
Abstract
Purpose
To evaluate circulating soluble α-klotho (sαKL) levels in GHD children before and after 12 months of GH treatment (GHT).
Methods
Auxological and basal metabolic parameters, oral glucose tolerance test for glucose and insulin levels, insulin sensitivity indices and klotho levels were evaluated before and after 12 months of follow-up in 58 GHD children and 56 healthy controls.
Results
At baseline, GHD children showed significantly lower growth velocity standard deviation score (SDS) (p < 0.001), bone/chronological age ratio (p < 0.001), GH peak and area under the curve (AUC) after arginine test (ARG) (both p < 0.001) and glucagon stimulation test (GST) (p < 0.001 and 0.048, respectively), IGF-1 (p < 0.001), with higher BMI (SDS) (p < 0.001), WC (SDS) (p = 0.003) and sαKL (p < 0.001) than controls. After 12 months of GHT, GHD children showed a significant increase in height (SDS) (p < 0.001), growth velocity (SDS) (p < 0.001), bone/chronological age ratio (p < 0.001) IGF-1 (p < 0.001), fasting insulin (p < 0.001), Homa-IR (p < 0.001) and sαKL (p < 0.001) with a concomitant decrease in BMI (SDS) (p = 0.002) and WC (SDS) (p = 0.038) than baseline. At ROC curve analysis, we identified a sαKL cut-off to discriminate controls and GHD children of 1764.4 pg/mL in females and 1339.4 pg/mL in males.
At multivariate analysis, the independent variables significantly associated with sαKL levels after 12 months of GHT were the oral disposition index (p = 0.004, β = 0.327) and IGF-1 (p = 0.019, β = 0.313).
Conclusions
Gender-related sαKL may be used as a marker of GHD combined to GH and IGF-1. Insulin and IGF-1 are independently associated with sαKL values after 12 months of GHT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Growth hormone deficiency (GHD) affects about 1 out of 4000 children [1]. GHD is characterized by short stature, altered body composition (reduced muscle mass and increased adiposity) [2, 3] as well as metabolic alterations (increase in fat mass) [4,5,6], and recombinant human (rh) GH treatment (GHT) may result in a reversal of these effects [7, 8].
The diagnosis of GHD is based on many factors including clinical, auxological, and biochemical parameters. As GH is secreted in a pulsatile way, basal GH measurements are useless, and the secretion is assessed using stimulation tests [9].
Children with normal GH secretion and GHD frequently show superimposable peak GH concentrations [9]. Only a minority of children with idiopathic GHD remain GH deficient after discontinuation of GHT [10,11,12,13], and about 20% of healthy children may test “deficient” if a single stimulation test is used [14]. For these reasons, two stimulation tests are recommended to assess the diagnosis of GHD [14]. In addition, serum biomarkers (e.g., IGF-1, IGFBP-3) are not fully accurate in distinguishing between GH sufficient and deficient children [15].
A strong relationship between klotho protein and GH/IGF-1 system has been demonstrated [16].
The Klotho gene was first identified in 1997 as an anti-aging agent [17]. An impairment in its genetic expression has been associated with short lifespan, infertility, atherosclerosis, metabolic syndrome, skin atrophy, osteoporosis, and emphysema in mice [17], while over-expression leads to a longer life span [18, 19]. The Klotho gene encodes the alpha klotho (α-klotho) protein composed by an intracellular and transmembrane domain and an extracellular domain [20, 21]. The transmembrane form (mKL) is a co-receptor of fibroblast growth factor 23 (FGF23), which regulates calcium and phosphorus homeostasis [20, 22]. From the cleavage of the extracellular domain mediated by proteases ADAM, can be obtained the soluble form (sαKL), a circulating molecule with endocrine functions [23]. Several effects of sαKL have been reported on insulin physiology, inhibition of insulin/IGF-1 receptor phosphorylation and signalling events, such as tyrosine phosphorylation of insulin receptor substrates and phosphoinositide 3-kinase, thereby blocking insulin and IGF-1 signalling [18, 24].
In the current study, we evaluated sαKL levels in a cohort of GHD children at diagnosis, and during GH treatment (GHT) to assess its potential diagnostic role in GHD.
Materials and methods
We prospectively studied 58 pre-pubertal children (32 males and 26 females, mean age 7.8 ± 1.7 years) with isolated idiopathic GHD, consecutively admitted to the Endocrinology Section of the University of Palermo during the years 2016–2018 and treated with rhGH for at least 12 months. Fifty-six healthy short children, matched for sex (36 M, 20 F), age (mean age 8.4 ± 1.9 years) and pubertal status, were recruited among patients referred for assessment of short stature as a control group and followed up at baseline and after 12 months. Both controls and GHD children were pre-pubertal during the observation period.
Exclusion criteria were the following: having a multiple pituitary hormone deficiency or panhypopituitarism, not having completed the 12 months of follow-up and pubertal onset before and during treatment. The diagnosis of GHD was assessed according the criteria of the GH Research Society [9]. Both the patients and the controls underwent two stimulation tests (arginine and glucagon) for their short stature or reduced height velocity SDS. Controls had a GH response > 10 μg/L to both stimulation tests.
All patients with GHD received replacement recombinant human GH therapy once daily at bedtime with a pen injection system. Children were treated with an initial mean daily dose of 0.025 mg/kg from baseline until the sixth month and a mean dose of 0.028 mg/kg from months 6 to 12. The decision to change the dose of rhGH therapy was based on the biochemical and auxological parameters, as previously reported [25]. During the entire follow-up IGF-1 levels were maintained within the normal range for age (81.3–255.3 mcg/L for males and 85.9–323 mcg/L for females).
Study protocol
At baseline and after 12 months of follow-up in controls and GHD children body height, growth velocity, body mass index (BMI), waist circumference (WC) [expressed as Standard Deviation Score (SDS)] and bone/chronological age ratio were evaluated.
The arginine test (ARG) and glucagon stimulation test (GST) were performed at baseline as provocative tests to diagnose GHD and the areas under the curve (AUC) of GH (AUCGH) were calculated. GHD was defined when GH peak was < 10 μg/L after two provocative tests. Brain MRI was performed in all children with a GH response < 10 μg/L after the two stimulation tests. No pituitary abnormalities were detected in the group of patients enrolled.
Fasting blood glucose and insulin, hemoglobin A1c (HbA1c), total and high-density lipoprotein (HDL) cholesterol and triglycerides, IGF-1 and sαKL were assayed. Low-density lipoprotein (LDL) cholesterol levels were calculated by the following formula: total cholesterol – (HDL cholesterol − triglycerides/5). We performed an oral glucose tolerance test (OGTT), with 1.75 g/kg body weight of glucose, with blood samples collection every 30 min up to 2 h for glucose and insulin measurements.
The homeostasis model assessment estimate of insulin resistance (Homa-IR) [(fasting glucose × fasting insulin)/22.5] [26], the Matsuda index of insulin sensitivity (ISI-Matsuda) [10,000/glucose (mg/dL) × insulin (mU/mL) × glucose mean × insulin mean] [27], the oral disposition index (DIo) [(ΔInsulin 0–30/ΔGlucose 0–30) × (1/fasting insulin)] [28] and the area under the curve for insulin (AUC2h insulinemia) and glucose (AUC2h glycaemia) were calculated.
The population evaluated in the current study was different from that enrolled in another study of our group [25].
The study was approved from the Ethics Committee of the Policlinico Paolo Giaccone Hospital, University of Palermo, in agreement with the ethical standards of the local committee on human experimentation (institutional and national) and with the Declaration of Helsinki (1964). At the time of hospitalization, all patients and their parents gave informed written consent to the study and for scientific use of the data.
Hormone and biochemical assays
Biochemical parameters were measured with standard methods previously reported [25].
Serum samples for sαKL concentration were analyzed using a commercial solid phase sandwich ELISA (Enzyme-linked Immunosorbent Assay) assay kit (cat.27998, Immuno-Biological Laboratoires Co., Fujioka-Shi, Japan). The normal measurement range was 93.75–6000 pg/mL [29]. Samples were assayed following the manufacturer’s instructions.
Statistical analysis
The Statistical Package for Social Sciences SPSS version 19 was used for data analysis. Baseline characteristics were presented as mean ± standard deviation (SD) for continuous variables, while rates and proportions were calculated for categorical data. Normality of distribution for quantitative variables was assessed with the Kolmogorov–Smirnov test. The differences between the two independent groups (GHD children vs. controls) were evaluated by Student’s t test, while the differences between paired continuous variables (before and after 12 months of follow-up in GHD children and controls) were analyzed by the paired t-Test. ROC curve analysis was performed to identify the sαKL cut-off differentiating children with GHD from healthy controls.
The independent variables associated with the dependent variable (sαKL) after 12 months of GHT were evaluated by multivariate analysis. A p value < 0.05 was considered statistically significant.
Results
The clinical, hormonal, and metabolic parameters of control subjects and GHD children at diagnosis and after 12 months of GHT are shown in Table 1.
At baseline, GHD children showed significantly lower bone/chronological age ratio (p < 0.001), GH peak and AUC after ARG (both p < 0.001) and GST (p < 0.001 and 0.048, respectively), IGF-1 (p < 0.001) and sαKL (p = 0.001), with higher BMI (SDS) (p < 0.001) and WC (SDS) (p = 0.003) than controls (Table 1).
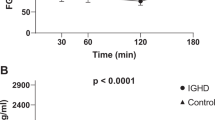
After 12 months of follow-up, controls had a significant increase in height (SDS) (p < 0.001), BMI (SDS) (p = 0.002), WC (SDS) (p < 0.001), height velocity (SDS) (p < 0.001) and sαKL (p = 0.030; females p = 0.045 and males p = 0.024) (Table 2) than baseline. After 12 months of GHT, GHD children showed a significant increase in height (SDS) (p < 0.001), growth velocity (SDS) (p < 0.001), bone/chronological age ratio (p < 0.001), IGF-1 (p < 0.001), fasting insulin (p < 0.001), Homa-IR (p < 0.001) and sαKL (p < 0.001; females p = 0.004 and males p = 0.001) levels, with a concomitant decrease in BMI (SDS) (p = 0.002) and WC (SDS) (p = 0.038) compared to baseline (Table 2). A comparison between sαKL levels at baseline and after 12 months of treatment in males and females with GHD was performed, showing that females with GHD had higher sαKL values than males (Fig. 1). No other differences between females and males were observed in controls and GHD (data not shown).

Comparison of sαKL levels between male and female children with GHD at baseline and after 12 months of treatment
The comparison between GHD children and controls at 12 months showed that GHD children had significantly higher WC (SDS) (p < 0.001), fasting glucose (p < 0.001), fasting insulin (p < 0.001), HOMA-IR (p < 0.001), sαKL (p < 0.001; females and males p < 0.001) levels, than controls (Table 2).
In addition, a ROC curve analysis was performed to identify the sαKL cut-off differentiating GHD children from controls. The sαKL cut-off of 1764.4 pg/mL discriminated female controls and GHD children with 83.3% sensitivity and 62.5% specificity, and the area under the curve was 0.667 (Fig. 2). The sαKL cut-off of 1339.4 pg/mL differentiated male controls and GHD children with a sensitivity of 72.7% and a specificity of 81%; the area under the curve was 0.828 (Fig. 2).

ROC curve analysis of sαKL cut-off in females and males to discriminate controls from GHD
At multivariate analysis, the independent variables significantly associated with sαKL levels after 12 months of GHT were the oral disposition index (p = 0.004, β = 0.327) and IGF-1 (p = 0.019, β = 0.313) (Fig. 3).

Independent variables associated with sαKL levels after 12 months of GH treatment at multivariate analysis
Discussion
In the current study, we evaluated the effects of GHT on sαKL levels in a paediatric cohort of GHD children and healthy controls. As expected from previous reports [30, 31], sαKL was lower at diagnosis in patients with GHD compared to healthy controls and increased after GHT.
Lower sαKL values have been reported in children with GHD compared with controls [30]. Interestingly, patients with organic GHD had lower sαKL levels than idiopathic GHD and GH-sufficient participants [31]. By contrast, some authors did not find differences in sαKL in the diagnosis of GHD compared to short stature, showing superimposable values in the two groups, likely due to the small sample included in the study and maybe to the lack of information on FGF-23 [32].
In the current study we found that the sαKL cut-off to discriminate healthy controls from GHD was 1764.4 pg/mL with a sensitivity of 83.3% and a specificity of 62.5% and 1339.4 pg/mL, with a sensitivity of 72.7% and a specificity of 81% in females and males, respectively.
As expected, IGF-1 was independently associated with sαKL levels. A positive correlation of sαKL and IGF-1 values has been widely demonstrated with interesting close reciprocal regulation [32, 33]. IGF-1 appears to stimulate klotho secretion [30, 34], whereas klotho inhibits IGF-1 signalling and activation of the hormone receptor [19]. A significant increase in IGF-1 and IGFBP3 after intraperitoneal injections of klotho has been reported in mice [35], which seems to contradict the inhibition of klotho on pituitary secretion of GH. However, in GH3-cultured cells it has been reported that klotho induces GH secretion by activation of the ERK1/2 pathway [35]. In these cultured cells cotreatment of klotho and bFGF further increased ERK1/2 phosphorylation, while inhibition of ERK1/2 favours the klotho-induced inhibition of GH release in normal pituitaries [35]. In brief, α-klotho stimulates GH secretion at the expense of ERK1/2 phosphorylation and blocks the inhibitory effect of IGF-1 on GH secretion in GH-secreting adenoma cultures [35].
In addition, we found a gender difference in sαKL. To our knowledge, this is the first time that this has been reported in humans, because till now it has only been shown in animals [36]. However, further studies are required to confirm our results.
Interestingly, we also found that insulin secretion expressed by the oral disposition index, a composite measure of β-cell function, which estimates the ability of β-cells to produce insulin adjusted for insulin sensitivity, was independently associated with sαKL levels. Insulin has been demonstrated to stimulate α-klotho [37, 38]. Recently, an exacerbation and aggravation of insulin resistance in sαKL deficiency was reported in patients with type 2 diabetes mellitus, while overexpression of sαKL was associated with increased insulin sensitivity [39]. On the other side, klotho might induce insulin resistance in adipocytes, preventing insulin effects on promotion of GLUT4 plasma membrane translocation, and attenuating intracellular insulin signalling through main mediators, such as Akt, GSK3β, and PFKf3β [40]. However, detailed information about the relationship between sαKL and insulin and the possible role of rhGH is still lacking and needs to be further investigated.
GHT has been suggested to impair insulin sensitivity, even though many studies have investigated insulin sensitivity by HOMA-IR, a basal index not enough reliable to assess insulin sensitivity [41].
The increase in Homa-IR, may just represent an expected consequence of GH-induced basal hyperinsulinemia and currently very few studies have investigated different indices [8]. In the current study, we found an increase in HOMA-IR, but no changes in ISI Matsuda and DIo were found after 12 months of GHT in GHD children.
A limitation of the study is that we do not have full information about the nutrition and physical activity of the children. The strength of the study is relevant number of children enrolled, with a very homogeneous sample (all children were pre-pubertal).
In conclusion, the findings of this study suggest that sαKL may be used as a marker of GHD combined with IGF-1 and GH. Direct GH measurements alone are useless due to the pulsatile nature of GH secretion, while IGF-1 levels alone are unsatisfactory as well as being influenced by age, gender (oestrogens), race, genetics, liver function, nutritional status, portal insulin, thyroid hormones, and concomitant inflammatory disease [42]. A gender-related cut-off of sαKL to discriminate controls from GHD children was identified. Insulin and IGF-1 are independently associated with sαKL values after 12 months of GHT, supporting the interesting relationship between sαKL levels and insulin/IGF-1 signalling.
However, further larger prospective studies are needed to confirm our results.
References
Stochholm K, Gravholt CH, Laursen T, Jørgensen JO, Laurberg P, Andersen M, Kristensen LØ, Feldt-Rasmussen U, Christiansen JS, Frydenberg M, Green A (2006) Incidence of GH deficiency—a nationwide study. Eur J Endocrinol 155(1):61–71. https://doi.org/10.1530/eje.1.02191
Boot AM, Engels MA, Boerma GJ, Krenning EP, De Muinck Keizer-Schrama SM (1997) Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab 82(8):2423–2428. https://doi.org/10.1210/jcem.82.8.4149
Matusik P, Klesiewicz M, Klos K, Stasiulewicz M, Barylak A, Nazarkiewicz P, Malecka-Tendera E (2016) Baseline body composition in prepubertal short stature children with severe and moderate growth hormone deficiency. Int J Endocrinol 2016:4563721. https://doi.org/10.1155/2016/4563721
Lanes R, Soros A, Gunczler P, Paoli M, Carrillo E, Villaroel O, Palacios A (2006) Growth hormone deficiency, low levels of adiponectin, and unfavorable plasma lipid and lipoproteins. J Pediatr 149(3):324–329. https://doi.org/10.1016/j.jpeds.2006.05.010
Capalbo D, MattaceRaso G, Esposito A, Di Mase R, Barbieri F, Meli R, Bruzzese D, Salerno M (2014) Cluster of cardiometabolic risk factors in children with GH deficiency: a prospective, case–control study. Clin Endocrinol (Oxf) 80(6):773–928. https://doi.org/10.1111/cen.12393
Ciresi A, Pizzolanti G, Leotta M, Guarnotta V, Teresi G, Giordano C (2016) Resistin, visfatin, leptin and omentin are differently related to hormonal and metabolic parameters in growth hormone-deficient children. J Endocrinol Invest 39(9):1023–1030. https://doi.org/10.1007/s40618-016-0475-z
Elbornsson M, Götherström G, Bosæus I, Bengtsson BA, Johannsson G, Svensson J (2013) Fifteen years of GH replacement improves body composition and cardiovascular risk factors. Eur J Endocrinol 168(5):745–753. https://doi.org/10.1530/EJE-12-1083
Ciresi A, Guarnotta V, Pizzolanti G, Giordano C (2018) Comparison between euglycemic hyperinsulinemic clamp and surrogate indices of insulin sensitivity in children with growth hormone deficiency. Growth Horm IGF Res 39:40–44. https://doi.org/10.1016/j.ghr.2017.12.007
Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, Rossi WC, Feudtner C, Murad MH, Drug and Therapeutics Committee and Ethics Committee of the Pediatric Endocrine Society (2016) Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr 86:361–397. https://doi.org/10.1159/000452150
Vuralli D, Gonc EN, Ozon ZA, Alikasifoglu A, Kandemir N (2017) Clinical and laboratory parameters predicting a requirement for the reevaluation of growth hormone status during growth hormone treatment: retesting early in the course of GH treatment. Growth Horm IGF Res 34:31–37. https://doi.org/10.1016/j.ghir.2017.05.003
Meazza C, Gertosio C, Pagani S, Pilotta A, Tinelli C, Buzi F, Farello G, Genoni G, Bona G, Bozzola M (2017) Is retesting in growth hormone deficient children really useful? Minerva Endocrinol 42:325–330. https://doi.org/10.23736/S0391-1977.16.02510-4
Dreismann L, Schweizer R, Blumenstock G, Weber K, Binder G (2016) Evaluation of the GHRH-arginine retest for young adolescents with childhood-onset GH deficiency. Growth Horm IGF Res 27:28–32. https://doi.org/10.1016/j.ghir.2016.02.001
Patti G, Noli S, Capalbo D, Allegri AME, Napoli F, Cappa M, Ubertini GM, Gallizia A, Notarnicola S, Ibba A, Crocco M, Parodi S, Salerno M, Loche S, Garré ML, Tornari E, Maghnie M, Di Iorgi N (2019) Accuracy and limitations of the growth hormone (GH) releasing hormone-arginine retesting in young adults with childhood-onset GH deficiency. Front Endocrinol (Lausanne) 10:525. https://doi.org/10.3389/fendo.2019.00525
Chesover AD, Dattani MT (2016) Evaluation of growth hormone stimulation testing in children. Clin Endocrinol (Oxf) 84:708–714. https://doi.org/10.1111/cen.13035
Hokken-Koelega ACS (2011) Diagnostic workup of the short child. Horm Res Paediatr 76(Suppl 3):6–9. https://doi.org/10.1159/000330136
Sato T, Komaba H, Nagatani T, Watanabe T, Kishida Y, Fukagawa M (2018) The pituitary is a candidate organ that modulates circulating klotho levels. J Endocr Soc 3(1):52–61. https://doi.org/10.1210/js.2018-00223
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Lida A, Shiraki-lida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390(6655):45–51. https://doi.org/10.1038/36285
Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-OM, (2005) Suppression of aging in mice by the hormone Klotho. Science 309(5742):1829–1833. https://doi.org/10.1126/scienze.1112766
Olauson H, Mencke R, Hillebrands JL, Larsson TE (2017) Tissue expression and source of circulating αKlotho. Bone 100:19–35. https://doi.org/10.1016/j.bone.2017.03.043
Dalton GD, Xie J, An SW, Huang CL (2017) New insights into the mechanism of action of soluble Klotho. Front Endocrinol 8:323. https://doi.org/10.3389/fendo.2017.00323
Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y (1998) Identification of the Human Klotho gene and its two transcripts encoding membrane and secreted Klotho protein. Biochem Biophys Res Commun 242(3):626–630. https://doi.org/10.1006/bbrc.1997.8019
Razzaque MS (2014) Bone–kidney axis in systemic phosphate turnover. Arch Biochem Biophys 561:154–158. https://doi.org/10.1016/j.abb.2014.06.031
Wang Y, Sun Z (2009) Current understanding of Klotho. Ageing Res Rev 8(1):43–51. https://doi.org/10.1016/j.arr.2008.10.002
Razzaque MS (2009) The FGF23-Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol 5(11):611–619. https://doi.org/10.1038/nrendo.2009.196
Ciresi A, Pizzolanti G, Guarnotta V, Giordano C (2019) Circulating irisin levels in children with GH deficiency before and after 1 year of GH treatment. J Clin Endocrinol Metab 104(3):801–808. https://doi.org/10.1210/jc.2018-01440
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28(7):412–419. https://doi.org/10.1007/BF00280883
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22(9):1462–1470. https://doi.org/10.2337/diacare.22.9.1462
Utzschneider KM, Prigeon RL, Faulenbach MV, Tong J, Carr DB, Boyko EJ, Leonetti DL, McNeely MJ, Fujimoto WY, Kahn SE (2009) Oral disposition index predicts the development of future diabetes above and beyond fasting and 2-h glucose levels. Diabetes Care 32(2):335–341. https://doi.org/10.2337/dc08-1478
Yamazaki Y, Imura A, Urakawa I, Shimada T, Murakami J, Aono Y, Hasegawa H, Yamashita T, Nakatani K, Saito Y, Okamoto N, Kurumatani N, Namba N, Kitaoka T, Ozono K, Sakai T, Hataya H, Ichikawa S, Imel EA, Econs MJ, Nabeshima Y (2010) Establishment of sandwich ELISA for soluble alpha-Klotho measurement: age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem Biophys Res Commun 398(3):513–518. https://doi.org/10.1016/j.bbrc.2010.06.110
Rubinek T, Shahmoon S, Shabtay-Orbach A, Ben Ami M, Levy-Shraga Y, Mazor-Aronovitch K, Yeshayahu Y, Doolman R, Hemi R, Kanety H, Wolf I, Modan-Moses D (2016) Klotho response to treatment with growth hormone and the role of IGF-I as a mediator. Metabolism. https://doi.org/10.1016/j.metabol.2016.08.004
Wolf I, Shahmoon S, Ben Ami M, Levy-Shraga Y, Mazor-Aronovitch K, Pinhas-Hamiel O, Yeshayahu Y, Hemi R, Kanety H, Rubinek T, Modan-Moses D (2014) Association between decreased klotho blood levels and organic growth hormone deficiency in children with growth impairment. PLoS ONE 9(9):e107174. https://doi.org/10.1371/journal.pone.0107174
Meazza C, Elsedfy HH, Khalaf RI, Lupi F, Pagani S, Kholy ME, Tinelli C, Radetti G, Bozzola M (2017) Serum α-klotho levels are not informative for the evaluation of growth hormone secretion in short children. J Pediatr Endocrinol Metab 30(10):1055–1059. https://doi.org/10.1515/jpem-2016-0464
Devesa J (2021) The complex world of regulation of pituitary growth hormone secretion: the role of ghrelin, klotho, and nesfatins in it. Front Endocrinol 12:636403. https://doi.org/10.3389/fendo.2021.636403
Caicedo D, Díaz O, Devesa P, Devesa J (2018) Growth hormone (GH) and cardiovascular system. Int J Mol Sci 19(1):290. https://doi.org/10.3390/ijms19010290
Shahmoon S, Rubinfeld H, Wolf I, Cohen ZR, Hadami M, Shimon H, Rubinek T (2014) The aging suppressor klotho: a potential regulator of growth hormone secretion. Am J Physiol Endocrinol Metab 307(3):E326–E334. https://doi.org/10.1152/ajpendo.00090.2014
Behringer V, Stevens JMG, Deschner T, Sonnweber R, Hohmann G (2018) Aging and sex affect soluble alpha klotho levels in bonobos and chimpanzees. Front Zool 15:35. https://doi.org/10.1186/s12983-018-0282-9
Rutkowski JM, Pastor J, Sun K, Park SK, Bobulescu A, Chen CT, Moe OW, Scherer PE (2017) Adiponectin alters renal calcium and phosphate excretion through regulation of klotho expression. Kidney Int 91(2):324–337. https://doi.org/10.1016/j.kint.2016.09.016
Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR (2007) Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM 17. Proc Natl Acad Sci USA 104(50):19796–19801. https://doi.org/10.1073/pnas.0709805104
Gu H, Jiang W, You N, Huang X, Li Y, Peng X, Dong R, Wang Z, Zhu Y, Wu K, Li J, Zheng L (2020) Soluble klotho improves hepatic glucose and lipid homeostasis in type 2 diabetes. Mol Ther Methods Clin Dev 18:811–823. https://doi.org/10.1016/j.omtm.2020.08.002
Hasannejad M, Samsamshariat SZ, Esmaili A, Jahanian-Najafabadi A (2019) Klotho induces insulin resistance possibly through interference with GLUT4 translocation and activation of Akt, GSK3β, and PFKfβ3 in 3T3-L1 adipocyte cells. Res Pharm Sci 14(4):369–377. https://doi.org/10.4103/1735-5362.263627
Ciresi A, Giordano C (2018) Glucose metabolism in children with growth hormone deficiency. Front Endocrinol (Lausanne) 9:321. https://doi.org/10.3389/fendo.2018.00321
Neidert MC, Sze L, Zwimpfer C, Sarnthein J, Seifert B, Frei K, Leske H, Rushing EJ, Schmid C, Bernays RL (2013) Soluble α-klotho: a novel serum biomarker for the activity of GH-producing pituitary adenomas. Eur J Endocrinol 168(4):575–583. https://doi.org/10.1530/EJE-12-1045
Funding
Open access funding provided by Università degli Studi di Palermo within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by VG, GP, RP, SR and CB. The first draft of the manuscript was written by VG and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
The study was approved by the institutional ethics committee of the Policlinico Paolo Giaccone, University of Palermo, and was performed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Competing interests
The authors have nothing relevant financial or non-financial interests to disclose.
Disclosure statement
The authors have nothing to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised to add missing OASIS funding note.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guarnotta, V., Pizzolanti, G., Petrancosta, R. et al. Gender-specific soluble α-klotho levels as marker of GH deficiency in children: a case–control study. J Endocrinol Invest 45, 1247–1254 (2022). https://doi.org/10.1007/s40618-022-01757-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-022-01757-y