
Abstract
Purpose of review
Axial spondyloarthritis remains an area of significant unmet clinical need with only two immune pathways currently targeted by licenced therapies compared to other immune-mediated inflammatory joint disorders such as rheumatoid arthritis where a multitude of therapeutic options are available. This review will look at emerging therapeutic targets in axial spondyloarthritis beyond the neutralisation of IL-17A and TNF by monoclonal antibodies.
Recent findings
Several promising targets are in various stages of pre-clinical and clinical development in axial spondyloarthritis. These include small molecule approaches to target transcription factors, epigenetic modification and intracellular modulation of cytokine signalling by kinase inhibition. GM-CSF has also emerged as a potential driver of inflammation.
Summary
A number of novel and promising therapeutic options are in various stages of development in axial spondyloarthritis. The Janus kinase inhibitors have shown great promise in other immune-mediated inflammatory disorders and will be an exciting addition to the axial spondyloarthritis field as the first oral disease-modifying agents. GM-CSF blockade also shows great promise since antibodies for neutralising this cytokine are safe in patients and have shown efficacy in other immune-mediated inflammatory diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
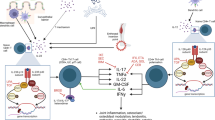
Axial spondyloarthritis (SpA) is an inflammatory disease affecting up to 0.5% of the population in North America and Europe and encompasses ankylosing spondylitis (AS) [1, 2]. The disease has a strong association with HLA-B27 inheritance [3] and typically causes inflammation in the spine and sacroiliac joints [4]. This inflammation can progress to new bone formation and eventual fusion of the spine in severe cases [5]. Despite recent advances, treatment options in this disease remain limited compared to rheumatoid arthritis. Physiotherapy and non-steroidal anti-inflammatory drugs (NSAID) are first-line therapies followed by biological anti-TNF, and more recently, anti-IL-17A therapies [6]. Currently, there are no approved therapies on the market for patients who are refractory to these two cytokine targets and thus a great unmet clinical need remains. There are several novel therapeutic approaches in development and this review will highlight some of these approaches beyond anti-IL-17A and anti-TNF. We will concentrate first on novel approaches to targeting “type 17 responses” (also referred to as type 3 immune responses), with a particular focus on the potential role of GM-CSF as a therapeutic target, before describing other approaches such as phosphodiesterase 4 inhibition.
Targeting ROR-γt, a key type 17 transcription factor
The role of the IL-23/IL-17A axis in SpA pathogenesis was first suggested following genome-wide association studies implicating polymorphisms of the IL-23 receptor [7], and has now been proven by the success of anti-IL-17A therapies in the treatment of in AS [8•]. Thus far, focus on this axis has predominantly been on the role of IL-17A secreted by CD4 T cells, but it is now appreciated that this axis involves numerous additional effector cytokines. In particular, Th17 cells have been shown to co-produce IL-17F, IL-22, IFNγ, IL-26 and GM-CSF [9, 10]. Additionally, the axis is not only limited to the CD4 compartment and other cell types including CD8 [11], mucosal-associated invariant T (MAIT) cells [12], γδ T cells [13] and innate-lymphoid cells [14] which are all capable of activating this inflammatory programme. All these cell types share expression of the retinoid orphan-related receptor-γt (ROR-γt) and thus targeting this transcription factor would be an attractive approach for switching off this inflammatory module. The first reports of ROR-γt inhibitors used digoxin to inhibit this transcription factor in murine cells [15] but the doses required to achieve this inhibition in human cells would be toxic. Over the years, a number of small molecule ROR-γt inhibitors have been developed and tested in several in vitro Th17-driven systems. One such report showed ROR-γt inhibitors to be capable of supressing Th17 signatures from psoriatic patient samples [16], whilst another report showed inhibition of both IL-17A and IL-17F by another ROR-γt inhibitor (SR2211) [17]. We developed an assay to expand Th17 cells from the peripheral blood and synovial fluid of SpA patients and showed a clear inhibition of IL-17A by two ROR-γt inhibitors (MRL-367 and MRL-248) [18••]. In addition to the inhibition of secreted IL-17A, we also demonstrated a downregulation of a number of inflammatory genes associated with the Th17 programme including IL-17F, IL-22, and IL-26. Despite the early promise of a number of ROR-γt inhibitors, recently, reports of thymic aberrations in Lewis rats treated with ROR-γt inhibitor compounds [19] have raised fears over long-term safety. It remains to be seen whether this safety concern is a class effect or specific to one compound, and at the time of writing, there is currently one registered trial on clinicaltrials.gov for a ROR-γt inhibitor ointment actively recruiting but no trials of systemic administration of ROR-γt inhibitors. If ROR-γt inhibition does not prove to be safe, there are likely other molecular targets in or on Th17 cells that are worthy of investigation in the context of SpA.
Epigenetic modulation of type 17 immune responses
Epigenetic modulation of key genes involved in Th17 differentiation and effector function, for example through the therapeutic targeting of bromodomains, is another area where novel therapies are currently being developed. Bromodomains recognise acetylated lysine residues in histones and thus play a very important role in the control of gene expression [20]. Pan-BET bromodomain inhibitors such as JQ1 have shown efficacy in Th17-driven murine models of inflammation including experimental autoimmune encephalomyelitis [21], but concerns remain about the specificity of such broad bromodomain inhibitors in clinical trials. In a study published by our group in collaboration with the Structural Genomics Consortium, we used a potent and very selective inhibitor for the bromodomains of the transcriptional coactivators CBP and its close relative p300 to inhibit Th17 responses from the peripheral blood of SpA patients [22•]. This approach was more selective compared to the pan-bromodomain inhibitor JQ1 and was successful in inhibiting IL-17A and GM-CSF but not IL-17F and IL-22. It is not clear if the sparing of IL-17F and IL-22 would be sufficient for switching off inflammation in an in vivo model, and the selective inhibitor CBP30 in its current form does not have the appropriate pharmacodynamics properties for such in vivo studies. Nevertheless, if safe and stable inhibitors could be delivered to inflammatory sites, this would be a promising therapeutic strategy.
JAK inhibition
The Janus kinase (JAK) system of signalling downstream of cytokine binding has over recent years been an area of great interest in the treatment of inflammatory disorders. Recent positive results seen in rheumatoid arthritis for tofacitinib, a JAK1/3 (plus some JAK2 activity) inhibitor [23], and baricitinib, a JAK1/2 inhibitor [24], have for the first time offered the promise of biologic-like efficacy with an oral small molecule inhibitor. In spondyloarthritis, we have tested the effects of tofacitinib on the Th17 responses of patients with SpA using patient-derived PBMCs in vitro and have showed a significant inhibition in the amount of secreted IL-17A, IL-17F, IL-22, and GM-CSF [25]. These results are in agreement with the results of the recently published, placebo-controlled trial in ankylosing spondylitis where tofacitinib met its primary ASAS20 endpoint compared to placebo in a phase II 16-week dose-ranging study [26••]. However, the response was only significant for the 5mg twice daily group (80.8% response), while in the 2mg and 10mg twice daily groups the response was not significant at 51.9% and 55.8% respectively, compared to a placebo response of 40.1%. Moreover, the data also went against expectations for those achieving the higher bar of an ASAS40 response with 42.3% of those on the lowest 2mg dose achieving an ASAS40 response versus 38.5% of those on the highest 10mg dose. Crucially, the Pfizer pipeline currently (Pfizer.com/pipeline) does not show plans for a phase III study for tofacitinib in SpA and there are no active trials registered on clinicaltrials.gov for either tofacitinib or baricitinib. We think that this is a promising area for future research, although further studies are needed to determine which key members of the JAK/STAT family are involved in SpA pathogenesis, and whether selective or broad targeting of these is most appropriate.
GM-CSF as a novel target in spondyloarthritis
GM-CSF is a haematopoietic growth factor and multifunctional cytokine [27]. Within the CD4 T cell compartment, GM-CSF production in the context of a Th17 response has been shown to be pathogenic in murine immune-mediated inflammatory responses [28, 29]. A more recent study showed neutralisation of GM-CSF can ameliorate inflammatory arthritis and lung inflammation in the SKG mouse model of SpA [30]. These immuno-modulatory properties of GM-CSF make it a novel therapeutic target in diseases characterised by immune dysregulation, such as RA and SpA.
GM-CSF gene expression can be induced by interleukin IL-1β, IL-6, TNF and endotoxin in a variety of cell types, including fibroblasts, endothelial cells, T cells, macrophages and epithelial cells [31]. GM-CSF exerts its effector function by binding to its cell surface receptor (GM-CSFR) which is coupled with JAK2, and the downstream mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signalling pathways [32, 33]. GM-CSFRs are predominantly expressed on myeloid cells but can also be found on some non-haematopoietic cells like fibroblasts [31].
In health, GM-CSF plays a critical role in haematopoiesis and in the immune response to injury and infection. GM-CSF shapes the interface between innate and adaptive immunity, by interacting with autocrine and paracrine cytokine networks [34] and by promoting myeloid cell survival, proliferation, activation and differentiation [35]. Furthermore, GM-CSF modulates subsequent adaptive immune responses by influencing antigen-presenting cell (APC) [36, 37] and T helper (Th) cell differentiation [38,39,40,41].
GM-CSF and its receptor have been shown to be overexpressed in synovial joints of patients with immune-mediated inflammatory arthritides like RA [36, 42] and SpA [43]. GM-CSF is thought to promote joint damage by recruiting granulocyte and macrophage precursors from activated bone marrow adjacent to synovial joints [44]. It then induces their differentiation into a more inflammatory phenotype and also activates monocyte-derived dendritic cell maturation [45]. In addition, GM-CSF causes the release of pro-inflammatory chemokines such as CCL17 [46]. GM-CSF also facilitates cartilage destruction and bone resorption by the induction of matrix metalloproteinases [47] and the osteoclast activating factor RANKL [48, 49]. Moreover, GM-CSF has been shown to be responsible for arthritic pain in mice independently of its pro-inflammatory characteristics and GM-CSF receptors have been shown to be expressed on nerve endings [50, 51].
Neutralising anti-GM-CSF monoclonal antibodies have been very effective in alleviating pain and in improving joint damage and inflammation in arthritic mice [30, 42]. This effect can be potentiated by concomitant blockade of IL-17A [52]. Furthermore, the benefits of GM-CSF blockade appear to be independent of IL-6 and TNF [30, 37, 53]. Five human monoclonal antibodies (mAbs) targeting the GM-CSF pathway are currently in phase I and II clinical trials in RA. Mavrilimumab, a high-affinity IgG4 monoclonal antibody (mAb) against the α-chain of GM-CSFR, has been evaluated in four phase II clinical trials [54, 55]. These comprise a total of 748 patients with moderate RA and an inadequate response to at least one disease-modifying anti-rheumatic drug (DMARD), usually methotrexate. Patients who failed one anti-TNF agent were also included. The ACR20 response improved significantly in a dose-dependent manner with mavrilimumab treatment (p < 0.001 when compared to placebo) [54]. Biomarkers in addition to pain and physical function scores also improved significantly [56]. The EARTH explorer II trial directly compared mavrilimumab with the anti-TNF agent golimumab in incomplete responders to DMARDs and/or one anti-TNF mAb. Whilst this study was not sufficiently powered to reach statistical significance, its findings nevertheless suggest that mavrilimumab is at least as effective as golimumab in these patients [57]. Moreover, benefits were maintained and no significant safety concerns were identified at 5-year follow-up in an open-label extension study comprising 442 patients [56]. Of note, phase I and II trials using mAbs that target GM-CSF directly, namely MOR103, namilumab and MORAb-022, are similarly encouraging [58]. Anti-GM-CSF/CSFR mAbs may therefore be a valuable novel biological treatment option; both as first-line therapy in anti-TNF naïve patients and as second-line therapy in anti-TNF non-responders. In addition, GM-CSF blockade may have significant analgesic benefits which would address a significant unmet need in inflammatory disorders. GM-CSF may be a very effective target in SpA based on the pre-clinical data from our group and others which show an expansion of GM-CSF-producing CD4 and CD8 cells in the blood and synovial fluid of patients with axial SpA [59••] compared to healthy donors and RA inflammatory control patients suggesting this pathway may be more relevant in SpA than RA.
Other therapeutic targets
Phosphodiesterase inhibition
One recent oral drug modulating inflammatory pathways is apremilast, a phosphodiesterase 4 (PDE4) inhibitor. PDE4 is the main class of phosphodiesterase expressed in T cells and inhibition of this enzyme leads to accumulation of cyclic AMP (cAMP). The persistence of cAMP in T cells after activation has been shown to decrease the amount of secreted IFNγ in vitro [60]. However, the role of cAMP in Th17 biology is less clear. Boniface and colleagues showed that cAMP was required for the differentiation of Th17 cells [61] in vitro and can be mimicked by the addition of an intracellular cAMP analogue [61], whilst the addition of apremilast to in-vitro T cell cultures broadly activated with anti-CD3 only was reported to decrease the amount of secreted IL-17A in the culture supernatants [62]. The clinical efficacy of apremilast has been somewhat mixed. In psoriasis [63] and psoriatic arthritis [64], apremilast has been shown to be effective in randomised placebo-controlled trials although the benefit in psoriatic arthritis seemed modest compared to biologic therapies with an ACR20 response rate of 38.1%. In ankylosing spondylitis, apremilast failed to meet its primary endpoint in a small phase II study but was associated with a numerically greater improvement from baseline for all clinical assessments [65]. A larger phase III study was undertaken, and results reported on clinicaltrial.gov show no difference in the ASAS20 primary outcome compared to placebo at 24 weeks. The failure of apremilast in ankylosing spondylitis may be due to patient heterogeneity or to the complexity of the underlying T cell biology and the exact role elevated cAMP levels play in Th17 differentiation and function.
ERAP1 or HLA-B27 inhibition?
A number of other potential therapeutic targets have been suggested by genetic studies of AS including genome-wide association studies. Endoplasmic reticulum aminopeptidase 1 (ERAP1) is strongly associated with predilection to AS [7], has effects on HLA-B27 cell biology and cell surface expression, with ERAP1 inhibition mitigating these effects [66]. Similarly, HLA-B27 itself may also be a potential therapeutic target. Whilst targeting the entirety of surface HLA-B27 might render individuals at risk of viral infections, it might be possible to target the free heavy chain or homodimeric forms expressed on the cell surface with specific monoclonal antibodies such as HD6 [67].
Conclusion
Despite recent advances, axial SpA still represents a therapeutic challenge with a significant unmet clinical need. Firstly, there is still no approved oral disease-modifying agent in axial SpA although several promising novel approaches are in the pipeline. Currently, our view is that the most promising oral agents may be the JAK inhibitors, which have shown efficacy in the clinical trial setting, although it remains to be determined which particular JAKs are most important in SpA. Secondly, there is currently a lack of therapeutic options for patients who are refractory to anti-TNF and anti-IL-17A therapies. Pre-clinical data suggests that blockade of GM-CSF may represent a new biological approach for these refractory patients and the safety and tolerability of GM-CSF blocking antibodies has already been shown in patients with other inflammatory diseases such as RA but a clinical trial in SpA is still needed.
References and Recommended Reading
Recently published papers of particular interest have been highlighted as: • Of importance •• Of major importance
Reveille JD. Epidemiology of spondyloarthritis in North America. Am J Med Sci. 2011;341:284–6.
Braun J, et al. Prevalence of spondylarthropathies in HLA-B27 positive and negative blood donors. Arthritis Rheum. 1998;41:58–67.
Bowness P. Hla-B27. Annu Rev Immunol. 2015;33:29–48.
Dougados M, Baeten D. Spondyloarthritis. Lancet. 2011;377:2127–37.
Tam L-S, Gu J, Yu D. Pathogenesis of ankylosing spondylitis. Nat Rev Rheumatol. 2010;6:399–405.
Braun J, et al. 2010 update of the ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2011;70:896–904.
Cortes A, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45:730–8.
• Baeten D, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373:2534–48. This was the definitive randomised controlled trial showing efficacy of anti-IL-17A inhibitors in patients with spondyloarthritis.
Wilson NJ. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7.
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–71.
Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, et al. Interleukin-17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol Hoboken NJ. 2014;66:1272–81.
Walker LJ, Kang YH, Smith MO, Tharmalingham H, Ramamurthy N, Fleming VM, et al. Human MAIT and CD8αα cells develop from a pool of type-17 precommitted CD8+ T cells. Blood. 2012;119:422–33.
Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating IL-17-secreting IL-23 receptor-positive γδ T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012;64(5):1420–9.
Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–89.
Huh JR, Leung MWL, Huang P, Ryan DA, Krout MR, Malapaka RRV, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature. 2011;472:486–90.
Skepner J, et al. Pharmacologic inhibition of RORγt regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol Baltim Md. 2014;1950(192):2564–75.
Melton AC, Melrose J, Alajoki L, Privat S, Cho H, Brown N, et al. Regulation of IL-17A production is distinct from IL-17F in a primary human cell co-culture model of T cell-mediated B cell activation. PLoS One. 2013;8:e58966.
•• de Wit J, et al. RORγt inhibitors suppress TH17 responses in inflammatory arthritis and inflammatory bowel disease. J Allergy Clin Immunol. 2016;137:960–3. This study was the first to demonstrate the potential role of small molecule ROR-γt inhibitors in blocking the type 17 pathway in axial spondyloarthritis.
Guntermann C, Piaia A, Hamel ML, Theil D, Rubic-Schneider T, del Rio-Espinola A, et al. Retinoic-acid-orphan-receptor-C inhibition suppresses Th17 cells and induces thymic aberrations. JCI Insight. 2017;2:e91127.
Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol. 2013;20:1040–6.
Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, Lora JM. BET bromodomain inhibition suppresses TH17-mediated pathology. J Exp Med. 2013;210:2181–90.
• Hammitzsch A, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci. 2015;112:10768–73. This was the first study to show the role of epigenetic modifying agents in modulating the Th17 pathway in axial spondyloarthritis.
van Vollenhoven RF, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:652–62.
Hammitzsch A, et al. OP0156 deciphering the in vitro therapeutic potential of JAK inhibitors in ankylosing spondylitis. Ann Rheum Dis. 2016;75:115.
•• Heijde, D. van der et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2017; Annrheumdis-2016-210322. doi: 10.1136/annrheumdis-2016-210322. This is the first clinical trial to demonstrate efficacy of JAK inhibitors in axial spondyloarthritis.
Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol. 2002;23:403–8.
Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163:1400–12.
El-Behi M, et al. The encephalitogenicity of TH17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–75.
Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol. 2014;193:849–59.
Griffin JD, et al. The biology of GM-CSF: regulation of production and interaction with its receptor. Int J Cell Cloning. 1990;8(Suppl 1):35–44-45.
Dijkers PF, van Dijk TB, de Groot RP, Raaijmakers JAM, Lammers JWJ, Koenderman L, et al. Regulation and function of protein kinase B and MAP kinase activation by the IL-5/GM-CSF/IL-3 receptor. Oncogene. 1999;18:3334–42.
Hamilton JA, Achuthan A. Colony stimulating factors and myeloid cell biology in health and disease. Trends Immunol. 2013;34:81–9.
Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. 2017;16:53–70.
Broughton SE, et al. Conformational changes in the GM-CSF receptor suggest a molecular mechanism for affinity conversion and receptor signaling. Struct Lond Engl. 2016;1993(24):1271–81.
Reynolds, G. et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. 2015;75(5). annrheumdis-2014-20657.
Zhang L, Fu J, Sheng K, Li Y, Song S, Li P, et al. Bone marrow CD11b(+)F4/80(+) dendritic cells ameliorate collagen-induced arthritis through modulating the balance between Treg and Th17. Int Immunopharmacol. 2015;25:96–105.
Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16:126–33.
Roeleveld DM, et al. A2.07 Antagonisticregulation of IL-17 and GM-CSF during cell development ex vivo and during experimental arthritis. Ann Rheum Dis. 2016;75:A18.
Danis VA, Franic GM, Rathjen DA, Brooks PM. Effects of granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, interferon-gamma (IFN-gamma), tumour necrosis factor-alpha (TNF-alpha) and IL-6 on the production of immunoreactive IL-1 and TNF-alpha by human monocytes. Clin Exp Immunol. 1991;85:143–50.
Witte K, et al. Novel Th-cell subsets within the Gm-csf producers in humans. Exp Dermatol. 2014;23:197.
Greven DEA, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. 2015;74(10):1924–30.
Al-Mossawi MH, Ridley A, Chen L, de Wit J, Bowness P. Role of lymphocytes producing GM-CSF in human spondyloarthritis. Lancet. 2017;389:S21.
Kotake S, Higaki M, Sato K, Himeno S, Morita H, Kim KJ, et al. Detection of myeloid precursors (granulocyte/macrophage colony forming units) in the bone marrow adjacent to rheumatoid arthritis joints. J Rheumatol. 1992;19:1511–6.
Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony-stimulating factor receptor α expression and its targeting in antigen-induced arthritis and inflammation. Arthritis Res Ther. 2016;18:287.
Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MWN, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. 2016;126:3453–66.
van Nieuwenhuijze AEM, et al. Complementary action of granulocyte macrophage colony-stimulating factor and interleukin-17A induces interleukin-23, receptor activator of nuclear factor-κB ligand, and matrix metalloproteinases and drives bone and cartilage pathology in experimental arthritis: rationale for combination therapy in rheumatoid arthritis. Arthritis Res Ther. 2015;17
Udagawa N, Kotake S, Kamatani N, Takahashi N, Suda T. The molecular mechanism of osteoclastogenesis in rheumatoid arthritis. Arthritis Res. 2002;4:281–9.
Balani D, et al. IL-17A inhibits osteoclast development by inducing the release of GM-CSF in osteoblast lineage cells. Bone. 2012;50:S88.
Cook AD, Pobjoy J, Steidl S, Dürr M, Braine EL, Turner AL, et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther. 2012;14:R199.
Schweizerhof M, Stösser S, Kurejova M, Njoo C, Gangadharan V, Agarwal N, et al. Hematopoietic colony–stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med. 2009;15:802–7.
Plater-Zyberk C, Joosten LAB, Helsen MMA, Koenders MI, Baeuerle PA, van den Berg WB. Combined blockade of granulocyte-macrophage colony stimulating factor and interleukin 17 pathways potently suppresses chronic destructive arthritis in a tumour necrosis factor α-independent mouse model. Ann Rheum Dis. 2009;68:721–8.
Plater-Zyberk C, Joosten LA, Helsen MM, Hepp J, Baeuerle PA, van den Berg W. GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Ann Rheum Dis. 2007;66:452–7.
Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis. 2013;72:1445–52.
Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F. Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-α, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis. 2011;70:1542–9.
Burmester GR, McInnes I, Kremer J, Miranda P, Korkosz M, Vencovsky J, et al. A randomised phase IIb study of mavrilimumab, a novel GM–CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis. annrheumdis-2016-210624. 2017;76:1020–30. https://doi.org/10.1136/annrheumdis-2016-210624.
Weinblatt M, et al. SAT0146 Earth Explorer 2, a phase IIB exploratory study evaluating efficacy and safety of mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor-alpha monoclonal antibody, and the tumor necrosis factor antagonist golimumab in rheumatoid arthritis. Ann Rheum Dis. 2016;75:717.2–18.
Huizinga TWJ, et al. SAT0210 first-in-patient study of namilumab, an anti-GM-CSF monoclonal antibody, in active rheumatoid arthritis: results of the Priora phase IB study. Ann Rheum Dis. 2015;74:733.
•• Al-Mossawi MH, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. 2017;8:1510. This study demonstrates the role of GM-CSF in patients with axial spondyloarthritis and provides a rational for trials of GM-CSF blockade.
Ozegbe P, Foey AD, Ahmed S, Williams RO. Impact of cAMP on the T-cell response to type II collagen. Immunology. 2004;111:35–40.
Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–48.
Schafer PH, Parton A, Capone L, Cedzik D, Brady H, Evans JF, et al. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014;26:2016–29.
Papp K, Reich K, Leonardi CL, Kircik L, Chimenti S, Langley RGB, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37–49.
Kavanaugh A, Mease PJ, Gomez-Reino JJ, Adebajo AO, Wollenhaupt J, Gladman DD, et al. Longterm (52-week) results of a phase III randomized, controlled trial of apremilast in patients with psoriatic arthritis. J Rheumatol. 2015;42:479–88.
Pathan E, Abraham S, van Rossen E, Withrington R, Keat A, Charles PJ, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in ankylosing spondylitis. Ann Rheum Dis. 2013;72:1475–80.
Chen L, Ridley A, Hammitzsch A, al-Mossawi MH, Bunting H, Georgiadis D, et al. Silencing or inhibition of endoplasmic reticulum aminopeptidase 1 (ERAP1) suppresses free heavy chain expression and Th17 responses in ankylosing spondylitis. Ann Rheum Dis. 2016;75:916–23.
Marroquin Belaunzaran O, Kleber S, Schauer S, Hausmann M, Nicholls F, van den Broek M, et al. HLA-B27-homodimer-specific antibody modulates the expansion of pro-inflammatory T-cells in HLA-B27 transgenic rats. PLoS One. 2015;10:e0130811.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
M. Hussein Al-Mossawi reports grants from UCB Pharma, outside the submitted work.
Paul Bowness reports grants from GSK, grants from Celgene, grants from Merck research laboratories, and personal fees from Pfizer, outside the submitted work.
Claudia Worth declares that she has no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Seronegative Arthritis
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Worth, C., Bowness, P. & Hussein Al-Mossawi, M. Novel Therapeutic Targets in Axial Spondyloarthritis. Curr Treat Options in Rheum 4, 174–182 (2018). https://doi.org/10.1007/s40674-018-0095-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-018-0095-1