleep apnea and comorbidities: a dangerous liaison. Multidiscip Respir Med. 2019;14:8.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work specifically was supported by the Deutsche Forschungsgemeinschaft (DFG, grant number: GR 1912/13-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
HJG has received travel grants and speaker’s honoraria from Fresenius Medical Care, Neuraxpharm, Servier, and Janssen Cilag as well as research funding from Fresenius Medical Care.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Sleep Apnea in the Golden Age
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Weihs, A., Frenzel, S. & Grabe, H.J. The Link Between Obstructive Sleep Apnoea and Neurodegeneration and Cognition. Curr Sleep Medicine Rep 7, 87–96 (2021). https://doi.org/10.1007/s40675-021-00210-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40675-021-00210-5