
Abstract
Background
Inflammation is central to the initiation of immune responses and to the pathogenesis of many diseases such as allergic contact dermatitis (ACD). ACD is an inflammatory skin disease caused by low molecular weight organic chemicals and metal ions. The immune system plays a decisive role. After protein binding, the triggering chemicals act as contact allergens that are recognized by specific T cells. Before this can happen, however, the chemicals must trigger inflammation in the skin, without which the adaptive immune system in particular is not activated.
Methods
In recent years, the inflammatory mechanisms of contact allergy have been studied at the cellular and molecular level in vivo and in vitro.
Results
Contact allergens activate the innate immune system and additionally cellular stress responses, which in interaction are responsible for skin inflammation. In this context, inflammation is required for both initial sensitization and elicitation of ACD.
Conclusion
Skin inflammation in ACD is orchestrated by the interplay of the innate immune system and cellular stress responses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Allergic contact dermatitis and inflammation
Allergic contact dermatitis (ACD) is a T-cell mediated skin disease caused by low molecular weight organic chemicals and metal ions capable of inducing inflammation [1, 2]. Protein reactivity of contact allergens is essential for this to occur. Subsequently, activation of contact allergen-specific T cells occurs, which then migrate into the inflamed skin and also contribute to inflammation through their effector function. Inflammation is central to pathogenesis [3, 4].
Inflammation is a response of the organism to a disruption of homeostasis, for example, by an infection, injury, or contact allergens. It is essential for the activation of tissue repair and regeneration [5, 6] and for the initiation of immune responses [7]. Maintenance of immunological homeostasis is an active process ensured by regulatory T cells and other immunoregulatory mechanisms. Nevertheless, to trigger immune responses, this constitutive brake of the immune system must be released. This requires a strong inflammatory stimulus.
Inflammatory reactions are triggered by so-called “danger signals” that activate sensors on and in cells that detect the disruption of homeostasis. These are, for example, so-called pattern recognition receptors (PRRs) of the innate immune system, which recognize, among other things, components of viruses and bacteria, so-called pathogen-associated molecular patterns (PAMPs), but also the body’s own pro-inflammatory molecules, so-called damage-associated molecular patterns (DAMPs). Such DAMPs are formed or released, for example, during cell stress and tissue damage and play an important role in many diseases associated with sterile inflammation [8]. Receptors for stress signals such as extracellular adenosine triphosphate (ATP), for example the purinergic receptor P2X7R in this case, also play an important role [9], or the receptors inositol-requiring kinase/endoribonuclease 1 (IRE1α), protein kinase activated by double-stranded RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) in the endoplasmic reticulum (ER) membrane, which recognize un- or misfolded proteins [10], are involved in inflammation. Ideally, the effector systems of inflammation can restore the state of homeostasis. If this is not the case, chronic inflammation and diseases may result.
Only after activation of the innate immune system can antigen-specific T cells and B cells be activated, which then also form memory cells. The majority of T cells are found in tissues. Tissue-resident memory cells thereby ensure long-lasting local immunity [7] and a rapid and efficient immune response, for example, in the case of re-infection. In the case of contact allergy, these T cells are involved in the recruitment of neutrophil granulocytes and are also responsible for the rapid flare-up of allergy upon renewed allergen contact [11,12,13].
Currently, three main types of inflammation are distinguished, which are characterized by specific mediator profiles [14,15,16,17]. Type I is associated with the production of IL-12 by APCs and the induction of Th1/Tc1 cells. Type II leads to Th2 induction with the involvement of TSLP, IL-25, and IL-33, and type III leads to Tc17/Th17 induction through IL-1β, IL‑6, and IL-23. However, there are also mixed forms as well as transitions between inflammation types, for example, during the course of a disease. In the mouse contact hypersensitivity (CHS) model, the in vivo model for ACD, type I/III inflammation dominates, leading to T cell polarization toward Tc1/Th1 and Tc17/Th17. Contact allergens are thus able to polarize the inflammatory response.
Allergic contact dermatitis
In patch tests, 5200 chemicals have been tested. Currently, an estimated 3000–3250 low molecular weight chemicals and metal ions have caused contact allergy (Anton de Groot, personal communication). The prerequisite for this is their chemical reactivity, which must allow protein binding. Only after covalent binding to amino acid side chains (organic chemicals such as fragrances, preservatives, dyes, etc.) or complex formation with amino acids such as histidine (metal ions such as nickel, cobalt, palladium) do the chemicals become recognizable by the adaptive immune system and, thus, become contact allergens. T cells recognize contact allergen-modified peptides on MHC molecules on the cell surface of the antigen-presenting cell with their T cell receptor after antigen processing, for example, in dendritic cells (DCs) [18]. Before this step can occur, the DCs must first be activated and mature in the skin by contact allergen-induced inflammation. The DCs then upregulate costimulatory molecules such as CD80/86, process haptenized proteins, and migrate to local lymph nodes. There, activation of naive allergen-specific T cells and their differentiation into Tc1/Th1 and Tc17/Th17 cells occurs, as well as programming for migration into the skin [19]. With the formation of allergen-specific T cells, sensitization is complete. After renewed allergen contact, the T cells circulating in the blood migrate into the inflamed skin and cause clinical symptoms of ACD such as erythema and eczema through their effector function. In the mouse model, recruitment of neutrophil granulocytes from the blood into the skin also plays an important role in this process [20].
Innate immune system and cellular stress in ACD
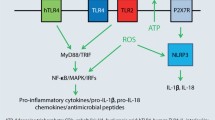
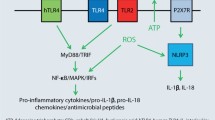
Contact allergen-induced inflammation is central to both sensitization and the elicitation of ACD. In viral or bacterial infection, inflammation proceeds via activation of PRRs. Thus, membrane Toll-like receptors (TLRs) and thereby NF-κB-dependent production of pro-inflammatory cytokines are activated. NOD-like receptors in the cytosol are also activated. Here, the NLRP3 inflammasome is important for caspase-1-dependent formation and secretion of mature IL-1β and IL-18 (Fig. 1).

Inflammation triggered by bacteria and viruses. Upon infection, bacteria or viruses activate the innate immune system through components known as pathogen-associated molecular patterns (PAMPs). These bind to pattern recognition receptors (PRRs) such as the Toll-like receptors (TLRs) and trigger signal transduction via a cascade of kinases leading to activation of the transcription factors NF-κB and IRF3/7 and the kinases p38, ERK, and MAPK. As a result, a number of proinflammatory cytokines are produced, including pro-IL-1β and pro-IL-18. As a consequence of infection, there is additional activation of the NLRP3 inflammasome by release of PAMPs and reactive oxygen species, as well as other factors, for example extracellular adenosin triphosphate (ATP). The resulting activated caspase‑1 then processes pro-IL-1β and pro-IL-18 to the mature and secreted forms of these pro-inflammatory cytokines. This reaction scheme is also activated analogously by a number of contact allergens (e.g., TNCB, oxazolone, PPD, citral, nickel) [18, 22, 23, 28,29,30, 32, 41]. (Figure created with Biorender.com)
We were able to show that this classical reaction scheme of an innate immune response to infection also applies in CHS. In addressing the question of how these small molecule chemicals activate the innate immune system, we demonstrated in the CHS model that mice with genetic defects for TLR4/IL-1Rβ2 or TLR2/4 are resistant to contact allergy [21]. We then identified hyaluronic acid (HA) fragmentation and biglycan release, both components of the extracellular matrix (ECM) of the skin, as factors responsible for TLR2/4 activation [22, 23]. HA fragments and biglycan have been described as TLR2/4 activators [24,25,26]. This highlights the important role of matrix–immune cell interaction and the role of endogenous ligands, so-called DAMPs for the activation of PRRs during inflammation [27]. Moreover, we demonstrated the triggering of cellular stress by contact allergens, leading to the release of ATP from and the formation of reactive oxygen species (ROS) in stressed cells [22, 28]. Extracellular ATP then activates the NLRP3 inflammasome via the purinergic receptor P2X7R, supported by ROS, which also promotes NF-κB activation and oxidative and p38 MAP kinase-dependent enzymatic degradation of HA by hyaluronidases [4]. Direct binding and activation of human TLR4 have been demonstrated for nickel, still the most common contact allergen for humans, but also for cobalt and palladium, and activation of the NLRP3 inflammasome has been demonstrated for nickel and ACD-causing chromium (VI) compounds [29,30,31]. The important potent contact allergen p-phenylenediamine (PPD), present for example in hair dyes, shows in vitro in NCTC2544 keratinocytes an induction of ROS and the release of HMGB1, a DAMP that can activate TLR4 [32].
Our work revealed that another cellular stress response is also centrally important for skin inflammation—the so-called unfolded protein response (UPR) resulting from ER stress [33]. The UPR plays an important role in the pathogenesis of cardiovascular and neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and amyotrophic lateral sclerosis, and is therefore a target for therapeutic intervention [34,35,36].
Activation of the UPR by contact allergens may be explained by the fact that chemical modification of proteins by contact allergen binding, as well as protein oxidation by ROS, can prevent the correct folding of proteins, leading to the accumulation of misfolded or unfolded proteins in the ER. This activates the UPR to restore protein homeostasis. The UPR contributes to inflammation, for example, through inflammasome activation [37]. ROS-dependent UPR induction has been shown for the contact allergen 2,4-dinitrofluorobenzene (DNFB) [38].
In the murine keratinocyte line Pam212 and human HaCaT keratinocytes, contact allergens such as oxazolone or TNCB or the irritant SDS trigger the UPR. While oxazolone tends to activate the IRE1α pathway, TNCB tends to induce PERK activation. The UPR also leads to activation of NF-κB as measured by its translocation to the nucleus. Interestingly, this is prevented by the use of IRE1α or PERK inhibitors. The inhibitors also reduce IL‑6 production and CD86 expression. In CHS, the UPR is efficiently activated. This is exemplified by the enhancement of a suboptimal CHS response by tunicamycin, an activator of the UPR. When a general UPR inhibitor, bile acid taurodeoxycholic acid (TUDCA), or IRE1α- or PERK-specific inhibitors are used, CHS can be significantly reduced [33]. Nickel is also capable of activating the UPR [39].
These results highlight the interaction, or “cross-talk”, of innate immune and cellular stress responses in inflammation (Fig. 2).

Orchestration of inflammation by innate immune and cellular stress responses. Contact allergens activate the innate immune system. Ni, Co, Pd ions bind directly to human TLR4, while organic chemicals activate TLR2/4 indirectly via ECM components such as HA fragments or biglycan or also TLR4 via HMGB1 from the nucleus. Cellular stress results in the production of ROS and the release of ATP. This conditions the activation of the NLRP3 inflammasome. ER stress leads to activation of the UPR, possibly as a result of protein oxidation by ROS and chemical modification by contact allergen binding. Inflammation is necessary for both sensitization and elicitation of ACD. (Figure created with Biorender.com)
Orchestration of inflammation—implications
The term orchestration describes the interaction of all elements of inflammation. Cross-talk is essential in order to exceed a critical threshold so that inflammation occurs. However, this also means that blocking any element can cause this critical threshold to be undershot. Experimentally, this can be seen in the CHS model at the cellular and molecular levels. If one eliminates mast cells or neutrophils [20, 40], CHS is prevented in both cases. If one uses TLR2/4- or NLRP3- or P2X7R-knockout mice, CHS is prevented in all three cases [20, 21, 41]. Blocking oxidative stress by topical antioxidants or inflammatory mediator action by P2X7R or IL-1R antagonists also prevents CHS [20, 21]. The use of inhibitors of the UPR also prevents CHS [33]. This shows that all elements of the orchestra are important for inflammation.
These findings explain why anti-inflammatory therapy can successfully prevent sensitization and triggering of CHS by modulating innate immune and cellular stress responses, as shown in the CHS model [22, 28, 33].
Heterologous stimulation and amplification of inflammation
In this context, it is important to understand that inflammation does not necessarily have to be triggered by the contact allergens and may be completely uncoupled from the antigen specificity of the T cells. This has far-reaching implications [42]. First, sensitization can occur even when a contact allergen can form T‑cell epitopes by protein binding but induces only weak or no inflammation. An inflammatory skin environment with the right type of inflammation, for example as a result of trauma or infection, can allow sensitization to occur (heterologous stimulation). Experimentally, we demonstrated that heterologous stimulation of inflammation in CHS-resistant mice by injection of the TLR9 agonist CpG-ODN or sensitization with CpG-ODN-treated DCs from CHS-resistant mice enables sensitization ([21] and unpublished data). The important contact allergen nickel cannot activate murine TLR4 because of a lack of binding sites. Therefore, to trigger CHS, nickel is combined with LPS to supplement the missing inflammatory signal via TLR4 [43].
The substance combination also affects the UPR. Thus, the combination of SDS with weak contact allergens or the combination of the weak contact allergens eugenol and resorcinol will trigger the UPR. The weak allergen/tolerogen DNTB can trigger the UPR by addition of the irritant SDS. These combinations clearly showed synergistic, not just additive, enhancement of the UPR [33] and thereby amplification of skin inflammation.
A good example of the consequence of a pre-existing inflammatory skin environment for the threshold value of triggering is shown by the so-called tear-off epicutaneous test. Here, repeated performance of tape tears on the skin can produce irritation with an increase in pro-inflammatory alarmins, which significantly increases patch test sensitivity [44, 45]. The experience from occupational dermatology that allergic contact dermatitis is often “grafted on” to irritant contact dermatitis caused, for example, by frequent hand disinfection or contact with cooling lubricants (so-called 2‑phase eczema), while the patch test on non-irritated skin is often negative, also illustrates how pre-existing inflammation increases sensitivity to contact allergens. This also illustrates that the severity of skin inflammation is a key parameter in ACD.
Because of these amplification mechanisms, it is understandable that the combination of several weak allergens or the combination of irritants and allergens can lead to the amplification of inflammation and thus to sensitization that would not occur against the single substance [42, 46]. This aspect is very important for the risk assessment of chemicals. As a rule, for cosmetics, only single substances are tested for contact allergenic potential in OECD guideline tests [47,48,49]. However, in consumer products such as perfumes, mixtures of several weak contact allergens such as fragrances and preservatives and irritants are then found, the combination of which can lead to amplification of inflammation beyond the critical threshold and thus to sensitization. Therefore, the testing of mixtures is quite reasonable.
High fat diet and ACD
Obesity is associated with underlying chronic inflammation [50]. It has recently been shown in a mouse model that feeding a high fat diet (HFD) leads to the enhancement of psoriasis-like inflammatory skin disease, which can be induced in the mouse model with the TLR7 agonist imiquimod [51]. This heterologous stimulation by dietary components leads to amplification and exacerbation of the disease. Here, especially saturated fatty acids play a proinflammatory role [52, 53].
In the CHS model, we investigated how feeding a HFD affects resistance to contact allergy in TLR2/4-deficient mice [54]. Feeding HFD for 4 weeks to 4‑week-old mice resistant to CHS completely abolished CHS resistance at 8 weeks of age. The weight of the mice and the size of the fat pads increased only marginally. The HFD-fed TLR2/4 mice respond to contact allergen sensitization and elicitation with a CHS response mediated by IFN‑γ producing CD8+-T cells like wild-type mice. In these, the HFD enhances the CHS response. HFD increased inflammatory parameters such as proinflammatory cytokines and the number of neutrophils migrating into the skin in CHS. Majewska-Szczepanik et al. observed exacerbation of CHS in wild-type mice by HFD feeding in the CHS model, which was associated with proinflammatory CD4+-T cells and DCs in skin-draining lymph nodes and spleen and dysbiosis of the gut microbiome. IL-17A-deficient mice were resistant to the effects of HFD [55].
These findings demonstrate the importance of heterologous inflammation initiation [42] and, more importantly, that there are alternative, TLR2/4-independent bypass mechanisms that also lead to sensitization to contact allergy. This has clinical relevance, as success and failure of therapy depend on whether the underlying mechanism has been identified. The clinical picture without information on the pathomechanism sometimes does not provide sufficient information to choose the appropriate therapy. Good examples are the mechanistically different endotypes of atopic dermatitis, chronic obstructive pulmonary disease (COPD), or asthma [56,57,58].
Conclusion
Inflammation is essential for the initiation of immune responses. In allergic contact dermatitis, the inflammatory response resembles an anti-infective response. The innate immune system and cellular stress responses interacting with each other are the basis of skin inflammation. In addition, the inflammatory pattern described here involving DAMPs, TLRs, NLRP3 inflammasome, ROS, ATP, and UPR is found in a great variety of diseases of different tissues and organs. Examples include psoriasis, Alzheimer’s disease, chronic obstructive pulmonary disease (COPD), myocarditis, nonalcoholic fatty liver disease (NAFLD), colitis, rheumatoid arthritis, and kidney disease associated with diabetes. Inflammation is of central importance. It remains to be elucidated how this basic module of inflammation is varied and modulated depending on each specific tissue microenvironment, as this common denominator does not yet explain how each disease develops.
Since inflammation driven by innate immune system and cellular stress responses is important not only initially but also for the maintenance of disease, anti-inflammatory therapy may be useful not only preventively but also at and after disease onset. For example, it is shown that early intervention (“early intervention”) may possibly prevent the establishment of a disease, or at least reduce the severity of disease [59,60,61,62]. A good example is the blockade of the cytokine IL-23, which is produced by cells of the innate immune system and plays an important role in psoriasis. The GUIDE study in psoriasis found that early intervention with the anti-IL-23 monoclonal antibody guselkumab had a beneficial effect on clinical outcome and disease progression. In psoriasis patients, anti-IL-23 therapy also resulted in a decrease in skin-resident Th17 memory cells (TRM 17) [63]. Additional studies in mouse models demonstrated the production of IL-17 by CD301+-DCs. IL-23 contributed to the proliferation, longevity, and skin retention of TRM 17 cells. Blockade of the IL-23 receptor following primary infection with Candida albicans-depleted CD69+ CD103+ TRM 17 from the skin [63].
Inflammation is the central initiator of immune responses and plays an important role in a wide variety of diseases. The study of inflammatory mechanisms is therefore crucial for the development of new, mechanism-based therapies. The principle of orchestration can be used to identify the optimal therapy. Combination therapy could be more efficient and have fewer side effects in this regard, as additive/synergistic effects are possible, the dose of single drugs can be reduced, and potential heterogeneity in response to a single drug can be covered [64,65,66].
The development of methods for early diagnosis via biomarkers is crucial in this context, especially because the evidence for therapeutic success of “early intervention” is increasing. Therefore, the innate immune system and cellular stress responses are very attractive as therapeutic targets, not least because the nature of the resulting inflammation determines the response patterns of the adaptive immune system and thus the disease type.
Abbreviations
- ACD:
-
Allergic contact dermatitis
- ATF6:
-
Activating transcription factor 6
- ATP:
-
Adenosine triphosphate
- CHS:
-
Contact hypersensitivity
- DAMP:
-
Damage-associated molecular pattern
- DNFB:
-
2,4-dinitrofluorobenzene
- DNTB:
-
2,4-dinitrothiocyanobenzene
- ECM:
-
Extracellular matrix
- ER:
-
Endoplasmic reticulum
- IRE1α:
-
Inositol-requiring kinase/endoribonuclease 1
- NLRP3:
-
NOD-like receptor family pyrin domain containing 3
- PAMP:
-
Pathogen-associated molecular patterns
- PERK:
-
Protein kinase activated by double-stranded RNA-like ER kinase
- PPD:
-
Para-phenylenediamine
- PRR:
-
Pattern recognition factor
- ROS:
-
Reactive oxygen species
- TLR:
-
Toll-like receptor
- TNCB:
-
2,4,6-trinitrochlorobenzene
- UPR:
-
Unfolded protein response
References
Martin SF, Rustemeyer T, Thyssen JP. Recent advances in understanding and managing contact dermatitis. F1000Research 2018;7:F1000 Faculty Rev-810
Scheinman PL, Vocanson M, Thyssen JP, Johansen JD, Nixon RL, Dear K, et al. Nat Rev Dis Primer. Contact Derm. 2021;7:38.
Meizlish ML, Franklin RA, Zhou X, Medzhitov R. Tissue Homeostasis and Inflammation. Annu Rev Immunol. 2021;39:557–81.
Esser PR, Huber M, Martin SF. Endoplasmic reticulum stress and the inflammatory response in allergic contact dermatitis. Eur J Immunol. 2023;53:e2249984.
Cooke JP. Inflammation and Its Role in Regeneration and Repair. Circ Res. 2019;124:1166–8.
Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science. 2017;356:1026–30.
Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–53.
Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–90.
Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci Basic Clin. 2015;191:117–23.
Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21:421–38.
Lefevre MA, Vocanson M, Nosbaum A. Role of tissue-resident memory T cells in the pathophysiology of allergic contact dermatitis. Curr Opin Allergy Clin Immunol. 2021;21:355–60.
Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, et al. Inhibitory checkpoint receptors control CD8+ resident memory T cells to prevent skin allergy. J Allergy Clin Immunol. 2019;143:2147–2157.e9.
Funch AB, Mraz V, Gadsbøll ASØ, Jee MH, Weber JF, Ødum N, et al. CD8+ tissue-resident memory T cells recruit neutrophils that are essential for flare-ups in contact dermatitis. Allergy. 2022;77:513–24.
Medzhitov R. The spectrum of inflammatory responses. Science. 2021;374:1070–5.
Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135:626–35.
Hilligan KL, Ronchese F. Antigen presentation by dendritic cells and their instruction of CD4+ T helper cell responses. Cell Mol Immunol. 2020;17:587–99.
Rankin LC, Artis D. Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell. 2018;173:554–67.
Martin SF, Esser PR, Schmucker S, Dietz L, Naisbitt DJ, Park BK, et al. T‑cell recognition of chemicals, protein allergens and drugs: towards the development of in vitro assays. Cell Mol Life Sci. 2010;67:4171–84.
Dudda JC, Lembo A, Bachtanian E, Huehn J, Siewert C, Hamann A, et al. Dendritic cells govern induction and reprogramming of polarized tissue-selective homing receptor patterns of T cells: important roles for soluble factors and tissue microenvironments. Eur J Immunol. 2005;35:1056–65.
Weber FC, Németh T, Csepregi JZ, Dudeck A, Roers A, Ozsvári B, et al. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J Exp Med. 2015;212:15–22.
Martin SF, Dudda JC, Bachtanian E, Lembo A, Liller S, Dürr C, et al. Toll-like receptor and IL-12 signaling control susceptibility to contact hypersensitivity. J Exp Med. 2008;205:2151–62.
Esser PR, Wölfle U, Dürr C, von Loewenich FD, Schempp CM, Freudenberg MA, et al. Contact sensitizers induce skin inflammation via ROS production and hyaluronic acid degradation. Plos One. 2012;7:e41340.
Esser PR, Zech A, Idzko M, Martin SF. Lack of biglycan reduces contact hypersensitivity in mice. Contact Derm. 2018;79:326–8.
Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84.
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9.
Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;284:24035–48.
Frevert CW, Felgenhauer J, Wygrecka M, Nastase MV, Schaefer L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J Histochem Cytochem. 2018;66:213–27.
Weber FC, Esser PR, Müller T, Ganesan J, Pellegatti P, Simon MM, et al. Lack of the purinergic receptor P2X[7] results in resistance to contact hypersensitivity. J Exp Med. 2010;207:2609–19.
Schmidt M, Raghavan B, Müller V, Vogl T, Fejer G, Tchaptchet S, et al. Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol. 2010;11:814–9.
Li X, Zhong F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase‑1 pathway. Inflammation. 2014;37:457–66.
Adam C, Wohlfarth J, Haußmann M, Sennefelder H, Rodin A, Maler M, et al. Allergy-Inducing Chromium Compounds Trigger Potent Innate Immune Stimulation Via ROS-Dependent Inflammasome Activation. J Invest Dermatol. 2017;137:367–76.
Galbiati V, Papale A, Galli CL, Marinovich M, Corsini E. Role of ROS and HMGB1 in contact allergen-induced IL-18 production in human keratinocytes. J Invest Dermatol. 2014;134:2719–27.
Gendrisch F, Völkel L, Fluck M, Apostolova P, Zeiser R, Jakob T, et al. IRE1 and PERK signaling regulates inflammatory responses in a murine model of contact hypersensitivity. Allergy. 2022;77:966–78.
Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13:477–91.
Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. 2021;18:499–521.
Hetz C, Axten JM, Patterson JB. Pharmacological targeting of the unfolded protein response for disease intervention. Nat Chem Biol. 2019;15:764–75.
Grootjans J, Kaser A, Kaufman RJ, Blumberg RS. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16:469–84.
Luís A, Martins JD, Silva A, Ferreira I, Cruz MT, Neves BM. Oxidative stress-dependent activation of the eIF2α-ATFr unfolded protein response branch by skin sensitizer 1‑fluoro‑2,4‑dinitrobenzene modulates dendritic-like cell maturation and inflammatory status in a biphasic manner. Free Radic Biol Med. 2014;77:217–29.
Yu M, Chen F, Wang H, Fu Q, Yan L, Chen Z, et al. Endoplasmic reticulum stress mediates nickel chloride-induced epithelial—mesenchymal transition and migration of human lung cancer A549 cells through Smad2/3 and p38 MAPK activation. Ecotoxicol Environ Saf. 2023;249:114398.
Dudeck A, Dudeck J, Scholten J, Petzold A, Surianarayanan S, Köhler A, et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity. 2011;34:973–84.
Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase‑1. Immunity. 2006;24:317–27.
Martin SF. Adaptation in the innate immune system and heterologous innate immunity. Cell Mol Life Sci. 2014;71:4115–30.
Sato N, Kinbara M, Kuroishi T, Kimura K, Iwakura Y, Ohtsu H et al. Lipopolysaccharide promotes and augments metal allergies in mice, dependent on innate immunity and histidine decarboxylase. Clin Exp Allergy J Br Soc Allergy Clin Immunol 2007;37:743–51
Dickel H, Kamphowe J, Geier J, Altmeyer P, Kuss O. Strip patch test vs. conventional patch test: investigation of dose-dependent test sensitivities in nickel- and chromium-sensitive subjects. J Eur Acad Dermatol Venereol JEADV 2009;23:1018–25
Dickel H, Gambichler T, Kamphowe J, Altmeyer P, Skrygan M. Standardized tape stripping prior to patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF‑α and IL-8/CXCL8 mRNA: new insights into the involvement of “alarmins”. Contact Derm. 2010;63:215–22.
Bonefeld CM, Geisler C, Gimenéz-Arnau E, Lepoittevin JP, Uter W, Johansen JD. Immunological, chemical and clinical aspects of exposure to mixtures of contact allergens. Contact Derm. 2017;77:133–42.
Ezendam J, Braakhuis HM, Vandebriel RJ. State of the art in non-animal approaches for skin sensitization testing: from individual test methods towards testing strategies. Arch Toxicol. 2016;90:2861–83.
Grundström G, Borrebaeck CAK. Skin Sensitization Testing-What’s Next? Int J Mol Sci. 2019;20:666.
Gądarowska D, Kalka J, Daniel-Wójcik A, Mrzyk I. Alternative Methods for Skin-Sensitization Assessment. Toxics. 2022;10:740.
Saltiel AR, Olefsky JM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. 2017;127:1–4.
Herbert D, Franz S, Popkova Y, Anderegg U, Schiller J, Schwede K, et al. High-Fat Diet Exacerbates Early Psoriatic Skin Inflammation Independent of Obesity: Saturated Fatty Acids as Key Players. J Invest Dermatol. 2018;138:1999–2009.
Fritsche KL. The science of fatty acids and inflammation. Adv Nutr Bethesda Md. 2015;6:293S–301S.
Ravaut G, Légiot A, Bergeron KF, Mounier C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int J Mol Sci. 2020;22:330.
Rühl-Muth AC, Maler MD, Esser PR, Martin SF. Feeding of a fat-enriched diet causes the loss of resistance to contact hypersensitivity. Contact Derm. 2021;85:398–406.
Majewska-Szczepanik M, Kowalczyk P, Marcińska K, Strzępa A, Lis GJ, Wong FS, et al. Obesity aggravates contact hypersensitivity reaction in mice. Contact Derm. 2022;87:28–39.
Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143:1–11.
Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74:1249–56.
Chupp GL, Kaur R, Mainardi A. New Therapies for Emerging Endotypes of Asthma. Annu Rev Med. 2020;71:289–302.
Carter LM, McGonagle D, Vital EM, Wittmann M. Applying Early Intervention Strategies to Autoimmune Skin Diseases. Is the Window of Opportunity Preclinical? A Dermato-Rheumatology Perspective. J Invest Dermatol. 2022;142:944–50.
Schäkel K, Reich K, Asadullah K, Pinter A, Jullien D, Weisenseel P et al. Early disease intervention with guselkumab in psoriasis leads to a higher rate of stable complete skin clearance [‘clinical super response’]: Week 28 results from the ongoing phase IIIb randomized, double-blind, parallel-group, GUIDE study. J Eur Acad Dermatol Venereol [Internet] [zitiert 2. August 2023];n/a[n/a]. Verfügbar unter: https://onlinelibrary.wiley.com/doi/abs/10.1111/jdv.19236
Boniface K, Seneschal J. Vitiligo as a skin memory disease: The need for early intervention with immunomodulating agents and a maintenance therapy to target resident memory T cells. Exp Dermatol. 2019;28:656–61.
Breedveld F. The value of early intervention in RA—a window of opportunity. Clin Rheumatol. 2011;30(Suppl 1):S33–9.
Whitley SK, Li M, Kashem SW, Hirai T, Igyártó BZ, Knizner K, et al. Local IL-23 is required for proliferation and retention of skin-resident memory TH17 cells. Sci Immunol. 2022;7:eabq3254.
Jia J, Zhu F, Ma X, Cao Z, Cao ZW, Li Y, et al. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111–28.
Cheng F, Kovács IA, Barabási AL. Network-based prediction of drug combinations. Nat Commun. 2019;10:1197.
Plana D, Palmer AC, Sorger PK. Independent Drug Action in Combination Therapy: Implications for Precision Oncology. Cancer Discov. 2022;12:5606–24.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S.F. Martin, A.-C. Rühl-Muth and P.R. Esser declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martin, S.F., Rühl-Muth, AC. & Esser, P.R. Orchestration of inflammation in contact allergy by innate immune and cellular stress responses. Allergo J Int 33, 41–48 (2024). https://doi.org/10.1007/s40629-023-00275-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-023-00275-4