
Abstract
The “epithelial barrier hypothesis” proposes that genetic predisposition to epithelial barrier damage, exposure to various epithelial barrier–damaging agents and chronic periepithelial inflammation are responsible for the development of allergic and autoimmune diseases. Particularly, the introduction of more than 200,000 new chemicals to our daily lives since the 1960s has played a major role in the pandemic increase of these diseases. The epithelial barrier constitutes the first line of physical, chemical, and immunological defence against external factors. A leaky epithelial barrier initiates the translocation of the microbiome from the surface of affected tissues to interepithelial and even deeper subepithelial areas. In tissues with a defective epithelial barrier, colonization of opportunistic pathogens, decreased microbiota biodiversity, local inflammation, and impaired regeneration and remodelling takes place. A dysregulated immune response against commensals and opportunistic pathogens starts. Migration of inflammatory cells to other tissues and their contribution to tissue injury and inflammation in the affected tissues are key events in the development and exacerbation of many chronic inflammatory diseases. Understanding the underlying factors that affect the integrity of epithelial barriers is essential to find preventive measures or effective treatments to restore its function. The aim of this review is to assess the origins of allergic and autoimmune diseases within the framework of the epithelial barrier hypothesis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The epithelial barrier is the primary contact between the host and the environment that plays a vital role in preserving the body’s structural and functional integrity by shielding the host against pathogen invasion and foreign substance infiltration [1]. Physiological epithelial barrier functions are critical, since an impaired epithelial barrier is linked to a variety of chronic diseases, including allergies, autoimmune and metabolic diseases.
Since the 1960s, there has been a pandemic increase in the prevalence of allergic and autoimmune diseases, affecting more than 2 billion individuals [2,3,4]. After 2000, a new wave of diseases emerged, including food allergies, eosinophilic esophagitis (EoE) and drug-induced anaphylaxis [5]. Environmental and genetic factors play a role in the pathogenesis of aforementioned diseases that are strongly associated with epithelial barrier defects [6, 7]. Studies on better understanding the epithelial barrier began with the first demonstration of keratinocyte apoptosis induced by activated T cells in the pathogenesis of atopic dermatitis (AD) [7, 8]. This was followed by the demonstration of epithelial barrier damage in asthma and chronic rhinosinusitis [6, 9, 10]. It was also shown that autophagy in intestinal epithelial cells acts to limit intestinal inflammation by protecting them from apoptosis during chronic colitis [11]. These diseases were characterized by apoptosis of highly activated local tissue epithelial cells with concomitant damage to the epithelial barrier. Spongiosis was evident in the skin of AD patients and epithelial desquamation was present in patients with asthma and sinusitis. The death of the highly activated epithelial cells ameliorated the local inflammatory burden. The damage of the epithelial barrier expulses local subepithelial inflammatory molecules to the lumens of mucosal surfaces. The spongiotic morphology facilitates the draining of the dermal inflammation away from the skin. Lesions of AD are characterized by an inflammatory signature and high transepidermal water loss. All of these events ameliorate the epithelial inflammatory burden related to the chronicity of these diseases with a continuous exacerbation and healing process.
The main functions of the mucosal barrier bring together the concepts of keep away, wash away and suppress that encompass multiple components of the immune system and tissue cell-related mechanisms [5]. The keep away function entails allergen ignorance by acting as a physical barrier to external stimuli with an increased basement membrane (lamina reticularis) thickness [12], and secretion of IgA antibodies and antimicrobial peptides [13, 14]. The wash away function is responsible to drain inflammatory cells, small molecule mediators and cytokines away from the site of inflammation by mechanisms including the opening of the epithelial barriers. The aim of this type of tissue response is to drain the inflammatory cells and mediators away from the inflammation area. For example in asthma cells and mediators are found in the sputum by using the wash away mechanisms. They have left the tissues and are removed from the body after being mixed with sputum due to the opening of the epithelial barriers and molecular drainage and are removed from the inflammatory area to decrease the inflammatory burden. Cough and ciliary movement contribute to the wash away function and together these mechanisms clear away the unwanted threads as microbes, allergens, pollutants as well as inflammatory cells and mediators and decrease the inflammatory burden [15]. The epithelial cells have a suppressive role and regulate inflammation by the generation of regulatory dendritic cells, regulatory T and B cells and their anti-inflammatory cytokines [12, 16].
The epithelial barriers of the skin, lungs and gut can be impaired by exposure to environmental and toxic compounds recently introduced by industrialization, urbanization and modern lifestyle, as proposed by the “epithelial barrier hypothesis” (Fig. 1; Box 1; [4]). The epithelial barrier hypothesis is a collective concept that incorporates prior hypotheses such as the “hygiene”, “old friends”, and “biodiversity” [17]. Following the damage of the epithelial barrier, opportunistic pathogens, such as S. aureus may colonize the leaky barrier areas. The dysbiotic microbiota then translocate to interepithelial and subepithelial sites, initiating a local or sometimes systemic immune and inflammatory response which is suspected as a culprit in many immune-related diseases. A cascade of events then follow to cause chronic periepithelial inflammation with barrier leakiness (Box 2; [4]). Currently, it is not fully understood whether the epithelial barrier damage occurs prior to or after the allergic/autoimmune response (Fig. 1). It is essential to understand the factors and molecular mechanisms that affect the epithelium barrier’s integrity and find preventative measures or effective therapies to restore its physiological function. The aim of this review is to account for the alarming increase in the prevalence of allergic and autoimmune diseases through the framework of the epithelial barrier hypothesis. The barrier-damaging environmental and genetic factors and the concomitant inflammatory response are highlighted. Finally, the interplay between the epithelial barrier and microbial dysbiosis is discussed as a key driving force of allergic and autoimmune diseases.

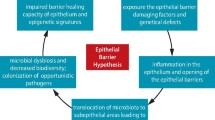
Epithelial barrier hypothesis: exposure to barrier-damaging agents or a genetic deficiency in barrier molecules leads to the colonization of opportunistic pathogens, translocation of the microbiome to the subepithelial area, immune response to dysbiotic microbiome and epithelial inflammation which causes chronic damage to the epithelial barrier integrity
Box 1 Epithelial barrier affecting factors and their associated diseases
-
A leaky gut epithelial barrier can contribute to the development of neurological diseases (e.g. multiple sclerosis) and joint diseases (e.g. rheumatoid arthritis) as the activated pathogenic immune cells in mucosal tissues with defective barriers can migrate to the affected organs [18, 19].
-
Barrier leakiness precedes diabetes, a disease associated with poor gut microbiota biodiversity and altered composition [20].
-
Epidemiological evidence collected from workers in detergent factories shows allergic and non-allergen-specific bronchial hyperreactivity in patients, allergen sensitizations [21].
-
Exposure to household or professional cleaning agents is found to be correlated with the incidence of rhinitis [22, 23].
-
Decreased occludin expression and barrier leakiness are observed when human and rat alveolar epithelial cells are exposed to diesel exhaust particulates [24, 25].
-
Particulate matter affects the distribution of occludin and the alveolar barrier as shown in ex vivo experiments with human and rat alveolar epithelial cells [26].
-
Respiratory barrier injury is promoted by ozone in mouse models [27].
-
Polystyrene microplastics affect the gut barrier as shown in mouse models [28].
-
Titanium dioxide nanoparticles which can be found in toothpaste amplify respiratory syncytial virus-induced airway epithelial barrier dysfunction [29].
-
Damage to the structure of hamster small intestine in vivo and the translocation of Escherichia coli across M‑cells in vitro are enhanced by emulsifiers [30].
-
Ocular allergy development promotes the cycle of allergic inflammation by facilitating paracellular transport of allergens, pathogens, pollutants and other harmful triggers [31].
Box 2 Stepwise events describing the epithelial barrier hypothesis
-
Genetic defects and/or exposure to epithelial-activating and barrier-damaging agents and opening of the skin and mucosal barriers.
-
Colonization of opportunistic pathogens, S. aureus, Moraxella, Haemophilus, Pneumococcus.
-
Translocation of microbiota to inter- and subepithelial areas.
-
Immune response to commensals and opportunistic pathogens, together with systemic inflammation.
-
Microbial dysbiosis and decreased biodiversity and commensals.
-
Chronic inflammation in the periepithelial area.
-
Defective epithelial barrier healing due to inflammation and epigenetic regulation of epithelial stem cells.
Factors affecting the epithelial barrier
Genetical factors: filaggrins and TJ polymorphisms
Studies have shown that environmental and genetic factors together play a role in the pathogenesis of diseases that are strongly associated with epithelial barrier defects such as AD [32] and inflammatory bowel disease (IBD) [33]. In addition, the fact that the strongest predictor of AD is a family history of allergy supports the importance of genetic factors in the pathogenesis of the disease [34].
Tight junctions (TJ), desmosome, and adherens junctions are the main structures involved in the formation of the mucosal epithelial barrier [35]. In addition, filaggrin protein, which binds to keratin fibres in epithelial cells in the epidermis, plays a role in the formation of the barrier [36]. One of the well-known genetic risk factors for the development of AD is loss-of-function mutations in the filaggrin gene (FLG) [37, 38]. The FLG protein is located in the stratum corneum and is essential for maintaining epidermal barrier integrity along with TJs in the stratum granulosum [32]. FLG loss-of-function mutations are associated with more severe AD [39] and food allergy development [40]. Furthermore, the presence of a FLG loss-of-function mutation predicts the development of asthma in AD patients who are sensitized to food allergens [41]. In addition, copy number variation in FLG affects the AD development risk [42]. Deficiencies in other epithelial barrier-associated proteins such as laminin alpha‑3 [43], TMEM79 [32], filaggrin‑2 [44] and late cornified envelope-like proline-rich 1 [45] genes were also found to be associated with AD.
The TJ is a continuous, circumferential belt-like intercellular adhesion complex that regulates paracellular passage. Mouse studies have shown that the knock-out of various TJ components such as claudins (CLDN) and zonula occludens (ZO) proteins cause defects in the blood–brain barrier [46], blood–testis barrier [47], skin barrier [48] and even fatal embryonic anomalies [49, 50]. Mutations in CLDN10B and CLND1 genes causing total absence or reduced protein expression of the respective claudin proteins leads to various syndromes associated with epidermal barrier impairment [51, 52]. The polymorphisms in the CLDN1 gene were found to be associated with AD [53] and contact dermatitis [54]. The localization of the TJs on the cell membrane is essential for forming an efficient epithelial barrier structure. This is evidenced by the deficiencies in protein sorter adaptors that cause epithelial barrier impairment [55]. Adaptor Related Protein Complex 1 Sigma 1 Subunit (AP1S1) mutations altered localization of TJ proteins ZO‑1 and claudin 3, resulting in an impaired intestinal epithelial barrier [56]. Single nucleotide polymorphisms of TJ-related proteins, membrane-associated guanylate kinase inverted (MAGI)-2 and MAGI3, are associated with IBD [57]. Moreover, loss-of-function mutations in the gene encoding desmosome protein desmoglein‑1 cause impaired epidermal integrity and barrier function, resulting in severe skin dermatitis, multiple allergies and metabolic wasting (SAM) syndrome characterized by severe dermatitis, multiple allergies and metabolic wasting [58]. A recent study demonstrated the association between desmosome proteins desmoplakin and periplakin variants with eosinophilic esophagitis [59]. Protocadherin 1 gene variants, an adherence junction protein, have been shown to predispose an individual to bronchial hyperresponsiveness [60]. Lastly, single nucleotide polymorphisms of mucin (MUC)-19 and MUC22 are associated with Crohn’s disease and MUC21 and MUC22 with ulcerative colitis [33].
Environmental factors
The external exposome is a term used to describe the collection of chemical substances and environmental pollutants we are exposed to starting from conception until death. There is mounting epidemiological evidence indicating that pollution, alterations in dietary habits and exposure to toxic substances, such as dishwasher and laundry detergents, household cleaners, diesel exhaust, microplastics, nanoparticles, and food emulsifiers, possess severe epithelial barrier-damaging properties [39, 61]. More than 200,000 new chemicals have become excessively common especially in developing countries after the 1960s due to modernization, urbanization and globalization. In parallel, allergic and autoimmune diseases have become one of the main threats for mankind and a significant healthcare burden [62].
Diesel exhaust particles (DEPs) harm the upper and lower respiratory tissues, causing serious damage to the nose, sinuses and lungs [24, 25]. Fukuoka et al. demonstrated that DEP disrupted TJs, thus leading to increased permeability of nasal epithelial cells [24] and facilitating the entry of allergens into subepithelial tissues causing the exacerbation of antigen-specific IgE-bearing mast-cell-mediated immediate allergic responses. Both in vitro and in vivo studies demonstrate that DEPs trigger the proliferation of eosinophils and increase secretion of interleukin (IL)-13 on lung epithelial cells causing fibrosis and tissue remodelling in the lungs [63]. Moreover, DEP stimulation on the primary human nasal epithelium modifies the basal secretome leading to inflammation with effusion of fluids [64, 65].
Laundry detergents and detergent residue after rinsing disrupt the TJ barrier integrity of human bronchial epithelial cells (HBEC). The toxicity of laundry detergents on HBECs has been demonstrated at relatively high concentrations (greater than 1:50,000 detergent dilution). While genes related to lipid metabolism, oxidative stress, cell survival, thymic stromal lymphopoietin (TSLP) and IL-33 are upregulated; genes related to cell adhesion, extracellular matrix organization and wound healing are downregulated in human bronchial cells after applying detergent at 1:50,000 dilution [23]. In the same study, the detergent residue remaining on clothes was demonstrated to be cytotoxic and induce epithelial barrier opening. Surfactants that are commonly used in various washing products and cosmetics, such as sodium lauryl sulfate and sodium benzenesulfonate, damage bronchial epithelial cultures even at 1:100,000 dilutions [22].
Ozone exposure plays a significant role in the pathogenesis of asthma and chronic obstructive pulmonary disease by disrupting the barrier integrity and inflammation. Similar to laundry detergents, ozone exposure also results in IL-33 upregulation in the human airways. IL-1α and IL-17A are also over-expressed and cause hyperinflammation after ozone-induced lung injury in humans and mice [66].
Polystyrene microplastics are cytotoxic on human airway cells at a relatively low dose after prolonged exposure or at an acute dose. TJ genes, such as ZO-1, are downregulated after microplastic exposure on the human airway inducing an inflammatory response and barrier dysfunction which can contribute to the development of chronic obstructive pulmonary disease [67].
Allergens
Proteolytic allergens cleave the TJs, enhancing epithelial permeability and disrupting barrier function. House dust mites (HDMs), Aspergillus fumigatus, and pollens are examples of allergens with intrinsic proteolytic activity. Antiviral airway responses were impaired in a mouse model of HDM-induced allergic bronchial inflammation by directly affecting toll-like receptor (TLR)-3 signalling [68]. Moreover, the nasal epithelial barrier function is compromised by the lower expression of epithelial cell TJ-related proteins, such as occludin and ZO‑1 [69]. Another study indicated that the inhalation of Aspergillus fumigatus allergen by naïve mice induced airway eosinophilia that was dependent on protease-activated receptor‑2 [70]. In addition, pollens have high molecular weight proteases with serine and/or aminopeptidase activity. It is generally accepted that transepithelial passage is increased due to the disruption of transmembrane junctional proteins by several allergen proteases [71].
Microbiota
A healthy, diverse gut microbiota is known to be essential in maintaining gut epithelial barrier integrity. However, a disturbed and dysbalanced gut microbiota is associated with increased gut barrier permeability and translocation of microbial and food antigens, beneath the epithelium, which leads to a systemic inflammation. Of relevance, children with food allergies show an early onset of intestinal dysbiosis and impaired gut barrier function [72]. Furthermore, in a mouse model of autoimmune chronic kidney disease, systemic inflammation was associated with an increase of gut proteobacteria, leading to dysbiosis, bacterial translocation to the liver and increased serum endotoxin levels. However, eradication of facultative anaerobic bacteria with an antibiotic cocktail, reduced systemic inflammation in these mice by preventing bacterial translocation and reducing serum endotoxin levels, highlighting a causative relationship between dysbiosis and systemic inflammation in chronic kidney disease [73].
Intestinal epithelial cells (IECs) orchestrate communication between the gut microbes and the innate and adaptive mucosal immune cells. IECs can sense molecular patterns of commensal and pathogenic microbes and therefore initiate an adequate immune response. Furthermore, IECs can secrete molecules such as antimicrobial peptides or serotonin, which directly affect the function and composition of the gut microbiota [74].
The gut microbiota interacts with the host through metabolites produced including short-chain fatty acids, secondary bile acids and polysaccharide A. Short-chain fatty acids induce Treg cell differentiation and strengthen epithelial barrier integrity by inducing mucus secretion from goblet cells and IL-22 secretion by innate lymphoid cells (ILCs) [72]. Secondary bile acids are known to inhibit IL-1β and IL‑6 gene expression in IECs, whereas polysaccharide A induces IL-10 secretion in CD4+ T cells [75]. Together, microbial-derived metabolites are important immunomodulators and major players in maintaining the gut epithelial barrier.
Food emulsifiers and dietary factors
Emulsifiers, also called surfactants, are amphipathic molecules, having both hydrophobic and hydrophilic regions that allow these molecules to interact with both oily and aqueous substances, and are commonly used in processed foods. There is accumulating evidence on the toxicity of emulsifiers as they increase TJ permeability [76, 77]. The detrimental impact of the widespread use of emulsifiers in processed food is now being widely studied [78, 79]. Ingestion of emulsifiers through processed food, ice cream and pastries has been speculated as an important factor behind the increase in inflammatory bowel disease (IBD) [78, 79]. The consumption of processed foods has been suggested to cause inflammatory changes leading to metabolic syndrome, particularly due to their high emulsifier content [80]. Food emulsifiers behave like surfactants and act similar to detergents. Interestingly, trace amounts have been demonstrated to increase bacterial translocation across epithelial cells [30, 81]. Several in vivo and in vitro studies have revealed such findings with increased weight gain and adiposity, changes in microbiome composition, elevated blood glucose, hyperphagia, hepatic steatosis and increased inflammatory markers. The use of emulsifiers in food products has significantly increased over the years to prevent phase separation and extend shelf life. Very recently, Khuda et al. reported that polysorbate-80, a well-known emulsifier, alters the function of TJs even at low cytotoxic levels [13]. Animal studies also showed that administration of polysorbate-80 and carboxymethylcellulose alter the microbiota composition and increase intestinal permeability [82, 83].
Gluten, the structural protein component of grains, has been reported to damage the epithelial barrier. Gliadin, the main fraction of gluten, has different effects on the gastrointestinal mucosa, including F‑actin content reduction, cell growth inhibition and apoptosis induction [84]. In addition, it was reported that gliadin induces a rapid increase in intestinal permeability by rearrangement of the cytoskeleton through the zonulin pathway and TJ opening [85]. Gluten was demonstrated to increase intestinal permeability and translocation of bacteria in a dextran sulfate sodium-induced colitis mice model. The impaired desmosomes and adherence junctions led to worsening of colitis [82].
Fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) are carbohydrates that are suspected to have a role in IBD symptoms [86]. In individuals with IBD, a high-FODMAP diet causes gastrointestinal symptoms [87]. Study shows that a high-FODMAP diet in mice promotes mast cell activation via lipopolysaccharide, which results in colonic barrier loss, and a low-FODMAP diet restores these pathophysiologic mucosal alterations [86].
Immune response to epithelial barrier damage
The immune response may vary depending on the site of the epithelial barrier (skin, upper and lower respiratory, oesophagus and gut). In general, epithelium-derived alarmins IL-25, IL-33 and TSLP, which are activated by allergens, pollutants, viruses, bacteria and toxins, lead to systemic inflammation characterized by activation of ILC2, exacerbated eosinophilia and IgE/IgG1 production and induce an immune response. Th2 cells and ILCs secrete type 2 cytokines such as IL‑4, IL‑5, IL‑9 and IL-13. The signature type 2 cytokines IL‑4 and IL-13 activate B cells to class-switch to IgE and also play a role in T‑cell and eosinophil migration to allergic inflammatory tissues and the inflammation begins (Fig. 2; [88, 89]). IL‑4 and IL-13 can also impair the epithelial barrier by reducing the expression of TJs and filaggrin. The dynamic crosstalk between the immune system, epithelial cells and their cytokines results in damage to skin and lung barriers [90].

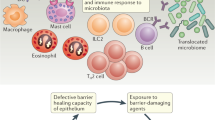
Immune response to the epithelial barrier damage. Environmental factors, allergens and microbiota composition affect epithelial barrier function. A disrupted skin barrier eases the entry of allergens and activates the innate immune response. Damaged epithelial cells produce alarmins such as IL-25, IL-33, and TSLP, followed by activation of ILC2 and Th2. Activated cells induce type 2 skewing and IgE production by B cells. Type 2 cytokines and degranulation of mast cells exacerbate the inflammation and further attenuate barrier function. ILC innate lymphoid cell, EOS eosinophil, MC mast cell, TSLP thymic stromal lymphopoietin
Type 2 immune responses in skin are typically induced by defects in genes that make the epidermal layers relatively intact to release alarmins such as IL-33 and TSLP [88]. After inflammation, T cell migration is observed in the skin epithelium with variable numbers of eosinophils [91].
In airway epithelia, the type 2 immune response ultimately leads to infiltration of lung tissue with Th2 cells, ILC2s, M2 macrophages and eosinophils unlike the skin epithelia [92, 93]. This promotes nitric oxide production and differentiation of epithelia, production of mucus as well as smooth muscle contraction, and extracellular matrix generation, activation, recruitment and survival of eosinophils [17]. Environmental influences may be mediated via epigenetic processes like deacetylation. Mechanistic studies have demonstrated that barrier leakiness in asthma is induced by TH2 cells, IL‑4 and IL-13 and histone deacetylase activity [94]. Moreover, increased histone deacetylase activity as a potential tissue-injury mechanism is found to be responsible for dysregulated epithelial cell repair, leading to defective epithelial barriers in allergic rhinitis [95].
Dysfunctions of intestinal epithelium lead to formation of several allergic, autoimmune and metabolic diseases. The majority of studies on the links between a leaky intestinal epithelial barrier, microbial dysbiosis, neuroinflammation, and neurodegeneration have been correlative, but a full causal relationship has yet to be shown [4].
Epithelial barrier impairment on the development of allergic diseases
Many diseases, such as AD, asthma, chronic rhinosinusitis and EoE are associated with local epithelial tissue barrier dysfunction in the affected organ [96]. The skin barrier is the frontline defence against exogeneous stimuli. Allergens passing through the skin barrier promote a local and systemic type 2-predominant inflammation, induced by IL‑4, IL‑5, IL-13 and IgE [88]. The allergen engages dendritic/Langerhans cells in the skin, activating a local skin and systemic inflammatory response [39, 88]. Several invasive and non-invasive methods have been developed to study the skin barrier function. Non-invasive methods include the measurement of skin hydration, colorimetry, skin surface pH, and sebometry. Recently, electrical impedance spectroscopy (EIS) has been introduced as a non-invasive tool to assess the integrity of the epidermal barrier in vivo [97, 98], as demonstrated in a mouse model of AD. After experimentally damaging the skin barrier by tape stripping and epicutaneous application of proteases and cholera toxin, a significant reduction in skin electrical impedance was observed, which is inversely correlated with transepidermal water loss, a well-known indicator of skin barrier function [97]. The clinical potential of EIS has been further investigated in patients with AD. Using artificial intelligence-based models, EIS can distinguish between the skin of controls without AD and the non-lesional skin of AD patients with good specificity and sensitivity. In addition, EIS can assess skin lesion healing in response to treatment, correlating with disease score, pruritus scores and inflammatory serum biomarkers that are significantly upregulated in patients and associated with inflammatory pathways that can affect the epithelial barrier [98].
An impaired airway epithelial barrier is a common characteristic of many asthmatic patients. The asthma susceptibility genes are expressed in the airway epithelium (e.g. IL1RL1, IL-33, TSLP). There is increasing evidence indicating that multiple genetic variants induced by environmental factors that increase the risk of asthma development regulate proteins associated with airway epithelial function [99]. Simultaneously, reduced expression of the TJ protein molecules occludin and ZO‑1 has been found in nasal polyp biopsy specimens from patients with chronic rhinosinusitis [100]. In the mid-1960s, anionic surfactants and enzymes were added to laundry detergents to improve cleaning. However, these substances pose serious challenges to the epithelial barrier integrity of the skin and mucous membranes. Many cases of occupational asthma and rhinitis associated with workplace exposure to detergent enzymes and related occupational cases have been reported [101]. Studies in AD patients have shown that the expression of the TJ proteins claudin‑1 and -23 is significantly reduced and epithelial barrier function is significantly impaired [53].
In addition, abnormal inflammatory or developmental signalling pathways may contribute to the establishment and worsening of epithelial barrier defects in allergies. It has been found that in EoE, pathological epithelial remodelling responses and an activated inflammatory response lead to an increase in the overall metabolic demand of the oesophageal epithelium. Thus, the oxygen-sensing transcription factor hypoxia-inducible factor (HIF)-1α plays an important role in maintaining the barrier; at the same time, high HIF has been shown to have a protective effect [102].
Epithelial barrier effects on the development of autoimmune diseases
There is mounting evidence indicating that damaged epithelial barriers, its leakage and the resulting disturbance of the bacterial microflora composition contribute to the development of autoimmune diseases such as type I diabetes, multiple sclerosis, rheumatoid arthritis, ankylosing spondylitis, IBD and systemic lupus erythematosus [18, 19].
Intestinal membrane leakage is often discussed in the context of the epithelial barrier and autoimmune diseases. In healthy individuals, the integrity of the intestinal epithelium is preserved. Usually, bacteria that live in the gut do not translocate to internal organs. However, if the intestinal barrier is compromised, the intestinal commensals are exposed to the host’s immune system in various organs. These observations have been well described in a murine model which described the link between intestinal barrier disruption and the pathogenesis of lupus. Intestinal epithelial TJ proteins are underexpressed in lupus-susceptible mice. In such individuals, commensal bacteria in the gut are transferred to the systemic tissues, activating antigen-presenting cells, which in turn migrate to the mesenteric lymph nodes. Subsequently, CD4+ T cells are activated, and inflammatory cytokines such as IL‑6 are released. In rodents, IL‑6 plays a vital role in the progression of lupus by inducing the production of autoantibodies by B cells and inhibiting the activity of regulatory T cells. Similar observations have also been made in humans. Dysbiosis, caused by a leaky gut, creates an inflammatory environment that results in an intense immune defence promoting the development of the disease or aggravating symptoms. Recognition of microbes or microbial products by lamina propria defences induces escalation of pro-inflammatory cytokines, including IFN‑γ, TNF‑α, IL‑1 and IL-13. These cytokines can aggravate the damage to the intestinal mucosa as in the case of IBD. Moreover, increased Th1 and Th17 cytokines and increased inflammatory activity of macrophages and dendritic cells can damage many tissues as observed in systemic lupus erythematosus [4, 103].
Experimental autoimmune encephalomyelitis (EAE) is a model of multiple sclerosis in rodents such that demyelination is produced by injection of brain extracts or proteins of central nervous system such as myelin basic protein. Moreover, pro-inflammatory cytokines can damage the blood–brain barrier by disrupting the myelin sheath, which directly contributes to the development of EAE. Studies also indicate that the relationship between the tightness of the intestinal epithelial membrane and the development of EAE is more complex because the intestinal barrier breakdown occurs depending on the model sometimes shortly after the induction of EAE and sometimes before. It has also been shown that the degree of intestinal barrier breakdown is associated with the severity of EAE and with the immune dysfunction of the nervous system at many levels, from local to central. Interestingly, improving the tightness of the intestinal barrier in a mouse model ameliorates and postpones the development of EAE, which indicates a close relationship on the brain–gut axis. This is attributed to the intestinal nervous system, made up of the ganglia of the intestinal neurons and glial cells, which are capable of releasing important mediators influencing repair, cell proliferation, epithelial differentiation and TJ changes, regulating intestinal permeability and providing a communication pathway between the gut microenvironment and the central nervous system. [18, 104, 105].
Celiac disease is also an immune-mediated disease of the small bowel with histopathology showing dramatic changes in mucosal structure. A number of groups have reported an increase in the number of apoptotic intestinal epithelial cells in the peritoneal mucosa [106]. In addition, one-path impedance spectroscopy measurements of patients with celiac disease indicated an impaired epithelial barrier function. It is indicated that these patients can benefit from the zonulin-targeting drug, larazotide, as a supplement to a gluten-free diet [107]. Elevated serum zonulin levels signify a leaky intestinal barrier, which facilitates microbial dysbiosis and inflammation. It is also found that gut barrier leakiness is linked to rheumatoid arthritis development with the migration of the inflammatory cells from the gut to joints and the prevention of arthritis by healing epithelial barriers. The use of zonulin antagonists or therapies to restore the intestinal barrier integrity helps to inhibit the symptoms of certain diseases such as rheumatoid arthritis or ankylosing spondylitis. Several findings support the hypothesis that the onsets of rheumatoid arthritis and ankylosing spondylitis are associated with the intestinal microflora and zonulin levels, as cell wall fragments of various intestinal bacteria are arthritogenic. Current medical reports indicate the effectiveness of treatment with the use of larazotide acetate, a zonulin antagonist, which restores the integrity of the intestinal barrier and effectively inhibits the development of diseases by delaying the initial stage of arthritis [19, 108].
Translocation of microorganisms into the pancreatic lymph nodes has been observed, initiating pro-inflammatory cytokines IL‑6 and TNF, thus enhancing the immune response of the Th1/Th17 type, which induces pancreatic cell damage, leading to pancreatic islet inflammation. These events are associated with the onset of type I diabetes as confirmed in a diabetic-resistant mouse model. However, diabetes is a much more complex disease as epithelial barriers are also regulated by metabolic mechanisms. It has been shown that high glucose levels in tissue due to insulin resistance may negatively affect TJs by reducing the expression of protein genes responsible for their formation. When discussing the relationship between epithelial barrier continuity and autoimmune diseases, the haptoglobin protein zonulin, the human analogue of the Vibrio cholerae occludens toxin, should be mentioned. In a rodent model, a zonulin inhibitor has been demonstrated to reverse the symptoms of diabetes and microbial translocation in the intestine. Increased concentration of this protein was associated with the opening of tight junctions and dysbiosis, indirectly affecting the blood–brain barrier, which is also related to multiple sclerosis as discussed above [18, 19, 105].
Conclusion
A defective epithelial barrier facilitates the absorption of foreign substances, inducing an inflammatory response, and subsequent systemic atopic reactions. Deciphering the underlying mechanisms involved in epithelial barrier disruption is vital for building novel strategies for the prevention and treatment of allergic and autoimmune diseases. Experimental models, organoids/spheroid models [109] and organ-on-a-chip systems should be developed to study the passage of allergens across a leaky epithelial barrier. There is clear evidence connecting barrier leakiness and allergic and autoimmune diseases, which can have systemic implications (Table 1). Several potential solutions have been proposed for reducing the incidence of diseases linked to a defective epithelial barrier: avoidance or dose control of all aforementioned substances, development of less toxic products, identification of biomarkers of a leaky barrier, novel therapies for restoring tissue-specific barrier components, inhibiting bacterial translocation, avoiding opportunistic pathogen colonization and interventions through diet and the microbiome.
References
Georas SN, Rezaee F. Epithelial barrier function: at the front line of asthma immunology and allergic airway inflammation. J Allergy Clin Immunol. 2014;134(3):509–20.
Platts-Mills TA. The allergy epidemics: 1870–2010. J Allergy Clin Immunol. 2015;136(1):3–13.
Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347(12):911–20.
Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21(11):739–51.
Akdis CA. Allergy and hypersensitivity: mechanisms of allergic disease. Curr Opin Immunol. 2006;18(6):718–26.
Trautmann A, Schmid-Grendelmeier P, Krüger K, et al. T cells and eosinophils cooperate in the induction of bronchial epithelial cell apoptosis in asthma. J Allergy Clin Immunol. 2002;109(2):329–37.
Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106(1):25–35.
Zimmermann M, Koreck A, Meyer N, et al. TNF-like weak inducer of apoptosis (TWEAK) and TNF‑α cooperate in the induction of keratinocyte apoptosis. J Allergy Clin Immunol. 2011;127(1):200–207, 207.e201–210.
Basinski TM, Holzmann D, Eiwegger T, et al. Dual nature of T cell-epithelium interaction in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124(1):74–80.e71–78.
Solarewicz-Madejek K, Basinski TM, Crameri R, et al. T cells and eosinophils in bronchial smooth muscle cell death in asthma. Clin Exp Allergy. 2009;39(6):845–55.
Pott J, Maloy KJ. Epithelial autophagy controls chronic colitis by reducing TNF-induced apoptosis. Autophagy. 2018;14(8):1460–1.
McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the Smad signaling pathway. J Immunol. 2005;174(9):5774–80.
Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307(5717):1920–5.
Laitinen LA, Laitinen A, Haahtela T. Airway mucosal inflammation even in patients with newly diagnosed asthma. Am Rev Respir Dis. 1993;147(3):697–704.
Erjefält JS, Uller L, Malm-Erjefält M, Persson CG. Rapid and efficient clearance of airway tissue granulocytes through transepithelial migration. Thorax. 2004;59(2):136–43.
Jutel M, Akdis M, Budak F, et al. IL-10 and TGF-beta cooperate in the regulatory T cell response to mucosal allergens in normal immunity and specific immunotherapy. Eur J Immunol. 2003;33(5):1205–14.
Pat Y, Ogulur I. The epithelial barrier hypothesis: a 20-year journey. Allergy. 2021;76(11):3560–2.
Abdelhamid L, Luo XM. Retinoic acid, leaky gut, and autoimmune diseases. Nutrients. 2018. https://doi.org/10.3390/nu10081016.
Tajik N, Frech M, Schulz O, et al. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat Commun. 2020;11(1):1995.
Sharma S, Tripathi P. Gut microbiome and type 2 diabetes: where we are and where to go? J Nutr Biochem. 2019;63:101–8.
Brant A, Hole A, Cannon J, et al. Occupational asthma caused by cellulase and lipase in the detergent industry. Occup Environ Med. 2004;61(9):793–5.
Xian M, Wawrzyniak P, Rückert B, et al. Anionic surfactants and commercial detergents decrease tight junction barrier integrity in human keratinocytes. J Allergy Clin Immunol. 2016;138(3):890–893.e899.
Wang M, Tan G, Eljaszewicz A, et al. Laundry detergents and detergent residue after rinsing directly disrupt tight junction barrier integrity in human bronchial epithelial cells. J Allergy Clin Immunol. 2019;143(5):1892–903.
Fukuoka A, Matsushita K, Morikawa T, Takano H, Yoshimoto T. Diesel exhaust particles exacerbate allergic rhinitis in mice by disrupting the nasal epithelial barrier. Clin Exp Allergy. 2016;46(1):142–52.
Manzo ND, Slade R, Richards JH, McGee JK, Martin LD, Dye JA. Susceptibility of inflamed alveolar and airway epithelial cells to injury induced by diesel exhaust particles of varying organic carbon content. J Toxicol Environ Health A. 2010;73(8):565–80.
Caraballo JC, Yshii C, Westphal W, Moninger T, Comellas AP. Ambient particulate matter affects occludin distribution and increases alveolar transepithelial electrical conductance. Respirology. 2011;16(2):340–9.
Michaudel C, Mackowiak C, Maillet I, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol. 2018;142(3):942–58.
Jin Y, Lu L, Tu W, Luo T, Fu Z. Impacts of polystyrene microplastic on the gut barrier, microbiota and metabolism of mice. Sci Total Environ. 2019;649:308–17.
Smallcombe CC, Harford TJ, Linfield DT, et al. Titanium dioxide nanoparticles exaggerate respiratory syncytial virus-induced airway epithelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2020;319(3):L481–L96.
Roberts CL, Keita AV, Duncan SH, et al. Translocation of Crohn’s disease Escherichia coli across M‑cells: contrasting effects of soluble plant fibres and emulsifiers. Gut. 2010;59(10):1331–9.
Singh N, Diebold Y, Sahu SK, Leonardi A. Epithelial barrier dysfunction in ocular allergy. Allergy. 2021. https://doi.org/10.1111/all.15174.
Bin L, Leung DYM. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin Immunol. 2016;12(1):52.
Vancamelbeke M, Vanuytsel T, Farré R, et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23(10):1718–29.
Apfelbacher CJ, Diepgen TL, Schmitt J. Determinants of eczema: population-based cross-sectional study in Germany. Allergy. 2011;66(2):206–13.
Adil MS, Narayanan SP, Somanath PR. Cell-cell junctions: structure and regulation in physiology and pathology. Tissue Barriers. 2021;9(1):1848212.
Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122(Pt 9):1285–94.
Smith FJ, Irvine AD, Terron-Kwiatkowski A, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38(3):337–42.
Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39(5):650–4.
Mitamura Y, Ogulur I, Pat Y, et al. Dysregulation of the epithelial barrier by environmental and other exogenous factors. Contact Derm. 2021;85(6):615–26.
Venkataraman D, Soto-Ramírez N, Kurukulaaratchy RJ, et al. Filaggrin loss-of-function mutations are associated with food allergy in childhood and adolescence. J Allergy Clin Immunol. 2014;134(4):876–882.e874.
Marenholz I, Kerscher T, Bauerfeind A, et al. An interaction between filaggrin mutations and early food sensitization improves the prediction of childhood asthma. J Allergy Clin Immunol. 2009;123(4):911–6.
Brown SJ, Kroboth K, Sandilands A, et al. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Invest Dermatol. 2012;132(1):98–104.
Stemmler S, Parwez Q, Petrasch-Parwez E, Epplen JT, Hoffjan S. Association of variation in the LAMA3 gene, encoding the alpha-chain of laminin 5, with atopic dermatitis in a German case-control cohort. BMC Dermatol. 2014;14:17.
Margolis DJ, Gupta J, Apter AJ, et al. Filaggrin‑2 variation is associated with more persistent atopic dermatitis in African American subjects. J Allergy Clin Immunol. 2014;133(3):784–9.
Trzeciak M, Wesserling M, Bandurski T, Glen J, Nowicki R, Pawelczyk T. Association of a single nucleotide polymorphism in a late cornified envelope-like proline-rich 1 gene (LELP1) with atopic dermatitis. Acta Derm Venereol. 2016;96(4):459–63.
Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147(1):185–94.
Gow A, Southwood CM, Li JS, et al. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell. 1999;99(6):649–59.
Furuse M, Hata M, Furuse K, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156(6):1099–111.
Katsuno T, Umeda K, Matsui T, et al. Deficiency of zonula occludens‑1 causes embryonic lethal phenotype associated with defected yolk sac angiogenesis and apoptosis of embryonic cells. Mol Biol Cell. 2008;19(6):2465–75.
Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO‑2, but Not ZO‑3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28(5):1669–78.
Hadj-Rabia S, Baala L, Vabres P, et al. Claudin‑1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: a tight junction disease. Gastroenterology. 2004;127(5):1386–90.
Hadj-Rabia S, Brideau G, Al-Sarraj Y, et al. Multiplex epithelium dysfunction due to CLDN10 mutation: the HELIX syndrome. Genet Med. 2018;20(2):190–201.
De Benedetto A, Rafaels NM, McGirt LY, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol. 2011;127(3):773–786.e771–777.
Ross-Hansen K, Linneberg A, Johansen JD, et al. The role of glutathione S‑transferase and claudin‑1 gene polymorphisms in contact sensitization: a cross-sectional study. Br J Dermatol. 2013;168(4):762–70.
Young CA, Rorke EA, Adhikary G, Xu W, Eckert RL. Loss of epidermal AP1 transcription factor function reduces filaggrin level, alters chemokine expression and produces an ichthyosis-related phenotype. Cell Death Dis. 2017;8(6):e2840.
Klee KMC, Janecke AR, Civan HA, et al. AP1S1 missense mutations cause a congenital enteropathy via an epithelial barrier defect. Hum Genet. 2020;139(10):1247–59.
Norén E, Almer S, Söderman J. Genetic variation and expression levels of tight junction genes identifies association between MAGI3 and inflammatory bowel disease. BMC Gastroenterol. 2017;17(1):68.
Samuelov L, Sarig O, Harmon RM, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45(10):1244–8.
Shoda T, Kaufman KM, Wen T, et al. Desmoplakin and periplakin genetically and functionally contribute to eosinophilic esophagitis. Nat Commun. 2021;12(1):6795.
Koppelman GH, Meyers DA, Howard TD, et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am J Respir Crit Care Med. 2009;180(10):929–35.
Zhu TH, Zhu TR, Tran KA, Sivamani RK, Shi VY. Epithelial barrier dysfunctions in atopic dermatitis: a skin–gut–lung model linking microbiome alteration and immune dysregulation. Br J Dermatol. 2018;179(3):570–81.
Celebi Sozener Z, Ozturk Ozdel B, Cerci P, et al. Epithelial barrier hypothesis: Effect of external exposome on microbiome and epithelial barriers in allergic disease. Allergy. 2022. https://doi.org/10.1111/all.15240.
Niranjan R, Subramanian M, Panneer D, Ojha SK. Eosinophils restrict diesel exhaust particles induced cell proliferation of lung epithelial A549 cells, vial Interleukin-13 mediated mechanisms: implications for tissue remodelling and fibrosis. Comb Chem High Throughput Screen. 2022. https://doi.org/10.2174/1386207325666220105150655.
Kim N, Han D, Wang IJ, et al. Altered secretome by diesel exhaust particles and lipopolysaccharide in primary human nasal epithelium. J Allergy Clin Immunol. 2022. https://doi.org/10.1016/j.jaci.2021.12.793.
Smyth T, Veazey J, Eliseeva S, Chalupa D, Elder A, Georas SN. Diesel exhaust particle exposure reduces expression of the epithelial tight junction protein Tricellulin. Part Fibre Toxicol. 2020;17(1):52.
Sokolowska M, Quesniaux VFJ, Akdis CA, Chung KF, Ryffel B, Togbe D. Acute respiratory barrier disruption by ozone exposure in mice. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.02169.
Dong C‑D, Chen C‑W, Chen Y‑C, Chen H‑H, Lee J‑S, Lin C‑H. Polystyrene microplastic particles: In vitro pulmonary toxicity assessment. J Hazard Mater. 2020;385:121575.
Akbarshahi H, Menzel M, Ramu S, Mahmutovic Persson I, Bjermer L, Uller L. House dust mite impairs antiviral response in asthma exacerbation models through its effects on TLR3. Allergy. 2018;73(5):1053–63.
Steelant B, Farré R, Wawrzyniak P, et al. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens‑1 expression. J Allergy Clin Immunol. 2016;137(4):1043–1053.e1045.
Hiraishi Y, Yamaguchi S, Yoshizaki T, et al. IL-33, IL-25 and TSLP contribute to development of fungal-associated protease-induced innate-type airway inflammation. Sci Rep. 2018;8(1):18052.
Vinhas R, Cortes L, Cardoso I, et al. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy. 2011;66(8):1088–98.
Stephen-Victor E, Crestani E, Chatila TA. Dietary and microbial determinants in food allergy. Immunity. 2020;53(2):277–89.
Andersen K, Kesper MS, Marschner JA, et al. Intestinal dysbiosis, barrier dysfunction, and bacterial translocation account for CKD-related systemic inflammation. J Am Soc Nephrol. 2017;28(1):76–83.
Soderholm AT, Pedicord VA. Intestinal epithelial cells: at the interface of the microbiota and mucosal immunity. Immunology. 2019;158(4):267–80.
Meng X, Zhou HY, Shen HH, et al. Microbe-metabolite-host axis, two-way action in the pathogenesis and treatment of human autoimmunity. Autoimmun Rev. 2019;18(5):455–75.
Chassaing B, Van de Wiele T, De Bodt J, Marzorati M, Gewirtz AT. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut. 2017;66(8):1414–27.
Zhu YT, Yuan YZ, Feng QP, et al. Food emulsifier polysorbate 80 promotes the intestinal absorption of mono-2-ethylhexyl phthalate by disturbing intestinal barrier. Toxicol Appl Pharmacol. 2021;414:115411.
Roberts CL, Rushworth SL, Richman E, Rhodes JM. Hypothesis: Increased consumption of emulsifiers as an explanation for the rising incidence of Crohn’s disease. J Crohns Colitis. 2013;7(4):338–41.
Aguayo-Patron SV, Calderon de la Barca AM. Old fashioned vs. ultra-processed-based current diets: possible implication in the increased susceptibility to type 1 diabetes and celiac disease in childhood. Foods. 2017. https://doi.org/10.3390/foods6110100.
Laster J, Bonnes SL, Rocha J. Increased use of emulsifiers in processed foods and the links to obesity. Curr Gastroenterol Rep. 2019;21(11):61.
Keita AV, Alkaissi LY, Holm EB, et al. Enhanced E. coli LF82 translocation through follicle-associated epithelium in Crohn’s disease is dependent on long polar fimbriae and CEACAM6 expression, and increases paracellular permeability. J Crohns Colitis. 2019. https://doi.org/10.1093/ecco-jcc/jjz144.
Khoshbin K, Camilleri M. Effects of dietary components on intestinal permeability in health and disease. Am J Physiol Gastrointest Liver Physiol. 2020;319(5):G589–G608.
Chassaing B, Koren O, Goodrich JK, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519(7541):92–6.
Sapone A, Lammers KM, Casolaro V, et al. Divergence of gut permeability and mucosal immune gene expression in two gluten-associated conditions: celiac disease and gluten sensitivity. BMC Med. 2011;9:23.
Clemente MG, De Virgiliis S, Kang JS, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52(2):218–23.
Singh P, Grabauskas G, Zhou SY, Gao J, Zhang Y, Owyang C. High FODMAP diet causes barrier loss via lipopolysaccharide-mediated mast cell activation. JCI Insight. 2021. https://doi.org/10.1172/jci.insight.146529.
Ong DK, Mitchell SB, Barrett JS, et al. Manipulation of dietary short chain carbohydrates alters the pattern of gas production and genesis of symptoms in irritable bowel syndrome. J Gastroenterol Hepatol. 2010;25(8):1366–73.
Akdis CA, Arkwright PD, Brüggen MC, et al. Type 2 immunity in the skin and lungs. Allergy. 2020;75(7):1582–605.
Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. 2018;18(2):121–33.
Sugita K, Soyka MB, Wawrzyniak P, et al. Outside-in hypothesis revisited: the role of microbial, epithelial, and immune interactions. Ann Allergy Asthma Immunol. 2020;125(5):517–27.
Guttman-Yassky E, Krueger JG, Lebwohl MG. Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Exp Dermatol. 2018;27(4):409–17.
Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2018;18(7):454–66.
Komlósi ZI, van de Veen W, Kovács N, et al. Cellular and molecular mechanisms of allergic asthma. Mol Aspects Med. 2021. https://doi.org/10.1016/j.mam.2021.100995.
Wawrzyniak P, Wawrzyniak M, Wanke K, et al. Regulation of bronchial epithelial barrier integrity by type 2 cytokines and histone deacetylases in asthmatic patients. J Allergy Clin Immunol. 2017;139(1):93–103.
Steelant B, Wawrzyniak P, Martens K, et al. Blocking histone deacetylase activity as a novel target for epithelial barrier defects in patients with allergic rhinitis. J Allergy Clin Immunol. 2019;144(5):1242–1253.e1247.
Akdis CA. The epithelial barrier hypothesis proposes a comprehensive understanding of the origins of allergic and other chronic noncommunicable diseases. J Allergy Clin Immunol. 2022;149(1):41–4.
Rinaldi AO, Morita H, Wawrzyniak P, et al. Direct assessment of skin epithelial barrier by electrical impedance spectroscopy. Allergy. 2019;74(10):1934–44.
Rinaldi AO, Korsfeldt A, Ward S, et al. Electrical impedance spectroscopy for the characterization of skin barrier in atopic dermatitis. Allergy. 2021;76(10):3066–79.
Heijink IH, Kuchibhotla VNS, Roffel MP, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. 2020;75(8):1902–17.
Soyka MB, Wawrzyniak P, Eiwegger T, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN‑γ and IL‑4. J Allergy Clin Immunol. 2012;130(5):1087–1096.e1010.
Adisesh A, Murphy E, Barber CM, Ayres JG. Occupational asthma and rhinitis due to detergent enzymes in healthcare. OCCMED. 2011;61(5):364–9.
Masterson JC, Biette KA, Hammer JA, et al. Epithelial HIF-1α/claudin‑1 axis regulates barrier dysfunction in eosinophilic esophagitis. J Clin Invest. 2019;129(8):3224–35.
Mu Q, Zhang H, Liao X, et al. Control of lupus nephritis by changes of gut microbiota. Microbiome. 2017;5(1):73.
Camara-Lemarroy CR, Silva C, Greenfield J, Liu WQ, Metz LM, Yong VW. Biomarkers of intestinal barrier function in multiple sclerosis are associated with disease activity. Mult Scler. 2020;26(11):1340–50.
Buscarinu MC, Fornasiero A, Romano S, et al. The contribution of gut barrier changes to multiple sclerosis pathophysiology. Front Immunol. 2019;10:1916.
Schumann M, Siegmund B, Schulzke JD, Fromm M. Celiac disease: role of the epithelial barrier. Cell Mol Gastroenterol Hepatol. 2017;3(2):150–62.
Leffler DA, Kelly CP, Green PH, et al. Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology. 2015;148(7):1311–1319 e1316.
Ciccia F, Guggino G, Rizzo A, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis. 2017;76(6):1123–32.
Pat Y, Rückert B, et al. Differentiation of bronchial epithelial spheroids in the presence of IL-13 recapitulates characteristic features of asthmatic airway epithelia. Allergy. 2022. https://doi.org/10.1111/all.15279.
Funding
The authors have received no funding for this article.
Funding
Open access funding provided by University of Zurich
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
C.A. Akdis has received research grants from the Swiss National Science Foundation, Christine Kühne-Center for Allergy Research and Education, European Commission Horizon’s 2020 Framework Programme “CURE”, Novartis Research Institutes, GlaxoSmithKline and AstraZeneca. He took part in the advisory board and received research grants from GlaxoSmithKline, Sanofi/Regeneron, SciBase and Novartis. He is the Editor-in-Chief of Allergy. D. Yazici, I. Ogulur, O. Kucukkase, M. Li, A.O. Rinaldi, Y. Pat, A. Wallimann, S. Wawrocki, Z. Celebi Sozener, B. Buyuktiryaki, C. Sackesen, M. Akdis and Y. Mitamura declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yazici, D., Ogulur, I., Kucukkase, O. et al. Epithelial barrier hypothesis and the development of allergic and autoimmune diseases. Allergo J Int 31, 91–102 (2022). https://doi.org/10.1007/s40629-022-00211-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40629-022-00211-y