
Abstract
Thyroid hormones are essential for skeletal development and are important regulators of bone maintenance in adults. Childhood hypothyroidism causes delayed skeletal development, retarded linear growth and impaired bone mineral accrual. Epiphyseal dysgenesis is evidenced by classic features of stippled epiphyses on X-ray. In severe cases, post-natal growth arrest results in a complex skeletal dysplasia. Thyroid hormone replacement stimulates catch-up growth and bone maturation, but recovery may be incomplete dependent on the duration and severity of hypothyroidism prior to treatment. A severe phenotype characteristic of hypothyroidism occurs in children with resistance to thyroid hormone due to mutations affecting THRA encoding thyroid hormone receptor α (TRα). Discovery of this rare condition recapitulated animal studies demonstrating that TRα mediates thyroid hormone action in the skeleton. In adults, thyrotoxicosis is well known to cause severe osteoporosis and fracture, but cases are rare because of prompt diagnosis and treatment. Recent data, however, indicate that subclinical hyperthyroidism is associated with low bone mineral density (BMD) and an increased risk of fracture. Population studies have also shown that variation in thyroid status within the reference range in post-menopausal women is associated with altered BMD and fracture risk. Thus, thyroid status at the upper end of the euthyroid reference range is associated with low BMD and increased risk of osteoporotic fragility fracture. Overall, extensive data demonstrate that euthyroid status is required for normal post-natal growth and bone mineral accrual, and is fundamental for maintenance of adult bone structure and strength.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Bone cells
Chondrocytes, osteoblasts, osteocytes and osteoclasts comprise the four major cell types in the skeleton. Cartilage-forming chondrocytes, bone-forming osteoblasts and terminally differentiated osteocytes are derived from mesenchyme whereas bone-resorbing multinucleated osteoclasts differentiate from precursor cells of the monocyte/macrophage lineage. Chondrocytes and osteoblasts are directly responsive to thyroid hormone and, whilst osteoclast activity is also sensitive to changes in thyroid status, it remains uncertain whether osteoclasts are direct target cells or whether the effects of thyroid hormone on bone resorption are indirect and mediated via primary thyroid hormone actions in other cell types. The effects of thyroid hormone on osteocyte function are unknown [1].
Bone formation and growth
During skeletal development, bone formation occurs via two distinct processes (Fig. 1). The flat bones of the skull vault and pelvis, together with the lateral third of the clavicle, form by intramembranous ossification in which mesenchyme progenitor cells condense and differentiate directly into osteoblasts [2]. Long bones and vertebrae, by contrast, form on an intermediary cartilage template by endochondral ossification after mesenchyme precursors differentiate to chondrocytes [3]. Epiphyseal growth plates form at the ends of developing long bones and contain chondrocytes that organize into distinct reserve, proliferative, pre-hypertrophic and hypertrophic zones (Fig. 1). Progression of reserve zone progenitor cells through the proliferative and hypertrophic zones is accompanied by a large increase in cell volume that is responsible for linear growth. Differentiating chondrocytes secrete a cartilage matrix that is rich in collagen types II and X. They ultimately undergo apoptosis and release growth factors and cytokines that stimulate vascular invasion and the migration of osteoblasts and osteoclasts, which model and mineralize the developing bone during growth [4]. Linear growth progresses until puberty when the growth plates fuse, but bone mineral accrual continues until peak bone mass is achieved during the third decade [5].

Intramembranous and endochondral ossification. a Skull vault stained with alizarin red (bone) and alcian blue (cartilage) from post-natal day 1 showing anterior and posterior fontanelles and sutures. Schematic representation of intramembranous bone formation at a skull suture. b Proximal tibial growth pate section at post-natal day 21 stained with alcian blue (cartilage) and van Gieson (bone matrix, red). Schematic representation of endochondral ossification showing chondrocyte proliferation, differentiation and apoptosis within the growth plate
Adult bone maintenance
In adults, the structure and function of the skeleton are maintained by a process of continuous repair, mediated by the bone remodelling cycle (Fig. 2) [6]. Osteocytes are terminally differentiated cells of the osteoblast lineage that become entombed within mature bone. They sense mechanical strain and bone micro-damage, and orchestrate the activities of bone-resorbing osteoclasts and bone-forming osteoblasts within a coordinated cycle of bone resorption and formation [7]. Bone resorption and formation are coupled in time and space, and so targeted bone modelling and remodelling enables the adult skeleton to repair and replace old and damaged tissue in response to injury and mechanical loading, or react quickly to the demands of mineral homeostasis [8]. Uncoupling of these processes leads to accelerated bone loss in osteoporosis or accumulation of bone in osteopetrosis.

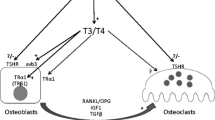
Bone remodelling cycle. The bone remodelling cycle is orchestrated by osteocytes that are entombed within the bone structure. Bone remodelling is initiated by changes in mechanical load, structural micro-damage or exposure to systemic or paracrine factors. Monocyte/macrophage precursors differentiate to mature osteoclasts and resorb bone. Differentiation is induced by macrophage colony-stimulating factor (M-CSF) and receptor activator of NFkB ligand (RANKL) and inhibited by osteoprotegerin (OPG). Following resorption, osteoblastic progenitors are recruited, synthesize an osteoid matrix and regulate its mineralization to form new bone and thus repair the defect
Thyroid hormone action
Thyroid hormones have important effects on skeletal development, linear growth and the maintenance of adult bone mass and strength. The thyroid gland mainly secretes thyroxine (3,5,3′,5′-l-tetraiodothyronine, T4), and the circulating level of T4 is approximately fourfold higher than the concentration of the biologically active hormone 3,5,3′-l-triiodothyronine (T3) [9]. A classic endocrine negative feedback loop (Fig. 3) maintains an inverse relationship between the circulating concentrations of thyroid hormones and thyroid stimulating hormone (thyrotropin, TSH), thus establishing the hypothalamic–pituitary–thyroid (HPT) axis set-point [10, 11].

Hypothalamic–pituitary–thyroid axis. The thyroid gland secretes the pro-hormone T4 and the active hormone T3 and circulating concentrations are regulated by a classical endocrine negative feedback loop that maintains an inverse physiological relationship between TSH, and T4 and T3
T4 and T3 enter target cells via active transport involving several specific transporter proteins, including monocarboxylate transporters 8 and 10 (MCT8, MCT10), organic anion transporter protein 1c1 (OATP1c1), and the l-type amino acid transporters 1 and 2 (LAT1, LAT2) [12]. Once inside the target cell, T4 and T3 are metabolized by either the activating type 2 iodothyronine deiodinase (DIO2) or the inactivating type 3 enzyme (DIO3). DIO2 catalyses 5′-deiodination of T4 to generate the active hormone T3, whereas DIO3 catalyses removal of the 5-iodine atom from T4 or T3 to generate the inactive metabolites, reverse T3 (3,3′,5′-l-triiodothyronine, rT3) or 3,3′-diiodotyrosine (T2), respectively. Thus, the balance of DIO2 and DIO3 activities regulates the intracellular supply of the active hormone, T3 [9, 13]. T3 then enters the nucleus where it binds and activates either thyroid hormone receptor α or β (TRα, TRβ) (Fig. 4). TRs function as hormone-dependent transcription factors that repress target gene expression in the absence of hormone and stimulate gene transcription in response to T3 binding [14]. TRβ is the main receptor expressed in the hypothalamus and pituitary where it mediates negative feedback control of the HPT axis, whereas TRα is the main receptor expressed in the skeleton and mediates T3 action in bone and cartilage [1].

Thyroid hormone action in bone cells. Thyroid hormones enter T3 target cells via specific membrane transporters. The relative activities of the type 2 and type 3 deiodinases (D2 and D3) are regulated to ensure optimal intracellular T3 availability resulting in the displacement of the co-repressor and the binding of the co-activator and thus the physiological transcriptional activity of TRα1
Skeletal consequences of thyroid disease in children
Hypothyroidism
Hypothyroidism in childhood is relatively common and causes delayed skeletal development, growth retardation and short stature with impaired bone maturation due to defective endochondral ossification. Impaired intramembranous ossification results in delayed closure of the fontanelles, persistently patent skull sutures and a typically flat nasal bridge and broad face. In severe undiagnosed cases, there is complete post-natal growth arrest and skeletal dysplasia with characteristic X-ray features that include stippled epiphyses reflecting epiphyseal dysgenesis, congenital hip dislocation, vertebral immaturity, scoliosis, patent fontanelles and sutures with delayed tooth eruption [1, 15].
Prompt treatment of children with thyroid hormone replacement induces a period of rapid “catch-up” growth in which skeletal maturation and bone age are also accelerated. Ultimately, normal adult height and bone mineral density (BMD) can be expected [16]. However, predicted adult height may not always be achieved and in such cases the deficit correlates with the severity of hypothyroidism and its duration prior to commencement of thyroid hormone replacement [17].
Hyperthyroidism
Thyrotoxicosis in children is relatively rare and causes accelerated intramembranous and endochondral ossification and an increase in linear growth rate [1]. Paradoxically, the accompanying advancement in bone age may result in premature fusion of the growth plates and early cessation of growth leading to persistent short stature [18, 19]. In severe thyrotoxicosis in young children, early closure of the cranial sutures can result in craniosynostosis with neurological sequelae. Untreated hyperthyroidism during pregnancy is also associated with craniosynostosis and may be a causative risk factor [20].
Summary of skeletal consequences of thyroid disease in children
Overall, T3 acts via TRα in chondrocytes and osteoblasts to regulate intramembranous and endochondral ossification and control the rate of linear growth and bone maturation and mineralization. Hypothyroidism in childhood causes delayed skeletal development and mineralization but prompt treatment with thyroid hormone initiates “catch-up” growth and stimulates bone maturation. Thyrotoxicosis, by contrast, accelerates skeletal development and bone mineral deposition, but may also result in short stature. Thus, the opposing skeletal consequences of childhood hypothyroidism and thyrotoxicosis, together with the rapid response of the hypothyroid skeleton to thyroid hormone replacement, demonstrate that normal euthyroid status during growth and adolescence is essential to establish peak bone mass and strength in early adulthood [1].
Skeletal consequences of thyroid disease in adults
A large number of studies investigating the influence of thyroid disease on the adult skeleton have been published and reviewed. However, overall interpretation of findings from these studies is limited by heterogeneous study design, confounding factors, lack of power and differing end-point analyses [1]. Nevertheless, large studies including extensive individual participant meta-analyses have been performed and this review will concentrate on the larger and more recent studies that have strengthened our understanding.
Hypothyroidism
Early histomorphometry analysis demonstrated that hypothyroidism results in low bone turnover with decreased osteoblastic bone formation and reduced osteoclastic bone resorption. The prolonged bone remodelling cycle includes a longer period of secondary mineralization resulting in a net increase in bone mineralization and mass without a change in bone volume [21, 22]. These changes are very slow, and long-term prospective follow-up of untreated hypothyroid patients would be required to demonstrate clinically significant changes in BMD by non-invasive imaging techniques. Thus, there are no good clinical data that demonstrate the skeletal consequences of hypothyroidism in adults.
Hyperthyroidism
Hyperthyroidism, in contrast, causes high bone turnover with an increased frequency of initiation of bone remodelling sites together with increased bone resorption and formation rates. The remodelling time is shortened with imbalance between resorption and formation that results in a net loss of approximately 10% of bone per remodelling cycle [23, 24]. Thus, hyperthyroidism is an established cause of high bone turnover with accelerated bone loss leading to osteoporosis and increased fracture susceptibility. Severe osteoporosis resulting from untreated thyrotoxicosis is now rare because of early diagnosis and treatment, although undiagnosed hyperthyroidism is an important risk factor for secondary bone loss and osteoporosis in patients presenting to hospital with fracture [25, 26]. Accordingly, population studies have demonstrated an increased risk of fracture in thyrotoxicosis especially in post-menopausal women [27,28,29,30,31].
Subclinical thyroid disease
Subclinical hypothyroidism is defined biochemically as the occurrence of circulating concentrations of T4 and T3 within their normal reference ranges in the presence of a TSH level elevated above its reference range. Conversely, subclinical hyperthyroidism occurs when T4 and T3 levels are within their reference ranges, but the TSH concentration is suppressed below its reference range. The effect of subclinical hypothyroidism on bone mineralization and fracture susceptibility has not been studied extensively [1], but a recent individual participant meta-analysis of data from 70,298 subjects from 13 prospective cohort studies consisting of 762,401 person-years follow-up demonstrated no association with fracture [28].
More studies have investigated the relationship between subclinical hyperthyroidism and BMD or fracture risk [1] and large population studies have suggested an increase in bone turnover, decrease in BMD and an increased risk of fracture, especially in post-menopausal women, although heterogeneity between studies prevented firm conclusions in several meta-analyses and reviews [32,33,34,35,36,37,38]. Nevertheless, the recent large individual participant meta-analysis of data from 70,298 subjects demonstrated that a TSH value below 0.01 mU/L was associated with a 2- and 3.5-fold increased risk of hip and spine fractures, respectively. Overall, subclinical hyperthyroidism was associated with bone loss and fracture, particularly in individuals with endogenous disease [28]. Furthermore, a register-based cohort of 222,138 subjects with a normal TSH level plus 9217 individuals with a low TSH were followed up for 7.5 years. This study demonstrated an exponential association between the duration of thyrotoxicosis and fracture. In euthyroid individuals, the risk of fracture increased with each standard deviation unit decrease in TSH (hazard ratio 1.45, p < 0.001 for hip fracture; HR 1.32, p < 0.001 for major osteoporotic fracture) [39].
Variation within the euthyroid reference range
These findings suggest the possibility that thyroid status even across the normal reference range is a continuous variable related to BMD and strength. A number of studies have been published to address this issue, but data have been conflicting, largely because of heterogeneity especially with regard to the age and gender of cohorts and differences in study size [40,41,42,43,44,45,46,47,48,49,50,51]. Recently, an individual participant meta-analysis was undertaken that included data from 56,835 subjects (n = 2565 with hip fracture) from 12 prospective cohort studies consisting of 659,059 person-years follow-up. This study demonstrated that lower TSH and higher fT4 within the reference range were associated with 22–25% increased risk of hip fractures [52]. Taken together, these studies indicate that thyroid status at within the upper end of the euthyroid reference range is associated with low BMD and an increased risk of fracture [1].
TSH suppression therapy for thyroid cancer
Thyroid cancer cells express TSH receptor, and TSH stimulates cell proliferation, iodine uptake and thyroglobulin secretion from tumour cells. This is also true in metastatic cells in about 65% of cases. There is, therefore, a good rationale for TSH suppression therapy using thyroxine. Treatment has been shown to reduce recurrence and disease-specific mortality in retrospective studies [53, 54], whilst a prospective non-randomized study demonstrated that a lesser degree of TSH suppression is an independent predictor of disease progression in patients at high-risk of recurrent disease [55].
In pre-menopausal women, three systematic literature reviews showed that suppressive treatment with T4 had no effect on BMD [32, 35, 36] and two meta-analyses demonstrated no effect on BMD at the hip, lumbar spine or radius [34, 37]. No fracture data are available.
In post-menopausal women, three systematic literature reviews demonstrated conflicting effects of suppressive treatment with T4 on BMD [32, 35, 36], while two meta-analyses showed BMD was reduced by 7% at the lumbar spine (CI 4–10%) and 5% at the femur (CI 2–8%) [34, 37]. No prospective fracture data are available, but hospital admission data for patients with fracture demonstrated that the incidence of admissions for fracture was 2.5× higher in patients with TSH <0.05 mU/l [29]. In patients with hyperthyroidism, there was a 3× increase in hip and 4× increase in vertebral fracture if TSH was suppressed <0.01 mU/l [27].
Overall, post-menopausal women on suppressive doses of T4 may be at risk of bone loss and osteoporosis [1].
Summary of skeletal consequences of thyroid disease in adults
Thyroid hormones act via TRα in osteoblasts, but their actions in osteocytes and osteoclasts have not been defined [1]. Thyroid hormones stimulate adult bone turnover via increased osteoclastic bone resorption. In euthyroid subjects, variation across the reference ranges for thyroid hormones and TSH is associated with bone loss. Thus, thyroid status at the upper end of the normal reference range is associated with lower BMD and an increased risk of fragility fracture. Thyrotoxicosis is a well-established cause of high bone turnover osteoporosis, resulting in an increased susceptibility to fracture. This complication is now rare because of prompt diagnosis and treatment. Subclinical hyperthyroidism is also associated with an increased risk of fracture in both men and women, but especially those with endogenous disease. TSH suppression therapy in thyroid cancer may be associated with bone loss and fracture in post-menopausal women. There is potential for the use of bisphosphonates to prevent bone loss in post-menopausal women at risk of fracture who are receiving long-term TSH suppression therapy [56]. There are no prospective studies in this area but, although the adverse side-effect of atrial fibrillation in patients taking bisphosphonates is controversial, it could be important as exposure to excess thyroid hormones in the elderly is also associated with AF and cardiovascular mortality [57, 58]. Thus, despite preclinical studies supporting the potential use of bisphosphonates for prevention of bone loss in post-menopausal women at risk of fracture who are exposed to thyroid hormone excess [56], other anti-resorptive drugs may theoretically be used according to the latest treatment guidelines [59]. Nevertheless, targeted prospective clinical trials are still needed to provide a clear evidence base for therapeutic intervention.
Overall, and in contrast to the effects on the juvenile skeleton in which thyroid hormones are anabolic and stimulate bone growth and mineralization, T3 exerts catabolic actions in the adult skeleton and stimulates bone loss [1].
Skeletal consequences of genetic disorders of thyroid function
Mutations affecting TSH (TSHB) and TSH receptor (TSHR)
Loss-of-function mutations in TSHB, encoding the TSHβ subunit, result in biologically inactive TSH and congenital hypothyroidism (OMIM 275100). Loss- and gain-of-function mutations in TSHR, encoding the TSH receptor, result in TSH resistance with congenital hypothyroidism (OMIM 275200) or autosomal dominant hyperthyroidism (OMIM 609152), respectively. Few reports have documented the skeletal consequences of these genetic disorders because children are treated early and the long-term outcomes reflect the response to thyroid hormone supplementation or thyroidectomy/ablation [1]. Nevertheless, normal growth occurs in individuals with congenital hypothyroidism following proper thyroid hormone replacement [60,61,62], and improvement of skeletal developmental manifestations is seen in patients with autosomal dominant hyperthyroidism following thyroidectomy and normalization of thyroid status [63, 64]. Overall, the limited available studies demonstrate that persistent absence of TSH signalling during normal skeletal development following adequate thyroid hormone replacement does not affect bone mineralization in children, whilst constitutive activation of the TSHR is not detrimental for skeletal development and maturation in patients who have been treated and had their normal euthyroid status restored [1].
Mutations affecting thyroid hormone receptors
Resistance to thyroid hormone α (RTHα) (OMIM 614450)
The key role of TRα in the human skeleton has been exemplified by discovery of a new syndrome of resistance to thyroid hormone due to dominant-negative mutations in THRA (RTHα) [65]. The skeletal manifestations of this condition (OMIM 614450) are consistent with the characteristic consequences of congenital and juvenile hypothyroidism, and reflect impaired T3 action in cartilage and bone. To date, 15 THRA mutations in 11 separate codons affecting 29 individuals from 16 families have been described. Nine result in the expression of a mutant TRα1 protein alone, and six affect both TRα1 and TRα2 [66,67,68,69,70,71,72,73,74,75,76]. Affected individuals have normal serum TSH with low/normal T4, high/normal T3 concentrations and characteristically elevated fT3:fT4 and T3:rT3 ratios [77, 78].
Children with RTHα due to expression of mutant TRα1 alone have skeletal dysplasia manifest by variable features that include delayed bone age and tooth eruption, patent skull sutures with wormian bones and delayed closure of the fontanelles, macrocephaly, flattened nasal bridge, hypertelorism, disproportionate (sub-ischial) short stature mainly affecting the lower limbs, epiphyseal dysgenesis, acetabular hypoplasia, congenital hip dislocation, vertebral ossification defects and defective bone mineralization [66, 67, 71, 73]. Adults have been studied less comprehensively, but variable skeletal abnormalities include increased cortical bone mass, disproportionate short stature, macrocephaly with skull vault thickening and hearing loss due to otosclerosis [67, 70, 73, 74]. As the number of reported cases has increased, a phenotype–genotype correlation has emerged. Missense mutations that impair T3 binding and result in only mild or moderate dominant-negative activity of mutant TRα1 are associated with fewer and less severe abnormalities. By contrast, nonsense mutations resulting in the expression of a truncated TRα1 protein with absent T3 binding and potent dominant-negative activity result in marked skeletal dysplasia and developmental delay. Accordingly, T4 treatment of children harbouring severe mutations has had little effect on growth and skeletal development, whereas treatment of individuals with milder missense mutations has been more promising [66, 67, 70, 71, 73, 74, 76].
More recently, THRA mutations affecting both TRα1 and TRα2 have been described [67, 69, 72, 76]. Affected children and adults had a similar spectrum of abnormalities characteristic of the skeletal dysplasia and variable response to T4 treatment reported in individuals with mutations affecting TRα1 alone. In vitro analysis of the functional defects of mutant TRα1 proteins was also consistent with the phenotype–genotype correlation described above, while all studies showed that wild-type and mutant TRα2 proteins were transcriptionally inactive and had no dominant-negative activity [67, 69, 72, 76]. Nevertheless, a severe and atypical phenotype was described in a 27-year-old woman with an N359Y substitution affecting both TRα1 and TRα2 [68]. Her unique combination of skeletal abnormalities includes intrauterine growth retardation and failure to thrive, macrocephaly, hypertelorism, micrognathia, short and broad nose, agenesis of the clavicles and 12th ribs, elongated thorax, ovoid vertebrae, scoliosis, congenital hip dislocation, short limbs and dwarfism, unilateral humero-radial synostosis, elbow dislocation and syndactyly. The TRα1N359Y mutant protein had impaired T3 binding and transactivation function with moderate dominant-negative activity, while no clear functional abnormality of the TRα2N359Y mutant was identified [68, 79].
Together, these reports define a new genetic disorder, RTHα, characterized by profound and consistent developmental abnormalities of the skeleton that recapitulate findings in mice with dominant-negative Thra mutations [80,81,82,83,84,85]. Overall, similar phenotypes in patients with mutations affecting either TRα1 alone, or both TRα1 and TRα2, suggest TRα2 has little or no physiological role in the skeleton. However, the unique phenotype in the patient with the N359Y substitution affecting both TRα1 and TRα2, in whom no other de novo mutations could be identified by whole exome sequencing [68], raises the intriguing possibility that TRα2 may fulfil an unrecognized function during skeletal development.
Resistance to thyroid hormone β (RTHβ) (OMIM 188570)
RTHβ results from dominant-negative mutations of THRB that cause disruption of the HPT axis leading to a characteristic increase in TSH along with inappropriately normal or increased circulating T4 and T3 levels [86,87,88]. The clinical syndrome is complex with a mixed phenotype comprising hyperthyroid-like responses in some tissues and hypothyroid effects in others. These variable tissue responses depend on several factors including genetic background, the severity of the THRB mutation, the relative amounts of expressed mutant and wild-type TRβ proteins and tissue-specific differences in the ratio of TRα and TRβ protein expression. In addition to these confounding issues, patients may have received varying prior management including treatment with anti-thyroid drugs, thyroid hormone analogues or surgery. In this context, it is not surprising that descriptions of skeletal abnormalities in RTHβ are variable and largely restricted to case reports or case series. Long-term prospective studies in large families will be necessary to advance understanding [1]. Nevertheless, analysis of the skeletal consequences of mutations of Thrb in mouse models of RTHβ is consistent with a skeletal phenotype of accelerated bone development in juveniles and increased bone turnover with osteoporosis in adults that is due to increased T3 action in cartilage and bone [1, 83, 89,90,91].
References
Bassett JH, Williams GR (2016) Role of thyroid hormones in skeletal development and bone maintenance. Endocr Rev 37(2):135–187. doi:10.1210/er.2015-1106
Rice DP, Rice R (2008) Locate, condense, differentiate, grow and confront: developmental mechanisms controlling intramembranous bone and suture formation and function. Front Oral Biol 12:22–40. doi:10.1159/0000115030
Karsenty G, Kronenberg HM, Settembre C (2009) Genetic control of bone formation. Ann Rev Cell Dev Biol 25:629–648. doi:10.1146/annurev.cellbio.042308.113308
Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423(6937):332–336
Bonjour JP, Chevalley T (2014) Pubertal timing, bone acquisition, and risk of fracture throughout life. Endocr Rev 35(5):820–847. doi:10.1210/er.2014-1007
Raggatt LJ, Partridge NC (2010) Cellular and molecular mechanisms of bone remodeling. J Biol Chem 285(33):25103–25108. doi:10.1074/jbc.R109.041087
Bonewald LF (2011) The amazing osteocyte. J Bone Miner Res 26(2):229–238. doi:10.1002/jbmr.320
Khosla S, Westendorf JJ, Oursler MJ (2008) Building bone to reverse osteoporosis and repair fractures. J Clin Invest 118(2):421–428. doi:10.1172/JCI33612
Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR (2002) Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 23(1):38–89
Andersen S, Bruun NH, Pedersen KM, Laurberg P (2003) Biologic variation is important for interpretation of thyroid function tests. Thyroid 13(11):1069–1078
Bassett JH, Williams GR (2008) Critical role of the hypothalamic–pituitary–thyroid axis in bone. Bone 43(3):418–426
Visser WE, Friesema EC, Visser TJ (2011) Minireview: thyroid hormone transporters: the knowns and the unknowns. Mol Endocrinol 25(1):1–14
Williams AJ, Robson H, Kester MH, van Leeuwen JP, Shalet SM, Visser TJ, Williams GR (2008) Iodothyronine deiodinase enzyme activities in bone. Bone 43(1):126–134
Brent GA (2012) Mechanisms of thyroid hormone action. J Clin Invest 122(9):3035–3043. doi:10.1172/JCI60047
Huffmeier U, Tietze HU, Rauch A (2007) Severe skeletal dysplasia caused by undiagnosed hypothyroidism. Eur J Med Genet 50(3):209–215. doi:10.1016/j.ejmg.2007.02.002
Salerno M, Micillo M, Di Maio S, Capalbo D, Ferri P, Lettiero T, Tenore A (2001) Longitudinal growth, sexual maturation and final height in patients with congenital hypothyroidism detected by neonatal screening. Eur J Endocrinol 145(4):377–383
Rivkees SA, Bode HH, Crawford JD (1988) Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med 318(10):599–602. doi:10.1056/NEJM198803103181003
Segni M, Gorman CA (2001) The aftermath of childhood hyperthyroidism. J Pediatr Endocrinol Metab 14(Suppl 5):1277–1282
Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM (1999) Special features of Graves’ disease in early childhood. Thyroid 9(9):871–877
Rasmussen SA, Yazdy MM, Carmichael SL, Jamieson DJ, Canfield MA, Honein MA (2007) Maternal thyroid disease as a risk factor for craniosynostosis. Obstet Gynecol 110(2 Pt 1):369–377. doi:10.1097/01.AOG.0000270157.88896.76
Eriksen EF, Mosekilde L, Melsen F (1986) Kinetics of trabecular bone resorption and formation in hypothyroidism: evidence for a positive balance per remodeling cycle. Bone 7(2):101–108
Mosekilde L, Melsen F (1978) Morphometric and dynamic studies of bone changes in hypothyroidism. Acta Pathol Microbiol Scand 86(1):56–62
Mosekilde L, Eriksen EF, Charles P (1990) Effects of thyroid hormones on bone and mineral metabolism. Endocrinol Metab Clin N Am 19(1):35–63
Mosekilde L, Melsen F (1978) Effect of antithyroid treatment on calcium-phosphorus metabolism in hyperthyroidism. II: Bone histomorphometry. Acta Endocrinol (Cph) 87(4):751–758
Bours SP, van Geel TA, Geusens PP, Janssen MJ, Janzing HM, Hoffland GA, Willems PC, van den Bergh JP (2011) Contributors to secondary osteoporosis and metabolic bone diseases in patients presenting with a clinical fracture. J Clin Endocrinol Metab 96(5):1360–1367. doi:10.1210/jc.2010-2135
Patel KV, Brennan KL, Brennan ML, Jupiter DC, Shar A, Davis ML (2014) Association of a modified frailty index with mortality after femoral neck fracture in patients aged 60 years and older. Clin Orthop Relat Res 472(3):1010–1017. doi:10.1007/s11999-013-3334-7
Bauer DC, Ettinger B, Nevitt MC, Stone KL (2001) Risk for fracture in women with low serum levels of thyroid-stimulating hormone. Ann Intern Med 134(7):561–568
Blum MR, Bauer DC, Collet TH, Fink HA, Cappola AR, da Costa BR, Wirth CD, Peeters RP, Asvold BO, den Elzen WP, Luben RN, Imaizumi M, Bremner AP, Gogakos A, Eastell R, Kearney PM, Strotmeyer ES, Wallace ER, Hoff M, Ceresini G, Rivadeneira F, Uitterlinden AG, Stott DJ, Westendorp RG, Khaw KT, Langhammer A, Ferrucci L, Gussekloo J, Williams GR, Walsh JP, Juni P, Aujesky D, Rodondi N, Thyroid Studies Collaboration (2015) Subclinical thyroid dysfunction and fracture risk: a meta-analysis. JAMA 313(20):2055–2065. doi:10.1001/jama.2015.5161
Flynn RW, Bonellie SR, Jung RT, MacDonald TM, Morris AD, Leese GP (2010) Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. J Clin Endocrinol Metab 95(1):186–193
Vestergaard P, Mosekilde L (2002) Fractures in patients with hyperthyroidism and hypothyroidism: a nationwide follow-up study in 16,249 patients. Thyroid 12(5):411–419. doi:10.1089/105072502760043503
Vestergaard P, Rejnmark L, Mosekilde L (2005) Influence of hyper- and hypothyroidism, and the effects of treatment with antithyroid drugs and levothyroxine on fracture risk. Calcif Tissue Int 77(3):139–144. doi:10.1007/s00223-005-0068-x
Murphy E, Williams GR (2004) The thyroid and the skeleton. Clin Endocrinol (Oxf) 61(3):285–298
Wirth CD, Blum MR, da Costa BR, Baumgartner C, Collet TH, Medici M, Peeters RP, Aujesky D, Bauer DC, Rodondi N (2014) Subclinical thyroid dysfunction and the risk for fractures: a systematic review and meta-analysis. Ann Intern Med 161(3):189–199. doi:10.7326/M14-0125
Uzzan B, Campos J, Cucherat M, Nony P, Boissel JP, Perret GY (1996) Effects on bone mass of long term treatment with thyroid hormones: a meta-analysis. J Clin Endocrinol Metab 81(12):4278–4289
Quan ML, Pasieka JL, Rorstad O (2002) Bone mineral density in well-differentiated thyroid cancer patients treated with suppressive thyroxine: a systematic overview of the literature. J Surg Oncol 79(1):62–69
Heemstra KA, Hamdy NA, Romijn JA, Smit JW (2006) The effects of thyrotropin-suppressive therapy on bone metabolism in patients with well-differentiated thyroid carcinoma. Thyroid 16(6):583–591
Faber J, Galloe AM (1994) Changes in bone mass during prolonged subclinical hyperthyroidism due to l-thyroxine treatment: a meta-analysis. Eur J Endocrinol 130(4):350–356
Yang R, Yao L, Fang Y, Sun J, Guo T, Yang K, Tian L (2017) The relationship between subclinical thyroid dysfunction and the risk of fracture or low bone mineral density: a systematic review and meta-analysis of cohort studies. J Bone Miner Metab. doi:10.1007/s00774-017-0828-5
Abrahamsen B, Jorgensen HL, Laulund AS, Nybo M, Brix TH, Hegedus L (2014) Low serum thyrotropin level and duration of suppression as a predictor of major osteoporotic fractures-the OPENTHYRO register cohort. J Bone Miner Res 29(9):2040–2050. doi:10.1002/jbmr.2244
Murphy E, Gluer CC, Reid DM, Felsenberg D, Roux C, Eastell R, Williams GR (2010) Thyroid function within the upper normal range is associated with reduced bone mineral density and an increased risk of nonvertebral fractures in healthy euthyroid postmenopausal women. J Clin Endocrinol Metab 95(7):3173–3181. doi:10.1210/jc.2009-2630
Grimnes G, Emaus N, Joakimsen RM, Figenschau Y, Jorde R (2008) The relationship between serum TSH and bone mineral density in men and postmenopausal women: the Tromso study. Thyroid 18(11):1147–1155
Hwangbo Y, Kim JH, Kim SW, Park YJ, Park DJ, Kim SY, Shin CS, Cho NH (2015) High-normal free thyroxine levels are associated with low trabecular bone scores in euthyroid postmenopausal women. Osteoporos Int. doi:10.1007/s00198-015-3270-3
Kim DJ, Khang YH, Koh JM, Shong YK, Kim GS (2006) Low normal TSH levels are associated with low bone mineral density in healthy postmenopausal women. Clin Endocrinol (Oxf) 64(1):86–90
Lin JD, Pei D, Hsia TL, Wu CZ, Wang K, Chang YL, Hsu CH, Chen YL, Chen KW, Tang SH (2011) The relationship between thyroid function and bone mineral density in euthyroid healthy subjects in Taiwan. Endocr Res 36(1):1–8
Mazziotti G, Porcelli T, Patelli I, Vescovi PP, Giustina A (2010) Serum TSH values and risk of vertebral fractures in euthyroid post-menopausal women with low bone mineral density. Bone 46(3):747–751. doi:10.1016/j.bone.2009.10.031
Morris MS (2007) The association between serum thyroid-stimulating hormone in its reference range and bone status in postmenopausal American women. Bone 40(4):1128–1134
Noh HM, Park YS, Lee J, Lee W (2015) A cross-sectional study to examine the correlation between serum TSH levels and the osteoporosis of the lumbar spine in healthy women with normal thyroid function. Osteoporos Int 26(3):997–1003. doi:10.1007/s00198-014-2906-z
Roef G, Lapauw B, Goemaere S, Zmierczak H, Fiers T, Kaufman JM, Taes Y (2011) Thyroid hormone status within the physiological range affects bone mass and density in healthy men at the age of peak bone mass. Eur J Endocrinol 164(6):1027–1034
Svare A, Nilsen TI, Asvold BO, Forsmo S, Schei B, Bjoro T, Langhammer A (2013) Does thyroid function influence fracture risk? Prospective data from the HUNT2 study, Norway. Eur J Endocrinol 169(6):845–852. doi:10.1530/EJE-13-0546
van der Deure WM, Uitterlinden AG, Hofman A, Rivadeneira F, Pols HA, Peeters RP, Visser TJ (2008) Effects of serum TSH and FT4 levels and the TSHR-Asp727Glu polymorphism on bone: the Rotterdam Study. Clin Endocrinol (Oxf) 68(2):175–181
van Rijn LE, Pop VJ, Williams GR (2014) Low bone mineral density is related to high physiological levels of free thyroxine in peri-menopausal women. Eur J Endocrinol 170(3):461–468. doi:10.1530/EJE-13-0769
Aubert CE, Floriani C, Bauer DC, da Costa BR, Segna D, Blum MR, Collet TH, Fink HA, Cappola AR, Syrogiannouli L, Peeters RP, Asvold BO, den Elzen WP, Luben RN, Bremner AP, Gogakos A, Eastell R, Kearney PM, Hoff M, Le Blanc E, Ceresini G, Rivadeneira F, Uitterlinden AG, Khaw KT, Langhammer A, Stott DJ, Westendorp RG, Ferrucci L, Williams GR, Gussekloo J, Walsh JP, Aujesky D, Rodondi N, Thyroid Studies Collaboration (2017) Thyroid function tests in the reference range and fracture: individual participant analysis of prospective cohorts. J Clin Endocrinol Metab. doi:10.1210/jc.2017-00294
Mazzaferri EL, Jhiang SM (1994) Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med 97(5):418–428
Pujol P, Daures JP, Nsakala N, Baldet L, Bringer J, Jaffiol C (1996) Degree of thyrotropin suppression as a prognostic determinant in differentiated thyroid cancer. J Clin Endocrinol Metab 81(12):4318–4323. doi:10.1210/jcem.81.12.8954034
Cooper DS, Specker B, Ho M, Sperling M, Ladenson PW, Ross DS, Ain KB, Bigos ST, Brierley JD, Haugen BR, Klein I, Robbins J, Sherman SI, Taylor T, Maxon HR 3rd (1998) Thyrotropin suppression and disease progression in patients with differentiated thyroid cancer: results from the National Thyroid Cancer Treatment Cooperative Registry. Thyroid 8(9):737–744
Williams GR (2014) Is prophylactic anti-resorptive therapy required in thyroid cancer patients receiving TSH-suppressive treatment with thyroxine? J Endocrinol Invest 37(8):775–779. doi:10.1007/s40618-014-0110-9
Biondi B, Wartofsky L (2014) Treatment with thyroid hormone. Endocr Rev 35(3):433–512. doi:10.1210/er.2013-1083
Sharma A, Einstein AJ, Vallakati A, Arbab-Zadeh A, Walker MD, Mukherjee D, Homel P, Borer JS, Lichstein E (2014) Risk of atrial fibrillation with use of oral and intravenous bisphosphonates. Am J Cardiol 113(11):1815–1821. doi:10.1016/j.amjcard.2014.03.008
Vescini F, Attanasio R, Balestrieri A, Bandeira F, Bonadonna S, Camozzi V, Cassibba S, Cesareo R, Chiodini I, Francucci CM, Gianotti L, Grimaldi F, Guglielmi R, Madeo B, Marcocci C, Palermo A, Scillitani A, Vignali E, Rochira V, Zini M (2016) Italian association of clinical endocrinologists (AME) position statement: drug therapy of osteoporosis. J Endocrinol Invest 39(7):807–834. doi:10.1007/s40618-016-0434-8
Papadimitriou A, Papadimitriou DT, Papadopoulou A, Nicolaidou P, Fretzayas A (2007) Low TSH levels are not associated with osteoporosis in childhood. Eur J Endocrinol 157(2):221–223
Bretones P, Duprez L, Parma J, David M, Vassart G, Rodien P (2001) A familial case of congenital hypothyroidism caused by a homozygous mutation of the thyrotropin receptor gene. Thyroid 11(10):977–980. doi:10.1089/105072501753211064
Sunthornthepvarakui T, Gottschalk ME, Hayashi Y, Refetoff S (1995) Brief report: resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med 332(3):155–160. doi:10.1056/NEJM199501193320305
Chawla R, Alden TD, Bizhanova A, Kadakia R, Brickman W, Kopp PA (2015) Squamosal suture craniosynostosis due to hyperthyroidism caused by an activating thyrotropin receptor mutation (T632I). Thyroid. doi:10.1089/thy.2014.0503
Fuhrer D, Wonerow P, Willgerodt H, Paschke R (1997) Identification of a new thyrotropin receptor germline mutation (Leu629Phe) in a family with neonatal onset of autosomal dominant nonautoimmune hyperthyroidism. J Clin Endocrinol Metab 82(12):4234–4238. doi:10.1210/jcem.82.12.4405
Refetoff S, Bassett JH, Beck-Peccoz P, Bernal J, Brent G, Chatterjee K, De Groot LJ, Dumitrescu AM, Jameson JL, Kopp PA, Murata Y, Persani L, Samarut J, Weiss RE, Williams GR, Yen PM (2014) Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. J Clin Endocrinol Metab 99(3):768–770. doi:10.1210/jc.2013-3393
Bochukova E, Schoenmakers N, Agostini M, Schoenmakers E, Rajanayagam O, Keogh JM, Henning E, Reinemund J, Gevers E, Sarri M, Downes K, Offiah A, Albanese A, Halsall D, Schwabe JW, Bain M, Lindley K, Muntoni F, Khadem FV, Dattani M, Farooqi IS, Gurnell M, Chatterjee K (2012) A mutation in the thyroid hormone receptor alpha gene. N Engl J Med 366(3):243–249. doi:10.1056/NEJMoa1110296
Demir K, van Gucht AL, Buyukinan M, Catli G, Ayhan Y, Nijat Bas V, Dundar B, Ozkan B, Meima ME, Edward Visser W, Peeters RP, Visser TJ (2016) Diverse genotypes and phenotypes of three novel thyroid hormone receptor alpha mutations. J Clin Endocrinol Metab. doi:10.1210/jc.2016-1404
Espiard S, Savagner F, Flamant F, Vlaeminck-Guillem V, Guyot R, Munier M, d’Herbomez M, Bourguet W, Pinto G, Rose C, Rodien P, Wemeau JL (2015) A novel mutation in THRA gene associated with an atypical phenotype of resistance to thyroid hormone. J Clin Endocrinol Metab 100(8):2841–2848. doi:10.1210/jc.2015-1120
Moran C, Agostini M, Visser WE, Schoenmakers E, Schoenmakers N, Offiah AC, Poole K, Rajanayagam O, Lyons G, Halsall D, Gurnell M, Chrysis D, Efthymiadou A, Buchanan C, Aylwin S, Chatterjee KK (2014) Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)alpha1 and TRalpha2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol 2(8):619–626. doi:10.1016/S2213-8587(14)70111-1
Moran C, Schoenmakers N, Agostini M, Schoenmakers E, Offiah A, Kydd A, Kahaly G, Mohr-Kahaly S, Rajanayagam O, Lyons G, Wareham N, Halsall D, Dattani M, Hughes S, Gurnell M, Park SM, Chatterjee K (2013) An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. J Clin Endocrinol Metab. doi:10.1210/jc.2013-2215
Tylki-Szymanska A, Acuna-Hidalgo R, Krajewska-Walasek M, Lecka-Ambroziak A, Steehouwer M, Gilissen C, Brunner HG, Jurecka A, Rozdzynska-Swiatkowska A, Hoischen A, Chrzanowska KH (2015) Thyroid hormone resistance syndrome due to mutations in the thyroid hormone receptor alpha gene (THRA). J Med Genet 52(5):312–316. doi:10.1136/jmedgenet-2014-102936
van Gucht AL, Meima ME, Zwaveling-Soonawala N, Visser WE, Fliers E, Wennink JM, Henny C, Visser TJ, Peeters RP, van Trotsenburg AS (2016) Resistance to thyroid hormone alpha in an 18-month-old girl: clinical, therapeutic, and molecular characteristics. Thyroid 26(3):338–346. doi:10.1089/thy.2015.0463
van Mullem A, van Heerebeek R, Chrysis D, Visser E, Medici M, Andrikoula M, Tsatsoulis A, Peeters R, Visser TJ (2012) Clinical phenotype and mutant TRalpha1. N Engl J Med 366(15):1451–1453. doi:10.1056/NEJMc1113940
van Mullem AA, Chrysis D, Eythimiadou A, Chroni E, Tsatsoulis A, de Rijke YB, Visser WE, Visser TJ, Peeters RP (2013) Clinical phenotype of a new type of thyroid hormone resistance caused by a mutation of the TRalpha1 receptor: consequences of LT4 treatment. J Clin Endocrinol Metab 98(7):3029–3038. doi:10.1210/jc.2013-1050
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, Gazzellone MJ, D’Abate L, Deneault E, Howe JL, Liu RS, Thompson A, Zarrei M, Uddin M, Marshall CR, Ring RH, Zwaigenbaum L, Ray PN, Weksberg R, Carter MT, Fernandez BA, Roberts W, Szatmari P, Scherer SW (2015) Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 21(2):185–191. doi:10.1038/nm.3792
Moran C, Agostini M, McGowan A, Schoenmakers E, Fairall L, Lyons G, Rajanayagam O, Watson L, Offiah A, Barton J, Price S, Schwabe J, Chatterjee K (2017) Contrasting phenotypes in resistance to thyroid hormone alpha correlate with divergent properties of thyroid hormone receptor alpha1 mutant proteins. Thyroid 27(7):973–982. doi:10.1089/thy.2017.0157
Schoenmakers N, Moran C, Peeters RP, Visser T, Gurnell M, Chatterjee K (2013) Resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. Biochim Biophys Acta 1830(7):4004–4008. doi:10.1016/j.bbagen.2013.03.018
van Mullem AA, Visser TJ, Peeters RP (2014) Clinical consequences of mutations in thyroid hormone receptor-α1. Eur Thyroid J 3:17–24
Vlaeminck-Guillem V, Espiard S, Flamant F, Wemeau JL (2015) TRalpha receptor mutations extend the spectrum of syndromes of reduced sensitivity to thyroid hormone. Presse Med 44(11):1103–1112. doi:10.1016/j.lpm.2015.07.022
Bassett JH, Boyde A, Zikmund T, Evans H, Croucher PI, Zhu X, Park JW, Cheng SY, Williams GR (2014) Thyroid hormone receptor alpha mutation causes a severe and thyroxine-resistant skeletal dysplasia in female mice. Endocrinology 155(9):3699–3712. doi:10.1210/en.2013-2156
Bassett JH, Nordstrom K, Boyde A, Howell PG, Kelly S, Vennstrom B, Williams GR (2007) Thyroid status during skeletal development determines adult bone structure and mineralization. Mol Endocrinol 21(8):1893–1904. doi:10.1210/me.2007-0157
Desjardin C, Charles C, Benoist-Lasselin C, Riviere J, Gilles M, Chassande O, Morgenthaler C, Laloe D, Lecardonnel J, Flamant F, Legeai-Mallet L, Schibler L (2014) Chondrocytes play a major role in the stimulation of bone growth by thyroid hormone. Endocrinology. doi:10.1210/en.2014-1109
O’Shea PJ, Bassett JH, Sriskantharajah S, Ying H, Cheng SY, Williams GR (2005) Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor alpha1 or beta. Mol Endocrinol 19(12):3045–3059. doi:10.1210/me.2005-0224
Quignodon L, Vincent S, Winter H, Samarut J, Flamant F (2007) A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol Endocrinol 21(10):2350–2360. doi:10.1210/me.2007-0176
Williams GR (2014) Role of thyroid hormone receptor-alpha1 in endochondral ossification. Endocrinology 155(8):2747–2750. doi:10.1210/en.2014-1527
Dumitrescu AM, Refetoff S (2013) The syndromes of reduced sensitivity to thyroid hormone. Biochim Biophys Acta 1830(7):3987–4003. doi:10.1016/j.bbagen.2012.08.005
Refetoff S, DeWind LT, DeGroot LJ (1967) Familial syndrome combining deaf-mutism, stippled epiphyses, goiter and abnormally high PBI: possible target organ refractoriness to thyroid hormone. J Clin Endocrinol Metab 27(2):279–294
Sakurai A, Takeda K, Ain K, Ceccarelli P, Nakai A, Seino S, Bell GI, Refetoff S, DeGroot LJ (1989) Generalized resistance to thyroid hormone associated with a mutation in the ligand-binding domain of the human thyroid hormone receptor beta. Proc Natl Acad Sci USA 86(22):8977–8981
Bassett JH, O’Shea PJ, Sriskantharajah S, Rabier B, Boyde A, Howell PG, Weiss RE, Roux JP, Malaval L, Clement-Lacroix P, Samarut J, Chassande O, Williams GR (2007) Thyroid hormone excess rather than thyrotropin deficiency induces osteoporosis in hyperthyroidism. Mol Endocrinol 21(5):1095–1107
O’Shea PJ, Bassett JH, Cheng SY, Williams GR (2006) Characterization of skeletal phenotypes of TRalpha1 and TRbeta mutant mice: implications for tissue thyroid status and T3 target gene expression. Nucl Recept Signal 4:e011. doi:10.1621/nrs.04011
O’Shea PJ, Harvey CB, Suzuki H, Kaneshige M, Kaneshige K, Cheng SY, Williams GR (2003) A thyrotoxic skeletal phenotype of advanced bone formation in mice with resistance to thyroid hormone. Mol Endocrinol 17(7):1410–1424
Acknowledgements
G.R.W. and J.H.D.B. are funded by a Wellcome Trust Strategic Award (101123), Wellcome Trust Joint Investigator Award (110140 and 110141) and a European Commission Horizon 2020 Grant (666869, THYRAGE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
No informed consent.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Williams, G.R., Bassett, J.H.D. Thyroid diseases and bone health. J Endocrinol Invest 41, 99–109 (2018). https://doi.org/10.1007/s40618-017-0753-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-017-0753-4