
Abstract
Purpose
Salivary gland (SG) tissue and derived neoplasms may occur in the sellar region. As the current literature is mostly limited to case reports, the puzzling case of an inflammatory SG removed by transsphenoidal surgery (TS) and mimicking a prolactinoma prompted us to perform the first systematic review of these unusual conditions.
Methods
A systematic literature search was conducted according to the PRISMA guidelines. Forty-four individual cases—non-neoplastic enlarged salivary glands (NNESG, n = 15), primary benign (n = 7) and malignant (n = 8) ectopic salivary tumours (ST) and sellar metastasis from eutopic primary ST (n = 14)—were suitable for the analysis of clinical, radiological and pathological characteristics. Therapeutic outcome was reviewed as a secondary endpoint.
Results
All cases were diagnosed after surgery. NNESG commonly affected young and/or female patients, typically leading to headaches and hyperprolactinemia and originating close to the neurohypophysis. Submucosal SG should be excluded before concluding to an intrasellar NNESG after TS. No gender or age predominance was found for primary ectopic ST, which present as large tumors, with histological phenotypes similar to common ST. Hypopituitarism and diabetes insipidus were more frequent in ST than in NNESG. NNESG and benign ectopic ST rarely recur. Malignant ectopic ST should be distinguished from secondary localizations of eutopic ST reaching the sella by contiguity or metastatic spread; both share a frequent unfavorable outcome.
Conclusion
Sellar neoplasms derived from SG are rare but misleading conditions and pituitary dysfunction is likely to be more common than currently reported. Appropriate pathological evaluation and multidisciplinary approach are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ectopic salivary gland (SG) tissue may occur in different sites of the body: extra-cranially (larynx, gastrointestinal tract, middle ear, chest wall) [1,2,3,4,5,6] and intra-cranially, with sellar and extra-sellar localizations (e.g.: optic nerve sheath, cerebellopontine angle) [7,8,9]. Intrasellar ectopic SG rests are typically localized close to the neurohypophysis or in the pars intermedia, often communicating with the Rathke’s cleft [7], and maybe incidentally found at autopsy [10, 11]. Only a small minority come to clinical attention because of mass effects and/or endocrine dysfunction, in particular hyperprolactinemia [8, 12,13,14,15,16,17,18,19,20]. Symptomatic enlargement of ectopic SG rests may be non-neoplastic (NNESG) or due to benign or malignant salivary tumours (ST) that mimic other non-functioning lesions, and the diagnosis relies on pathology where surgery is indicated. In addition, because malignant ST derived from major or minor eutopic SG may reach the sella through local invasion or blood spread [21,22,23], an extra-sellar origin should be excluded before concluding to a primary ectopic SG malignancy [24]. Sellar salivary neoplasms represent an unusual challenge for specialists involved in the management of pituitary neuroendocrine tumours (Pit-NETs) [25] and other sellar/parasellar lesions.
A recent puzzling observation (illustrated in Fig. 1) prompted us to perform the first systematic review of the literature about sellar NNESG and ST, pointing out an additional diagnostic pitfall, i.e. an inflammatory submucosal SG mimicking a prolactinoma during transsphenoidal surgery (TS). Individual cases were classified into four groups: ectopic NNESG, benign and malignant ectopic ST (eST) and secondary localizations of eutopic ST. Clinical, neuroradiological, pathological characteristics, and therapeutic outcome were analysed. This review points out the importance of a multidisciplinary work-up to reach a correct diagnosis and optimize clinical management.

A puzzling case of sellar salivary gland (SG). A 19-year-old woman was referred in February 2019 because of a prolactinoma showing increasing pharmacological resistance. The diagnosis was made 3 years earlier in the setting of primary amenorrhea–galactorrhea and intermittent headache, plasma PRL 1763 ng/ml (N < 26.7) and a macroadenoma with a fluid hemorrhagic component at Magnetic Resonance Imaging (MRI) (A1, A2 coronal and sagittal T2-weighted). Menarche occurred within 5 months of treatment, with regular menses but an increasing and poorly tolerated drug requirement to obtain a sub-optimal control of hyperprolactinemia (CAB up to 3.5 mg/week). As MRI showed clear evidence of residual disease (B1, B2 pre-and post-Gadolinium coronal views), endoscopic TS was proposed. A small nodular lesion, consistent with a microadenoma, was removed. Unexpectedly, pathological examination revealed numerous groups of glandular berries composed of typical serous and mucinous cells, compatible with SG tissue, separated by a chronic inflammatory lymphoplasmacellular infiltrate (C1 hematoxylin–eosin). Immunostaining for lysozyme was positive in mucinous cells (C2). Bony spicules and flaps of respiratory mucosae were also present, with no evidence of pituitary cells. Immunostaining for PRL was negative (not shown). The first pathological diagnosis was NNESG. However, post-operative CAB withdrawal was followed by a progressive recurrence of symptomatic hyperprolactinemia (up to 245 ng/ml 4 months after surgery), with MRI evidence of residual/recurring disease. Careful revision of serial pre-operative imaging revealed in a single MRI study (2017) a small intrasphenoidal nodular lesion localized just beneath the adenomatous lesion, with spontaneous hypointensity in T2 (D1 coronal view) and hyperintensity in T1 before and after gadolinium (D2 sagittal view). This finding was consistent with a cystic SG, undergoing subsequent inflammation and shrinkage. The final diagnosis was a sub-mucosal SG, mimicking and masking the residual microprolactinoma during TS. As CAB was re-started up to the maximal well-tolerated dose (2.0 mg weekly) with an incomplete response (PRL 45 ng/ml), TS will be potentially re-considered if necessary.
Methods
A systematic review of case reports and case series was performed according to the Cochrane Collaboration and PRISMA statements [26, 27]. A literature search without limits was conducted on Medline and Scopus up to September 2020, including international and non-English literature, using the following keywords: ectopic salivary gland/ salivary gland/salivary gland tumour AND pituitary/sella/sphenoidal/sphenoid sinus. Cross-references were used to identify additional papers, allowing to retrieve six additional cases. Titles and abstracts of all papers were screened to assess their relevance. Duplicates, reviews, animal studies, in vitro studies and congress reports were excluded. Based on available abstracts and full texts, all the papers describing NNESG and benign or malignant sellar ST were analyzed. The following data were extracted for each paper: (1) first author, year of publication; (2) case demographics (gender, age), (3) symptoms, (4) endocrine abnormalities, in particular, PRL values, (5) neuro-radiological findings at MRI and/or computed tomography (CT): localization, size, invasion, signal intensity/density, contrast enhancement, (6) pre-operative and final pathological diagnosis, (7) treatment and, (8) where available, status at last follow-up (recurrence, progression, hormone replacement therapy, death).
Results
Overall, 1024 potentially relevant studies were found, 978 were excluded at first screening and 46 were selected for full-text assessment (Fig. 2). Thirty-five papers were finally retained (1963–2020): 32 in English language, 1 in French, and 2 papers in Japanese or Korean with detailed English abstracts and figures footnotes. Overall, 44 individual cases of symptomatic sellar NNESG and ST were described, including 14 secondary sellar localizations of primary eutopic ST. Because PitNETs were originally reported as pituitary adenomas (PA) in all papers, we elected to maintain this terminology to report the pre-operative diagnosis.

Flowchart of the literature eligibility assessment process
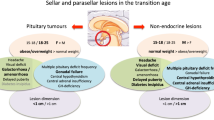
NNESG
Fifteen cases of NNESG were found (Table 1) [8, 12,13,14,15,16,17,18,19,20, 28]. Most patients were younger than 30 years (11/15, 73.3%), including a pediatric case. Most were females (12/15, 80%). The most frequent complaints were headache (12/15, 80%), visual symptoms—bitemporal hemianopsia, blurred vision, decreased visual acuity—(4/15, 26.6%), nausea (4/15, 26.6%), galactorrhea and menstrual irregularities (3/15, 20%). Endocrine dysfunction was frequent (8/15, 53.3%), including mild hyperprolactinemia (23.9–93.0 ng/ml, median 83.5) (3/15, 20%), growth hormone deficiency (2/15, 13.3%), panhypopituitarism (2/15, 13.3%) and/or central hypothyroidism (1/15, 6.7%). Pre-operative diabetes insipidus (DI) was present in 3/15 cases (20%). NNESG were variable in size (maximal dimension 0.6–4.6 cm, median 1.7). Intrasellar lesions were typically localized in the posterior pituitary (6/15, 40%), suprasellar extension was frequent (8/15, 53.3%), but hydrocephalus was rare (1/15, 6.7%) [28]. Lateral extension was uncommon (2/15, 13.3%), with cavernous sinus infiltration in the largest case [16]. Based on the neuroradiological description and/or pre-operative diagnosis, they presented as pseudo-tumorous solid lesions, with frequent cystic component(s) (7/15, 46.7%). At MRI, most lesions appeared as hyper- or iso-intense on T1 weighted imaging (T1) (7/15, 46.7% and 4/15 26.6%, respectively) but either hyper- or hypo-intense on T2 weighted imaging (T2) (4/15, 26.6% and 4/15, 26.6%, respectively), with inconstant contrast enhancement (4/15, 26.6%). Spontaneous hyperdensity was found at CT where available (n = 4). Pre-operative diagnosis was: PA (8/15, 53.3%), Rathke’s cleft cyst (RCC) (5/15, 30%), craniopharyngioma (2/15, 13.3%), exceptionally chordoma (the largest one) [16]. Two patients received bromocriptine but the lesion was unchanged despite PRL normalization [12, 20]. All patients were operated on, 80% through a TS route (12/15). Follow-up was limited (1–4 years, median 1 year, n = 10), but no recurrence or progression was reported, except a cystic relapse after 2 years, without pathological evidence of SG tissue [28]. At pathological examination, SG rests or cysts were found within or close to the posterior pituitary lobe, in 7 cases within the wall of a RCC (46.7%). NNESG were composed of acini with a low columnar or cuboidal epithelium, embedded in a fibrovascular stroma, without cellular atypia, with occasional inflammation (2/15, 13.3%). Anti-PGP immunoreactive nerve fibers were reported in one case, suggesting parasympathetic innervation [18]. Adjacent anterior pituitary cells were observed in a minority of cases (20%).
Sellar ST
The individual characteristics of sellar ST according to their pathological classification are shown in Table 2.
Primary benign ectopic ST
Seven cases of benign sellar eST have been reported [7, 29,30,31,32], with a majority of pleomorphic adenomas (4/7, 57.1%) and single reports of monomorphic adenoma, oncocytoma and adenomyoepithelioma. They manifested at any age in both genders (4M, 3F, 17–81 years-old, median 44), with visual symptoms in all cases and inconstant headache (2/7, 28.6%). General symptoms—such as muscle weakness, fatigue, weight loss, anorexia, nausea and/or vomiting—were also present in all patients, and endocrine dysfunction reported as hypopituitarism (4/7, 57.1%), hyperprolactinemia (2/7, 28.6%) and/or DI (3/7, 42.8%). Accordingly, benign eST were large (2.5–4.0 cm, median 3.0), with a suprasellar extension in all cases—up to the optic chiasm (5/7, 71.4%) or the hypothalamus (2/7, 28.6%). MRI signal was not reported, but contrast enhancement was frequent (3/7, 42.8%) and calcifications or pseudo-hemorrhage could be found at CT. The most frequent pre-operative diagnosis was PA (n = 5), but craniopharyngioma, chordoma and benign teratoma were also considered. All patients were operated on—TS (4/7, 57.1%) or transcranially (TC) (3/7, 42.8%). Based on a variable follow-up duration (0.4–14 years, median 2.5, n = 6) recurrences were rarely reported after complete surgical removal, but occurred 4 and 14 years after partial removal [29,30,31]. Four patients received radiotherapy [29,30,31]. Noteworthy, the first pathological diagnosis was inaccurate in 3 cases (benign teratoma, chordoma, craniopharyngioma) [30,31,32].
Primary malignant ectopic ST
Primary malignant eST were reported in 8 cases [24, 29, 33,34,35,36,37] and consisted of adenoid cystic carcinoma (2/8, 25%), myoepithelioma (2/8, 25%), epithelial–myoepithelial carcinoma (1/8, 12.5%), papillary mucinous adenocarcinoma (1/8, 12.5%), low-grade acinic cell carcinoma (1/8, 12.5%) and adenocarcinoma (1/8, 12.5%). All patients were adult (3M, 5F, 34–68 years, median 51.5) and had visual symptoms, with frequent oculomotor nerve palsy/diplopia (5/8, 62.5%). Endocrine dysfunction was reported as hypopituitarism (4/8, 50%)—including one case of acute adrenal insufficiency, hyperprolactinemia (3/8, 37.5%) and DI (2/8, 25%). Accordingly, tumours were large (2.0–3.8 cm, median 2.5), growing up to the optic chiasm (4/8, 50%) or the floor of the 3° ventricle (3/8, 37.5%). An invasive growth was reported in some cases, eroding inferiorly into the sphenoid and ethmoid sinuses (1/8, 12.5%) or extending in the cavernous sinus (2/8, 25%)—in one case reaching the middle fossa [34]. Contrast enhancement was inconstantly reported (4/8, 50%) and, according to limited detailed MRI (n = 2), the tumour was isointense on T1 and slightly hyper- or hypointense on T2 [34, 36]. Pre-operative diagnosis was PA (n = 3) [29, 34, 37], craniopharyngioma (n = 2) [35, 36], but also inflammatory granulomatosis/hypophysitis, metastatic brain tumour and primary tumour of the cavernous sinus [34]. All patients were operated on—half of them through a TC approach—and subsequently irradiated (20–54 Gy). Two patients also received chemotherapy (isofosfamide/BCNU or temozolomide) for an epithelial–myoepithelial carcinoma and an aggressive myoepithelioma, respectively [24, 35], with a poor response. Except for one case of papillary mucinous carcinoma [33], all patients showed disease progression within 4 years (4/8, 50%) or died (6/8, 75%, 3 post-operative deaths). In one case, neuropathological misdiagnosis of PA was reported [29].
Secondary malignant sellar ST
Secondary ST were reported in 14 patients, deriving in most cases from minor salivary glands situated in the sphenoid sinus (n = 6), nasopharynx (n = 2), palate (n = 1) and paranasal sinus (n = 1)—but also from the parotid gland (4/14, 28.6%) [21,22,23, 29, 38,39,40,41,42,43,44,45,46]. A majority were adenoid cystic carcinoma (ACC) (11/14, 78.6%), with single reports of ductal adenocarcinoma, mixed salivary tumour, and a monomorphic multi-recurrent parotid adenoma extending to the sella. Most primary ST reached the sella through the sphenoid sinus (9/14, 64.3%) or dural infiltration (1/14, 7.1%), but metastatic blood spread could occur (4/14, 28.6%). In 4 cases sellar involvement was delayed (5–26 years after the primary tumour) [22, 23, 40, 45]. Adults were affected at any age (6M, 8F, 28–85 years, median 45). Most patients had visual defects (10/14, 71.4%), cranial nerve palsy (7/14, 50%) with frequent diplopia (6/14, 42.8%) and/or trigeminal sensory symptoms (3/14, 21.4%). Endocrine dysfunction (hypopituitarism, hyperprolactinemia, DI, syndrome of inappropriate antidiuretic hormone secretion—SIADH) was documented in a minority of cases (5/14, 35%) despite suggestive symptoms in additional cases. Tumours were large, frequently invasive with skull base erosion (6/14, 42.8%) or cavernous sinus infiltration (5/14, 35.7%)., and inconstant suprasellar invasion (5/14, 35.7%). Pre-operative diagnosis was PA (n = 3) [22, 29, 43]—with an apoplectic presentation in one case [40]—but also craniopharyngioma, meningioma, neurinoma and metastasis [40, 45]. Surgery was proposed in all but one patient, who received first-line radiotherapy for tumour inoperability [21]. One patient who initially declined surgery was operated on 5 years after radiotherapy but the tumour had reached a considerable volume with extensive bone destruction and multiple cranial nerve palsy [43]. Patients underwent TS/endoscopic (n = 7), TC (n = 4) or undetermined (n = 2) surgery, and frequent post-operative radiotherapy (n = 8). Follow-up was available in 8 cases, 6 patients died within 4 years, in one case from early post-operative meningitis [43].
Discussion
This is the first systematic review on salivary diseases and neoplasia localized to the sellar region. The puzzling case of an apparently intrasellar NNESG removed during TS surgery prompted us to further analyse these conditions, which are not mentioned in exhaustive reviews on sellar/parasellar lesions [47, 48] or single-center experiences reporting rare sellar-suprasellar masses [47, 49, 50]. Indeed, nearly half of the reports were published in the last decade (17/35 papers), in particular those concerning NNESG (7/11 papers, 11/15 cases) and benign primary eST (4/5 papers, 4/7 cases). Strikingly, pre-operative neuroimaging was inconclusive or misleading in all cases. The heterogeneity of radiological descriptions, in part reflecting a variety of pathological histotypes [51], confirms the lack of strongly suggestive features, although cystic components of variable protein content were frequent in NNESG. Where present, DI or rapidly evolving symptoms may help to distinguish such conditions from non-secreting Pit-NETs, but usually suggest alternative diagnosis (craniopharyngiomas, hypophysitis or metastasis). Thus, similar to other rare lesions coming up as pathological surprises [52], they are extremely difficult to consider at the time of pre-operative evaluation. Of note, the pathological diagnosis may also be inaccurate at first observation.
Salivary rests in the pituitary are relatively common incidental findings at autopsy (3.4–8.8%) [10, 11]. This may be explained by pre-existing seromucous glands from the primitive oral cavity remaining in the Rathke’s pouch during migration and persisting during postnatal life [17], similar to ectopic pituitary tissue reported at various locations along its migratory path, including the roof of the nasopharynx [53]. Experimental studies also suggest that Rathke’s pouch components may occasionally differentiate into salivary and adenohypophyseal tissues during organogenesis [54] and that parotid gland tissue may trans-differentiate into pituitary hormone-producing cells under the influence of hypothalamic factors [55]. Embryological development would thus explain why NNESG are found close to the posterior pituitary lobe and sometimes within RCC’s wall [16,17,18]. In this latter case, the role of salivary remnants in the development of clinical symptoms is not always clear-cut, and some may be incidental findings in the setting of a symptomatic RCC rather than true NNESG [17]. Similarly, we recently observed incidental salivary rests adjacent to an apoplectic gonadotroph PitNET (Suppl Fig. 1), although in our experience this is extremely rare. Alternatively, active secretion from ectopic salivary rests within the cyst was proposed to contribute to RCC enlargement, possibly triggered by parasympathic innervation [18].
The mechanisms leading to NNESG are not fully elucidated but the development of mucinous cysts [14, 18, 19] and/or chronic inflammation [12, 19] are frequently observed. In normal conditions, the lack of neuro-vegetative innervation of the posterior pituitary may prevent the secretion of mucinous material, or local lymphatic/venous reabsorption may remove secretions [20]. Symptomatic NNESG may grow up into the opto-chiasmatic cistern, but exceptionally reach considerable dimensions or appear invasive [16]. Because they more frequently affect young (73.3%) and/or female patients (80%), who present with headache and symptomatic hyperprolactinemia [12,13,14,15,16,17,18,19,20], NNESG may represent a rare differential diagnosis of cystic sellar lesions in such patients. Hyperprolactinemia likely results from functional disruption of the physiological dopaminergic inhibition, although direct stimulation by EGF, which is abundantly produced by SG, may be hypothesized [56, 57]. This explains why hyperprolactinemia is moderate and promptly normalized by dopamine-agonists in the absence of tumour shrinkage [12, 20]. Less frequently, pre-operative DI or hypopituitarism are present—including growth retardation [15]—and require appropriate hormone replacement therapy. In our patient, the lateral localization of the lesion associated with post-operative recurrence of hyperprolactinemia lead us to reconsider our first diagnosis of intrasellar NNESG with surgical aspiration of prolactinoma cells escaping pathological examination. Furthermore, we found no previous observation of NNESG coexisting with a PitNET. Based on careful revision of serial pre-operative MRI, transient evidence of an intrasphenoidal cyst was noticed, placed on the TS route to the prolactinoma. As only pathological minor salivary glands are seen by radiological imaging [58, 59], we finally concluded for residual inflammation in a submucosal SG following spontaneous reabsorption of the cyst. This pitfall should therefore be considered in the presence of SG acini contaminating surgical fragments obtained by TS, as it may open the possibility of a second TS approach. Once made a definitive diagnosis of NNESG, recurrences have been exceptionally reported [28]. In such cases, a neoplastic origin may not be totally ruled out [28].
ST involving the sellar region have been reported more frequently than NNESG. Patients were adults of any age, mass effects were almost invariably present and amenorrhea–galactorrhea or hyperprolactinemia were occasionally reported. Malignant forms were characterized by a mild female predominance and a major frequency of ocular palsy and symptoms suggestive of hypopituitarism or DI. Similar to primary eutopic ST, they include a variety of histotypes [51, 58], and a minority of eST were found in association with or close to a RCC [7, 33].
Primary benign sellar eST were mostly represented by pleomorphic adenomas. Where specified, they presented as medium/large-sized heterogeneous sellar/suprasellar masses, sometimes with intratumoural hemorrhage or calcifications, potentially mimicking PA, craniopharyngioma, teratoma or chordoma. Hypopituitarism and DI were frequently documented in this group. Post-operative radiotherapy was often proposed due to the risk of recurrence after incomplete surgical removal, although but delayed regrowth could occur [31]. Long-term follow-up is therefore recommended.
Malignant ST usually presented as large, heterogeneous, typically invasive sellar/parasellar masses, and rapid progression could suggest pituitary metastasis or other malignancies. They included a variety of histotypes. Adenoid cystic carcinomas (ACC) accounted for nearly 80% of secondary forms versus 25% of primary malignant eST. With a few exceptions, both were diagnosed earlier (4th–5th decade) than common eutopic ACC (5th–7th decade), which are usually slowly growing [51]. This suggests that sellar ACC may have a different natural history. However, further information is needed to compare the prognosis of sellar ST with similar eutopic histotypes. Radical surgical resection followed by radiotherapy was the treatment of choice in most cases, although first-line (chemo-)radiotherapy was also proposed [33, 46]. Incomplete surgical removal favored tumour progression and/or metastatic spread, with a poor response to radiotherapy and chemotherapy [36]. Of note, one patient died 8 days after surgery for unexplained hypotension [33]. As pituitary function was poorly evaluated in these patients, hypopituitarism could be left untreated.
Conclusion
Sellar/parasellar lesions derived from SG tissues are rare but challenging conditions. An appropriate pathological characterization is essential for a correct multidisciplinary approach, which should consider and treat their frequent endocrine complications. Where required, hormone replacement therapy is essential to improve patient’s quality of life and prevent the risk of acute adrenal insufficiency. In addition to surveillance for the early recognition of ST recurrences or metastasis, life-long endocrinological follow-up is necessary for the presence of permanent dysfunction or after radiotherapy, which may induce delayed hypopituitarism. Multicenter collection and long-term follow-up would be useful to better define disease evolution and optimal clinical management of these unusual conditions.
References
Aby JL, Patel M, Sundram U, Benjamin LT (2014) Salivary gland choristoma (heterotopic salivary gland tissue) on the anterior chest wall of a newborn. Pediatr Dermatol 31(1):e36–e37. https://doi.org/10.1111/pde.12159
Chen S, Li Y (2015) Salivary gland choristoma of the middle ear. Ear Nose Throat J 94(2):E9–E12. https://doi.org/10.1177/014556131509400211
Lee JH, Kim S, Kim SJ (2018) Heterotopic salivary gland tissue at the hepatic flexure of the large intestine: a case report. Korean J Gastroenterol = Taehan Sohwagi Hakhoe chi 72(4):213–216. https://doi.org/10.4166/kjg.2018.72.4.213
Schulberg SP, Serouya S, Cho M, Gadangi PK (2020) Ectopic salivary gland found on rectal biopsy—a rare pathological diagnosis. Int J Colorectal Dis 35(5):967–969. https://doi.org/10.1007/s00384-020-03561-7
Tajima S, Yamauchi K, Higo R, Ikeda K (2018) A case of ectopic salivary gland of the larynx. Auris Nasus Larynx 45(3):633–636. https://doi.org/10.1016/j.anl.2017.08.001
Wang C, Chen L, Guo W, Zhu X, Liu Z (2014) Salivary gland choriostoma in the esophagus. Endoscopy. https://doi.org/10.1055/s-0034-1390842
Chimelli L, Gadelha MR, Une K, Carlos S, Pereira PJ, Santos JL, Filho PN, Duarte F (2000) Intra-sellar salivary gland-like pleomorphic adenoma arising within the wall of a Rathke’s cleft cyst. Pituitary 3(4):257–261. https://doi.org/10.1023/a:1012888315577
Hintz EB, Yeaney GA, Buchberger GK, Vates GE (2014) Intracranial salivary gland choristoma within optic nerve dural sheath: case report and review of the literature. World Neurosurg 81(5–6):842.e841-844.e841. https://doi.org/10.1016/j.wneu.2013.01.094
Rodriguez F, Scheithauer BW, Ockner DM, Giannini C (2004) Solitary fibrous tumor of the cerebellopontine angle with salivary gland heterotopia: a unique presentation. Am J Surg Pathol 28(1):139–142. https://doi.org/10.1097/00000478-200401000-00017
Rittierodt M, Hori A (2007) Pre-morbid morphological conditions of the human pituitary. Neuropathology 27(1):43–48. https://doi.org/10.1111/j.1440-1789.2006.00745.x
Schochet SS Jr, McCormick WF, Halmi NS (1974) Salivary gland rests in the human pituitary. Light and electron microscopical study. Arch Pathol 98(3):193–200
Chen CH, Hsu SS, Lai PH, Lo YS (2007) Intrasellar symptomatic salivary gland rest. J Chin Med Assoc 70(5):215–217. https://doi.org/10.1016/S1726-4901(09)70361-2
Hwang JH (2013) Pituitary symptomatic salivary gland rest cyst: case report. Brain Tumor Res Treat 1(1):54–56. https://doi.org/10.14791/btrt.2013.1.1.54
Kato T, Aida T, Abe H, Miyamachi K, Hida K, Taneda M, Ogata A (1988) Ectopic salivary gland within the pituitary gland. Case report. Neurol Medico-chirurgica 28(9):930–933. https://doi.org/10.2176/nmc.28.930
Kim TH, Park TJ, Kim HJ, Kim DJ, Chung Y-S, Lee KW, Lee TH, Kim HS, Cho KG (2007) A case report of symptomatic salivary gland rest within the pituitary gland. J Korean Endocr Soc 22(6):436. https://doi.org/10.3803/jkes.2007.22.6.436
Liu Z, Zhang Y, Feng R, Tian Z, Rao Y, Lu Y, Xu J (2019) Intrasellar symptomatic salivary gland: case series and literature review. Pituitary 22(6):640–646. https://doi.org/10.1007/s11102-019-01002-5
Ranucci V, Coli A, Marrucci E, Paolo MP, Della Pepa G, Anile C, Mangiola A (2013) Ectopic salivary gland tissue in a Rathke’s cleft cyst. Int J Clin Exp Pathol 6(7):1437–1440
Stefanits H, Matula C, Frischer JM, Furtner J, Hainfellner JA, Woehrer A (2013) Innervated ectopic salivary gland associated with Rathke’s cleft cyst clinically mimicking pituitary adenoma. Clin Neuropathol 32(3):171–175. https://doi.org/10.5414/NP300560
Tanaka Y, Kubo A, Ayabe J, Watanabe M, Maeda M, Tsuura Y, Tanaka Y (2015) Intrasellar symptomatic salivary gland rest with inflammations. World Neurosurg 84(1):189.e113–188.e113. https://doi.org/10.1016/j.wneu.2015.02.018
Tatter SB, Edgar MA, Klibanski A, Swearingen B (1995) Symptomatic salivary-rest cyst of the sella turcica. Acta Neurochir 135(3–4):150–153. https://doi.org/10.1007/BF02187760
Giridhar P, Mallick S, Laviraj MA, Bhasker S (2015) Adenoid cystic carcinoma sphenoid sinus with intracranial extension treated by radical radiotherapy: a rare case. Eur Arch Oto-rhino-laryngol 272(4):1037–1040. https://doi.org/10.1007/s00405-014-3441-4
Jd H, Retzlaff A, Sims J, O’Brien E, Giannini C, Huston J 3rd, Van Gompel JJ (2016) Adenoid cystic carcinoma metastatic to the pituitary: a case report and discussion of potential diagnostic value of magnetic resonance elastography in pituitary tumors. World Neurosurg 91:669.e611. https://doi.org/10.1016/j.wneu.2016.03.044
Kaur A, Harrigan MR, MeKeever PE, Ross DA (1999) Adenoid cystic carcinoma metastatic to the dura: report of two cases. J Neurooncol 44(3):267–273. https://doi.org/10.1023/a:1006352307507
Lavin V, Callipo F, Donofrio CA, Ellwood-Thompson R, Metcalf R, Djoukhadar I, Higham CE, Kearney T, Colaco R, Gnanalingham K, Roncaroli F (2020) Primary epithelial–myoepithelial carcinoma of the pituitary gland. Neuropathology. https://doi.org/10.1111/neup.12628
Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, Grossman A, Ikeda H, Inoshita N, Karavitaki N, Korbonits M, Laws ER, Lopes MB, Maartens N, McCutcheon IE, Mete O, Nishioka H, Raverot G, Roncaroli F, Saeger W, Syro LV, Vasiljevic A, Villa C, Wierinckx A, Trouillas J (2017) From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): an International Pituitary Pathology Club proposal. Endocr Relat Cancer 24(4):C5–C8. https://doi.org/10.1530/erc-17-0004
Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, Clarke M, Devereaux PJ, Kleijnen J, Moher D (2009) The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol 62(10):e1–e34. https://doi.org/10.1016/j.jclinepi.2009.06.006
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P (2009) Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol 62(10):1006–1012. https://doi.org/10.1016/j.jclinepi.2009.06.005
Kleinschmidt-DeMasters BK, Rosenblum MK, Kerr JM, Lillehei KO (2020) Cystic sellar salivary gland-like lesions. Clin Neuropathol 39(05):115–125. https://doi.org/10.5414/np301235
Hampton TA, Scheithauer BW, Rojiani AM, Kovacs K, Horvath E, Vogt P (1997) Salivary gland-like tumors of the sellar region. Am J Surg Pathol 21(4):424–434. https://doi.org/10.1097/00000478-199704000-00008
Rychly B, Kazakov DV, Danis D, Szep Z, Michal M (2010) Primary adenomyoepithelioma of the sellar region: a case report. Am J Surg Pathol 34(10):1550–1554. https://doi.org/10.1097/PAS.0b013e3181f0ac1b
Takahashi S, Mikami S, Akiyama T, Kawase T (2011) Intrasellar salivary gland-like pleomorphic adenoma: case report. Neurosurgery 68(2):E562–E565. https://doi.org/10.1227/NEU.0b013e3182020856(discussion E566)
Yao K, Duan Z, Bian Y, Wang M, Qi X (2014) Suprasellar salivary gland-like pleomorphic adenoma. Indian J Pathol Microbiol 57(1):136–138. https://doi.org/10.4103/0377-4929.130926
Gilcrease MZ, Delgado R, Albores-Saavedra J (1999) Intrasellar adenoid cystic carcinoma and papillary mucinous adenocarcinoma: two previously undescribed primary neoplasms at this site. Ann Diagn Pathol 3(3):141–147. https://doi.org/10.1016/s1092-9134(99)80041-0
Hong Y, Guo SX, Chen S, Klebe D, Zhang JM, Wu Q (2013) Rapid-developed primary malignant myoepithelioma in the cavernous sinus: a case report. BMC Neurol 13:40. https://doi.org/10.1186/1471-2377-13-40
Nieder C, Schneller F, Grosu AL, Peschel C, Molls M (2005) Radiotherapy and chemotherapy for myoepithelioma of the sellar region. Strahlentherapie und Onkologie: Organ der Deutschen Rontgengesellschaft [et al] 181(4):260–263. https://doi.org/10.1007/s00066-005-1356-0
Tsuyuguchi N, Ohata K, Goto T, Haque M, Hara M (2001) Intracranial adenoid cystic carcinoma of suprasellar region. Acta Neurochir 143(7):729–732. https://doi.org/10.1007/s007010170053
van Furth W, Smyth HS, Horvath E, Kovacs K, Salehi F, Cusimano MD (2007) Salivary gland-like tumor of the sella. Can J Neurol Sci Le journal canadien des sciences neurologiques 34(4):478–482. https://doi.org/10.1017/s0317167100007393
Dickhoff P, Wallace CJ, MacRae ME, Campbell WN (1993) Adenoid cystic carcinoma: an unusual sellar mass. Can Assoc Radiol J = Journal l’Association canadienne des radiologistes 44(5):393–395
Abdul-Hussein A, Morris PA, Markova T (2007) An unusual presentation of adenoid cystic carcinoma of the minor salivary glands with cranial nerve palsy: a case study. BMC Cancer 7:157. https://doi.org/10.1186/1471-2407-7-157
Kawamata T, Harashima S, Kubo O, Hori T (2006) Intrasellar remote metastasis from adenoid cystic carcinoma of parotid gland: case report. Endocr J 53(5):659–663. https://doi.org/10.1507/endocrj.k05-146
Koc K, Cabuk B, Uye B, Gok N, Ercin C, Ceylan S (2015) Adenoid cystic carcinoma metastatic to the sellar area: report of two cases. J Neurol Sci 32(1):193–198
McCutcheon IE, Kitagawa RH, Sherman SI, Bruner JM (2001) Adenocarcinoma of the salivary gland metastatic to the pituitary gland: case report. Neurosurgery 48(5):1161–1165. https://doi.org/10.1097/00006123-200105000-00044(discussion 1165–1166)
Taillens JP (1963) Nasopharyngeal mixed salivary tumor with pituitary and intracranial invasion. (Differential diagnosis from a chromophobe adenoma of the pituitary). Confin Neurol 23:313–322
Tripathy P, Dewan Y (2009) Endoscopic-assisted microscopic decompression of adenoid cystic carcinoma of paranasal sinus extending to the sella: a case report and review of literature. Neurol India 57(2):197–199. https://doi.org/10.4103/0028-3886.51295
Vincentelli F, Grisoli F, Leclercq TA, Ardaud B, Diaz-Vasquez P, Hassoun J (1986) Cylindromas of the base of the skull. Report of four cases. J Neurosurg 65(6):856–859. https://doi.org/10.3171/jns.1986.65.6.0856
Jahandideh H, Sanaie A, Ghadi A, Eskandarzadeh H (2018) Adenoid cystic carcinoma arising in the sphenoid sinus presenting with headache and diplopia: a case report. J Res Med Dent Sci 6(6):171–174
Abushamat LA, Kerr JM, Lopes MBS, Kleinschmidt-DeMasters BK (2019) Very unusual sellar/suprasellar region masses: a review. J Neuropathol Exp Neurol. https://doi.org/10.1093/jnen/nlz044
Gatto F, Perez-Rivas LG, Olarescu NC, Khandeva P, Chachlaki K, Trivellin G, Gahete MD, Cuny T (2020) Diagnosis and treatment of parasellar lesions. Neuroendocrinology. https://doi.org/10.1159/000506905
Thakkar K, Ramteke-Jadhav S, Kasaliwal R, Memon SS, Patil V, Thadani P, Lomte N, Sankhe S, Goel A, Epari S, Goel N, Lila A, Shah NS, Bandgar T (2020) Sellar surprises: a single-centre experience of unusual sellar masses. Endocr Connect 9(2):111–121. https://doi.org/10.1530/ec-19-0497
Petrakakis I, Pirayesh A, Krauss JK, Raab P, Hartmann C, Nakamura M (2016) The sellar and suprasellar region: a “hideaway” of rare lesions. Clinical aspects, imaging findings, surgical outcome and comparative analysis. Clin Neurol Neurosurg 149:154–165. https://doi.org/10.1016/j.clineuro.2016.08.011
Kessler AT, Bhatt AA (2018) Review of the major and minor salivary glands, Part 2: neoplasms and tumor-like lesions. J Clin Imaging Sci 8:48. https://doi.org/10.4103/jcis.JCIS_46_18
Chiloiro S, Giampietro A, Bianchi A, De Marinis L (2015) Clinical management of teratoma, a rare hypothalamic-pituitary neoplasia. Endocrine 53(3):636–642. https://doi.org/10.1007/s12020-015-0814-4
Sioshansi PC, Vezina G, Yaun AL, Miller MM, Bauman NM (2015) Considering the ectopic pituitary gland in evaluation of the nasopharyngeal mass. JAMA Otolaryngol Head Neck Surg 141(7):649. https://doi.org/10.1001/jamaoto.2015.0706
Kusakabe M, Sakakura T, Sano M, Nishizuka Y (1985) A pituitary-salivary mixed gland induced by tissue recombination of embryonic pituitary epithelium and embryonic submandibular gland mesenchyme in mice. Dev Biol 110(2):382–391. https://doi.org/10.1016/0012-1606(85)90097-1
Tresguerres JA, Ariznavarreta C, Granados B, Alvarez-Vega P, Fernandez-Mateos P, Gil-Loyzaga P, Alvarez-Buylla R (1999) Parotid gland tissue is able partially to assume pituitary functions under the influence of hypothalamic factors: in vivo and in vitro studies. J Endocrinol 160(2):205–216. https://doi.org/10.1677/joe.0.1600205
Denef C (2003) Paracrine control of lactotrope proliferation and differentiation. Trends Endocrinol Metab 14(4):188–195. https://doi.org/10.1016/s1043-2760(03)00057-2
Fisher DA, Lakshmanan J (1990) Metabolism and effects of epidermal growth factor and related growth factors in mammals. Endocr Rev 11(3):418–442. https://doi.org/10.1210/edrv-11-3-418
Kessler AT, Bhatt AA (2018) Review of the major and minor salivary glands, Part 1: anatomy, infectious, and inflammatory processes. J Clin Imaging Sci 8:47. https://doi.org/10.4103/jcis.JCIS_45_18
Cuthbertson DW, Raol N, Hicks J, Green L, Parke R (2015) Minor salivary gland basal cell adenocarcinoma. JAMA Otolaryngol Head Neck Surg 141(3):276. https://doi.org/10.1001/jamaoto.2014.3344
Acknowledgements
The illustrating case was presented at the meeting of the European Pituitary Pathology Group (EPPG) (Paris, France, December 2019) and the authors are grateful to EPPG members for the stimulating discussion.
Funding
Open access funding provided by Università degli Studi dell’Aquila within the CRUI-CARE Agreement. No funding was received for this study.
Author information
Authors and Affiliations
Contributions
The review was designed and written by TF and MLJR, who take full responsibility for the paper. TF and FGianno performed the systematic review with the contribution of MDA. The manuscript was critically revised and approved by CC, VE and FGiangaspero.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Consent to publication
Consent for publication has been obtained from the patient, including permission for the details/images to be available on the Internet and viewable by the general public.
Ethical statement
Institutional review board approval was not required.
Informed consent
Written informed consent was obtained from the patient for the scientific report of the illustrating case.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feola, T., Gianno, F., De Angelis, M. et al. Salivary gland tissues and derived primary and metastatic neoplasms: unusual pitfalls in the work-up of sellar lesions. A systematic review. J Endocrinol Invest 44, 2103–2122 (2021). https://doi.org/10.1007/s40618-021-01577-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-021-01577-6