
Abstract
Purpose of Review
Store-operated calcium entry (SOCE) is dysregulated in prostate cancer, contributing to increased cellular migration and proliferation and preventing cancer cell apoptosis. We here summarize findings on gene expression levels and functions of SOCE components, stromal interaction molecules (STIM1 and STIM2), and members of the Orai protein family (Orai1, 2, and 3) in prostate cancer. Moreover, we introduce new research models that promise to provide insights into whether dysregulated SOCE signaling has clinically relevant implications in terms of increasing the migration and invasion of prostate cancer cells.
Recent Findings
Recent reports on Orai1 and Orai3 expression levels and function were in part controversial probably due to the heterogeneous nature of prostate cancer. Lately, in prostate cancer cells, transient receptor melastatin 4 channel was shown to alter SOCE and play a role in migration and proliferation. We specifically highlight new cancer research models: a subpopulation of cells that show tumor initiation and metastatic potential in mice and zebrafish models.
Summary
This review focuses on SOCE component dysregulation in prostate cancer and analyzes several preclinical, cellular, and animal cancer research models.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Physiological calcium concentrations vary widely in different intra- and extracellular compartments. While the extracellular Ca2+ concentration is about 1.2 mM, the cytoplasmic Ca2+ concentration ranges from the 100 nM to μM range. In the endoplasmic reticulum (ER), which functions as an intracellular Ca2+ store, the Ca2+ concentration is up to ~ 500 μM. Changes in intracellular Ca2+ act as signals that drive various cellular functions, including gene expression and cell migration, proliferation, and apoptosis [1,2,3,4,5,6]. In addition, numerous Ca2+ transporting enzymes generate tightly regulated local Ca2+ subdomains that are important for enzymatic and cellular functions, especially cell migration [7, 8].
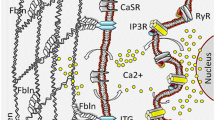
In order to mobilize Ca2+ for signaling, many cellular pathways lead to the production of inositol 1,4,5-trisphosphate (IP3) as a second messenger. IP3 binds to IP3 receptors in the ER membrane that release Ca2+ from the ER. Upon the drop in the Ca2+ concentration, Ca2+ dissociates from the EF motif of stromal interaction molecule 1 (STIM1). This leads to STIM1 clustering, to the recruitment of Orai1 channels, and subsequently to store-operated calcium entry (SOCE) [9,10,11] (Fig. 1). SOCE affects cellular functions such as gene expression and cell proliferation, apoptosis, and migration, and several studies report the dysregulation of SOCE in cancer [12,13,14,15]. Specifically, the dysregulation of distinct molecular components of SOCE, especially the STIM1 and Orai1 proteins and their homologues STIM2, Orai2, and Orai3, plays important roles in the pathophysiology of different types of cancer. For example, dysregulation of both STIM1 and Orai1 contribute to human glioblastoma invasion [16]. In colorectal cancer, decreased STIM2 protein levels contribute to decreased Ca2+ concentrations in intracellular Ca2+ stores, and elevated SOCE is associated with increased cell proliferation and invasion and with characteristics that are implicated in tumor cell survival [17]. In an acute myeloid leukemia cell line, Orai1-Orai2 complexes mediate Ca2+ influx, which is important for cell migration [18]. Furthermore, STIM2 and Orai1 are the predominant isoforms in melanoma. These proteins contribute to adaptive tanning, while their dysregulation leads to the proliferation and migration of melanoma cells [19,20,21,22]. In addition, Orai1 is upregulated in breast cancer upon stimulation of the membrane androgen receptor [23]. The role of Orai3 has been extensively investigated in different types of cancer including lung [24, 25], breast [26,27,28], and prostate cancer [29, 30].

Activation of SOCE and TRPM4 as feedback mechanism for SOCE. Upon receptor stimulation, IP3 is produced and binds to the IP3 receptor. Subsequently, Ca2+ is released from intracellular Ca2+ stores. The decrease in Ca2+ in the ER results in the dissociation of Ca2+ from STIM1 proteins, which cluster and recruit and activate Orai1 channels in the plasma membrane that mediate SOCE. The increase in intracellular Ca2+ activates TRPM4. The Na+ influx via TRPM4 is a negative feedback mechanism for SOCE (please see text for details)
Many proteins regulate SOCE, including α-SNAP, CRACR2A, golli, ORMDL3, SARAF, septins, STIMATE, and TMEM110 [31,32,33,34,35,36,37,38,39,40,41,42]. In addition to direct modulation of SOCE by its key players, other regulatory mechanisms can influence SOCE. These mechanisms include pH modulation [43,44,45] and post-translational modifications like STIM1 phosphorylation and Orai1 glycosylation [46,47,48,49,50]. Na+ influx via the transient receptor potential melastatin-4 channel (TRPM4) [51, 52] decreases the driving force for Ca2+, which also regulates SOCE.
This article summarizes what is known about the dysregulation of the key molecular players involved in SOCE (STIM1, STIM2, Orai1, Orai2, and Orai3) and the negative regulator TRPM4 in prostate cancer. In addition, we introduce prostate cancer research models that allow the selection of a subpopulation of metastasis-initiating cells (MICs) with high metastatic potential. Finally, we introduce novel mouse and zebrafish prostate cancer research models. Future investigation of SOCE in these models will shed light on SOCE as a possible therapeutic target in prostate cancer.
Gene Expression Levels of SOCE Components in Prostate Cancer
Table 1 summarizes studies [66] that compared gene expression in normal prostate tissue versus prostate carcinoma tissue. For Orai1, 5 out of 7 studies [53–57] reported a slight elevation in ORAI1 gene expression in prostate cancer tissue, only 2 out of 12 studies [53, 54] reported an elevation in ORAI2 gene expression. Notably, no changes in ORAI3 gene expression levels have been detected in prostate cancer [56, 58]. For STIM1, 2 of 16 studies [59, 67] showed slightly elevated levels of STIM1 gene expression in prostate cancer tissue, while 2 out of 5 studies [53, 54] reported elevated or slightly elevated gene expression levels for STIM2. These results suggest that some SOCE components may be dysregulated in prostate cancer.
Protein Expression Levels of Orai1 and Orai3
STIM1 and Orai1 expression levels are differentially regulated depending on the prostate cancer stage [68]. In early clinically localized cancer stages, STIM1 and Orai1 expression is increased, while in the later castration-resistant prostate cancer stages, their expression levels are decreased. These findings are consistent with the known role of Orai1 in cell migration [69]. Furthermore, low Orai1 expression may contribute to an apoptosis-resistant phenotype in prostate cancer cells [70].
The expression levels of STIM1 and Orai1 correlate with the expression level of the androgen receptor [68]. Notably, STIM1 expression is directly regulated by androgens, and thus androgens can directly decrease Ca2+ signaling via STIM1 [71]. Remarkably, in breast cancer, estrogen receptor-positive breast cancer cells are associated with elevated levels of Orai3, which is an estrogen receptor α-regulated Ca2+ channel [26, 28]. Interestingly, an estrogen receptor-α blocker reduces the viability of prostate cancer cells [72]. These studies imply that there may be a direct link between estrogen receptor-α antagonists, SOCE, and cell viability.
Dubois et al. reported that the Orai3 gene expression level was increased in 15 prostate cancer tissue samples compared to normal prostate tissue samples [30]. In that study, siRNA-based knockdown of Orai3 did not change SOCE. The study suggested that native Orai1 and Orai3 proteins form non-store-activated ion channels that are activated by arachidonic acid [73, 74].
A study by our group found a slightly decreased Orai3/Orai1 ratio in prostate cancer tissue compared to normal prostate tissue [29]. Submaximal activation with an endogenous stimulus of SOCE, dihydrotestosterone, decreased SOCE signals upon siRNA knockdown of Orai3. In addition, the Orai3/Orai1 ratio correlated with the pharmacological profile of SOCE channels. First, application of 2-APB, which blocks currents via Orai1 and enhances currents via Orai3 [75], resulted in stronger amplification of ICRAC in primary human prostate epithelial cells (hPECs) from healthy tissue compared to prostate cancer cells [29]. Second, reactive oxygen species (ROS) blocked SOCE and ICRAC to a greater extent in prostate cancer cells than in primary human prostate epithelial cells from healthy tissue [76]. Orai1 is sensitive to ROS [77, 78], and ROS production seems to be coupled to Orai1 [79, 80]. In contrast, Orai3 is insensitive to ROS due to the lack of the ROS sensor, cysteine-195, that is present in Orai1 [77]. Furthermore, STIM2 appears to contribute to the ROS profile of SOCE [81]. Taken together, these findings show that the Orai1/Orai3 ratio is higher in prostate cancer cells than in healthy tissue, which is consistent with the observation that Orai1 is elevated in early clinically localized cancer stage [68].
Studies of Orai3 expression level have produced inconsistent results, which may be due to the heterogeneous nature of prostate cancer. While our study focused on prostate cancer with Gleason scores of 6–8 [29], Dubois et al. excluded tumor tissue fragments that showed a mix of normal and tumoral tissue, which may have resulted in the selection of tissue samples from later stage prostate cancers [30]. To address this issue, the heterogeneity of prostate cancer must be taken into account. Future prostate cancer therapies may be personalized, with medicine that differentiates patients based on their genetic backgrounds and prostate cancer markers. Notably, great progress has been made in the development of tailored therapeutic approaches in prostate cancer [82].
TRPM4
TRPM4 is a negative regulator of SOCE (Fig. 1) that contributes to the migration of dendritic cells, mast cells, and vascular endothelial cells [52, 83,84,85,86,87]. TRPM4 expression is associated with immune disease [88] and several cardiac diseases [89,90,91,92,93,94,95,96,97,98,99,100], with proliferation of breast cancer cells [101], and with poor outcome in B cell lymphoma [102]. While the database Oncomine reports no differences or only slight differences in the gene expression levels of STIM1, STIM2, Orai1, Orai2, and Orai3, TRPM4 expression is reported to be elevated in 8 out of 9 studies that compared its expression in cancer tissue samples versus normal or benign prostate tissue (Table 1). In addition, TRPM4 is a cancer-driver gene in androgen-insensitive prostate cancer [103•], and TRPM4 protein expression is upregulated in human prostate cancer tissue [104•, 105•]. Patients with higher expression levels of TRPM4 in prostate cancer glands compared to matched benign glands have an increased risk of biochemical recurrence [104•]. We previously demonstrated that siRNA-based knockdown of TRPM4 increases SOCE (Fig. 1) and reduces cell migration in the prostate cancer cell lines DU145 and PC3 [105•, 106]. In addition, Sagredo et al. recently showed that TRPM4 knockdown significantly reduce the proliferation of PC3 cells [107]. Thus, TRPM4 represents an interesting putative target in prostate cancer therapy.
Future Cancer Research Models
The STIM and the Orai proteins are putative targets for cancer therapy [108, 109], and TRPM4 was more recently identified as a potential target for prostate cancer therapy. SOCE, and particularly the expression levels and functions of STIM and Orai proteins in prostate cancer, is complex and remains incompletely understood. Below, we introduce selected sophisticated prostate cancer research models, including cancer stem cell and mouse and zebrafish models that may increase our understanding.
Cancer Stem Cells as a Cellular System for Studying Human Prostate Cancers
The study of ion channels in selected cell subpopulations may be a key strategy for identifying the biological functions of these molecules in order to understand their roles in prostate cancer maintenance and progression. According to the so-called cancer stem cell (CSC) hypothesis, only a selected subpopulation of cells supports tumor initiation and metastasis. Moreover, current therapies may efficiently target more differentiated “bulk” tumor cells without affecting the tumor- and metastasis-initiating properties of CSCs or metastasis-initiating cells (MICs). Determining the molecular characteristics of selected CSCs and MICs may help researchers formulate strategies to block cancer progression and metastasis.
One potential strategy for identifying and isolating MICs involves measuring the aldehyde dehydrogenase (ALDH) enzyme activity with the ALDEFLUOR assay [110, 111••]. The assay defines two subpopulations of cells, namely ALDHlow and ALDHhigh cells. The ALDHhigh subpopulations that have been isolated from prostate cancer cell lines (e.g., C4-2B and PC-3M-Pro4) showed increased aggressiveness and invasion in vitro. Importantly, in PC-3M-Pro4Luc2 cells, the ALDHhigh subpopulation has much higher bone metastasis-initiating potential than the ALDHlow subpopulation. ALDH1A1 expression is associated with advanced clinical stage and unfavorable prognosis in hormone-naïve prostate cancer [112]. Recent studies show that bulk unsorted prostate cancer cell lines with different metastatic abilities can be distinguished based on the presence of specific ion channels [113]. Selected MICs, which are characterized by elevated ALDH activity, play a crucial role in tumor initiation and metastasis in human prostate cancer [111••]. Thus, this model represents a promising alternative to bulk and heterogeneous cell lines for assessing the contribution of SOCE components to prostate cancer.
Notably, the use of cellular models that have different metastatic characteristics shows promise as a way to investigate the roles of STIMs, Orais, and TRPM4 in the metastatic process. Despite the predominant blastic response at the level of bone metastasis in prostate cancer patients, a fraction of cases shows lytic lesions. Therefore, the use of different cell lines with distinct metastatic phenotypes (i.e., lytic vs. blastic) might provide new insights into the roles of SOCE components in prostate cancer bone metastasis. Blastic cell lines, such as VCaP and C4-2B, or lytic prostate cancer cell lines, such as PC3 and DU145, are examples of some available models that can be used to better understand the contribution of ion channel signaling during the formation of lytic and blastic bone lesions.
Mouse Models
The use of blastic and lytic cell lines in animal models of bone metastasis may elucidate the contributions of STIMs, Orais, and TRPM4 in preclinical settings [114]. Current mouse models of intra-osseous (IO) and intra-cardiac (IC) inoculation are excellent models for dissecting the metastatic cascade in the different steps of metastatic dissemination. IO inoculation of lytic or blastic cells could be used to study the roles of SOCE signaling in the growth of human prostate cancer cells in bone. Similarly, IC injection represents a state-of-the-art model for measuring the contribution of SOCE signaling in the metastatic cascade, i.e., in dissemination, survival in the circulation, extravasation, homing to the distant site, and establishment of a metastatic lesion. Furthermore, it was recently shown that these models represent a good source of information for understanding the metastatic process in terms of events in both the tumor and the host [115]. This approach allows the tumoral and stromal components to be studied separately in order to identify the contributions of ion channels to the maintenance of the supportive stroma at the metastatic site.
The Zebrafish Xenograft Model for the Study of Human Prostate Cancers
The zebrafish has long been used as a model for cancer research, first for studying chemically induced cancers [116, 117] and then, at the turn of the century, as a genetic cancer model [118]. However, it was only in the last decade that it made its debut as an animal system for the study of the xenotransplantation of mammalian cancer cells. Xenotransplantation can be defined as the process through which organs or tissues of one species are grafted or transplanted into another species. The method has long been used in cancer research, the first time by Green in 1938, who successfully transplanted adenocarcinomas from rabbit into a guinea pig eye [119]. Currently, this technique is used in mouse models to study the various phases of cancer development and progression. Haldi et al. were the first to describe xenografts of mammalian tumors into zebrafish embryos as a way to study tumor development [120]. This work was followed by that of Nicoli et al., who used xenografts of mammalian cancer cells in zebrafish embryos to analyze tumor-induced neoangiogenesis [121].
Following these initial studies of cancer xenografts in zebrafish embryos, many others used this model to study different aspects of tumor formation and cancer development. The zebrafish model allowed the investigation of the behavior of primary human tumors, such as pancreatic, colon, and stomach tumors [122], primary leukemia [123], and prostate cancer [124••], as well as their response to genetic [125] and chemical therapies [126]. In the case of primary human tumors, the zebrafish model could prove very useful, especially for the examination of the metastatic nature of a primary human tumor, as well as a tool for toxicological testing of potential anti-cancer drugs [127,127,129]. For example, in the study published by Bansal and colleagues [124••], they use the zebrafish xenograft transplantation model to access how frequently prostate tumor-initiating cells are found in different prostate cancer cell lines as well as in primary tumors.
The zebrafish model has unique advantages compared to, for example, the mouse model, especially when zebrafish embryos are used. In particular, a single female can produce over 100 eggs in 1 day, providing a large number of individuals for experimental and control groups from a single male/female couple. Within 25 h, the larvae are still less than 2 mm in size but already have an established vascular plexus with a beating heart, and all organ primordia are set. Importantly, the larvae can be kept transparent until later developmental stages. This transparency can be either genetically selected [130] or chemically induced [131] using quantum dots [124••], which has proved to be very useful for long-term in vivo imaging. Xenotransplanted cancer cells can then be followed over time either by fluorescent labeling with vital dyes that are stable throughout a few cell divisions [132] or by creating transgenic cancer cell lines that express a fluorescent protein associated with a gene of interest. The images are easy to acquire using a simple fluorescent stereomicroscope. High-resolution images can also be obtained using more advanced microscopy techniques, such as confocal, spinning-disk, two-photon, or light-sheet microscopy [133]. These more advanced methods not only allow researchers to track cells over time, but also enable 3D reconstructions, providing researchers with a way to analyze metastasis formation in vivo in four dimensions (time plus the x, y, and z planes). Even in adulthood, albino zebrafish strains such as Casper [130] are nearly fully transparent. This presents a unique opportunity to follow in vivo tumor development and progression. Furthermore, since embryos do not develop an adaptive immune system until 14 days post-fertilization [134], there is no need to suppress the immune system to facilitate the acceptance of foreign tissue when implanting xenografts. This provides researchers with an ideal system for studying tumor growth, invasion, and metastasis and how the microenvironment can affect these processes in a small organism [135]. This system allows rapid testing of drugs and genetic factors that might affect these processes [136] in order to identify ways to eradicate tumors [137, 138].
However, this model also has its limitations, and these must be taken into account when translating the acquired knowledge into other organisms, especially humans. Despite the similarity between the human and zebrafish genomes, zebrafish do not have many of the genes that are associated with cancer in humans [139]. This poses challenges in determining both the function of such genes and the molecular mechanisms that they might affect. Another major limitation is the difference in homeostatic temperatures between fish cell lines (usually grown at 28 °C) and mammalian cancer cell lines (37 °C). As a way to bridge this gap in physiological growth conditions, after xenograft implantation, zebrafish embryos can be grown at higher temperatures (32–35 °C). This allows the mammalian cells to grow and develop in an environment that is more similar to their natural state and has not been found to affect normal zebrafish development [120]. The benefits and limitations of the zebrafish xenograph model have been extensively analyzed in a recent review by Drabsch et al. [140].
In conclusion, although the zebrafish embryo xenograft model of cancer is not yet as well established as mouse models, it provides scientists with a rapid, cost-effective way to investigate tumor formation, microenvironment interactions, and new drug therapies, and it is an attractive option for testing personalized medicine. These benefits are reflected in the increasing number of cancer research publications that use the zebrafish as a model system (Fig. 2). Future studies of prostate cancer cell subpopulations using the zebrafish model are likely to give us valuable insights into prostate tumor development and metastatic behavior and will add to our understanding of the molecular mechanisms of SOCE.

Evolution of cancer publications using the zebrafish model. The data were obtained from a PubMed search for “cancer + zebrafish”
Conclusion
In the last decade, it has become increasingly clear that while SOCE signals impact the fates of prostate cancer cells, the underlying expression patterns and molecular mechanisms are complex and are not yet fully understood. Only a fraction of primary tumor cells are capable of recapitulating the neoplastic and malignant phenotype at a distant site, thus generating metastases. Therefore, selecting this subpopulation of cells from patient specimens and establishing cell lines that recapitulate the metastatic phenotype are promising as novel approaches to elucidate SOCE signaling in preclinical in vitro models. Cancer research models like the zebrafish model will add important insights in the in vivo setting. Additional studies are needed to investigate the dysregulation of ion channel signaling in more differentiated pathophysiological conditions using these novel cancer model systems.
Change history
20 June 2018
The article Store-Operated Ca2+ Entry as a Prostate Cancer Biomarker — a Riddle with Perspectives, written by Sven Kappel, Ines Joao Marques, Eugenio Zoni, Paulina Stokłosa, Christine Peinelt, Nadia Mercader, Marianna Kruithof-de Julio, and Anna Borgström, was originally published electronically.
20 June 2018
The article Store-Operated Ca2+ Entry as a Prostate Cancer Biomarker — a Riddle with Perspectives, written by Sven Kappel, Ines Joao Marques, Eugenio Zoni, Paulina Stokłosa, Christine Peinelt, Nadia Mercader, Marianna Kruithof-de Julio, and Anna Borgström, was originally published electronically.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65.
Collins SR, Meyer T. Calcium flickers lighting the way in chemotaxis? Dev Cell. 2009;16:160–1.
Parekh AB, Muallem S. Ca(2+) signalling and gene regulation. Cell Calcium. 2011;49:279.
De Smedt H, Verkhratsky A, Muallem S. Ca(2+) signaling mechanisms of cell survival and cell death: an introduction. Cell Calcium. 2011;50:207–10.
Wei C, Wang X, Zheng M, Cheng H. Calcium gradients underlying cell migration. Curr Opin Cell Biol. 2012;24:254–61.
Pinto MC, Kihara AH, Goulart VA, Tonelli FM, Gomes KN, Ulrich H, et al. Calcium signaling and cell proliferation. Cell Signal. 2015;27:2139–49.
Tsai FC, Kuo GH, Chang SW, Tsai PJ. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int. 2015;2015:409245.
Iamshanova O, Fiorio Pla A, Prevarskaya N. Molecular mechanisms of tumour invasion: regulation by calcium signals. J Physiol. 2017;595:3063–75.
Muik M, Schindl R, Fahrner M, Romanin C. Ca(2+) release-activated Ca(2+) (CRAC) current, structure, and function. Cell Mol Life Sci. 2012;69(24):4163–76.
Hogan PG, Rao A. Store-operated calcium entry: mechanisms and modulation. Biochem Biophys Res Commun. 2015;460:40–9.
Rosado JA. Calcium entry pathways in non-excitable cells. Preface. Adv Exp Med Biol. 2016;898:vii–viii.
Jardin I, Rosado JA. STIM and calcium channel complexes in cancer. Biochim Biophys Acta. 2016;1863:1418–26.
Villalobos C, Sobradillo D, Hernandez-Morales M, Nunez L. Remodeling of calcium entry pathways in cancer. Adv Exp Med Biol. 2016;898:449–66.
Chen YF, Hsu KF, Shen MR. The store-operated Ca(2+) entry-mediated signaling is important for cancer spread. Biochim Biophys Acta. 2016;1863:1427–35.
Frischauf I, Zayats V, Deix M, Hochreiter A, Jardin I, Muik M, et al. A calcium-accumulating region, CAR, in the channel Orai1 enhances Ca(2+) permeation and SOCE-induced gene transcription. Sci Signal. 2015;8:ra131.
Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, et al. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465:1249–60.
Sobradillo D, Hernandez-Morales M, Ubierna D, Moyer MP, Nunez L, Villalobos C. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J Biol Chem. 2014;289:28765–82.
Diez-Bello R, Jardin I, Salido GM, Rosado JA. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim Biophys Acta. 2016;1864(6):1064–70.
Stanisz H, Stark A, Kilch T, Schwarz EC, Muller CS, Peinelt C, et al. ORAI1 Ca(2+) channels control endothelin-1-induced mitogenesis and melanogenesis in primary human melanocytes. J Invest Dermatol. 2012;132:1443–51.
Bogeski I, Kilch T, Niemeyer BA. ROS and SOCE: recent advances and controversies in the regulation of STIM and Orai. J Physiol. 2012;590:4193–200.
Stanisz H, Saul S, Muller CS, Kappl R, Niemeyer BA, Vogt T, et al. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014;27:442–53.
Stanisz H, Vultur A, Herlyn M, Roesch A, Bogeski I. The role of Orai-STIM calcium channels in melanocytes and melanoma. J Physiol. 2016;594:2825–35.
Liu G, Honisch S, Liu G, Schmidt S, Alkahtani S, AlKahtane AA, et al. Up-regulation of Orai1 expression and store operated Ca(2+) entry following activation of membrane androgen receptors in MCF-7 breast tumor cells. BMC Cancer. 2015;15:995.
Ay AS, Benzerdjeb N, Sevestre H, Ahidouch A, Ouadid-Ahidouch H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS One. 2013;8:e72889.
Benzerdjeb N, Sevestre H, Ahidouch A, Ouadid-Ahidouch H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget. 2016;7:81588–97.
Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285:19173–83.
Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226:542–51.
Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, et al. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013;27:63–75.
Holzmann C, Kilch T, Kappel S, Armbruster A, Jung V, Stockle M, et al. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget. 2013;4:2096–107.
Dubois C, Vanden Abeele F, Lehen'kyi V, Gkika D, Guarmit B, Lepage G, et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell. 2014;26:19–32.
Walsh CM, Doherty MK, Tepikin AV, Burgoyne RD. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem J. 2010;430:453–60.
Palty R, Raveh A, Kaminsky I, Meller R, Reuveny E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell. 2012;149:425–38.
Carreras-Sureda A, Cantero-Recasens G, Rubio-Moscardo F, Kiefer K, Peinelt C, Niemeyer BA, et al. ORMDL3 modulates store-operated calcium entry and lymphocyte activation. Hum Mol Genet. 2013;22:519–30.
Miao Y, Miner C, Zhang L, Hanson PI, Dani A, Vig M. An essential and NSF independent role for alpha-SNAP in store-operated calcium entry. Elife. 2013;2:e00802.
Sharma S, Quintana A, Findlay GM, Mettlen M, Baust B, Jain M, et al. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature. 2013;499:238–42.
Quintana A, Rajanikanth V, Farber-Katz S, Gudlur A, Zhang C, Jing J, et al. TMEM110 regulates the maintenance and remodeling of mammalian ER-plasma membrane junctions competent for STIM-ORAI signaling. Proc Natl Acad Sci U S A. 2015;112:E7083–92.
Wilson LA, McKeown L, Tumova S, Li J, Beech DJ. Expression of a long variant of CRACR2A that belongs to the Rab GTPase protein family in endothelial cells. Biochem Biophys Res Commun. 2015;456:398–402.
Jing J, He L, Sun A, Quintana A, Ding Y, Ma G, et al. Proteomic mapping of ER-PM junctions identifies STIMATE as a regulator of Ca(2)(+) influx. Nat Cell Biol. 2015;17:1339–47.
Lopez JJ, Albarran L, Gomez LJ, Smani T, Salido GM, Rosado JA. Molecular modulators of store-operated calcium entry. Biochim Biophys Acta. 2016;1863:2037–43.
Li P, Miao Y, Dani A, Vig M. alpha-SNAP regulates dynamic, on-site assembly and calcium selectivity of Orai1 channels. Mol Biol Cell. 2016;27:2542–53.
Albarran L, Lopez JJ, Amor NB, Martin-Cano FE, Berna-Erro A, Smani T, et al. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci Rep. 2016;6:24452.
Albarran L, Regodon S, Salido GM, Lopez JJ, Rosado JA. Role of STIM1 in the surface expression of SARAF. Channels (Austin). 2017;11:84–8.
Glitsch M. Protons and Ca2+: ionic allies in tumor progression? Physiology (Bethesda). 2011;26:252–65.
Beck A, Fleig A, Penner R, Peinelt C. Regulation of endogenous and heterologous Ca(2+) release-activated Ca(2+) currents by pH. Cell Calcium. 2014;56:235–43.
Tsujikawa H, Yu AS, Xie J, Yue Z, Yang W, He Y, et al. Identification of key amino acid residues responsible for internal and external pH sensitivity of Orai1/STIM1 channels. Sci Rep. 2015;5:16747.
Lee HJ, Bae GU, Leem YE, Choi HK, Kang TM, Cho H, et al. Phosphorylation of Stim1 at serine 575 via netrin-2/Cdo-activated ERK1/2 is critical for the promyogenic function of Stim1. Mol Biol Cell. 2012;23:1376–87.
Smyth JT, Beg AM, Wu S, Putney JW Jr, Rusan NM. Phosphoregulation of STIM1 leads to exclusion of the endoplasmic reticulum from the mitotic spindle. Curr Biol. 2012;22:1487–93.
Putney JW. Alternative forms of the store-operated calcium entry mediators, STIM1 and Orai1. Curr Top Membr. 2013;71:109–23.
Dorr K, Kilch T, Kappel S, Alansary D, Schwar G, Niemeyer BA, et al. Cell type-specific glycosylation of Orai1 modulates store-operated Ca2+ entry. Sci Signal. 2016;9:ra25.
Yazbeck P, Tauseef M, Kruse K, Amin MR, Sheikh R, Feske S, et al. STIM1 phosphorylation at Y361 recruits Orai1 to STIM1 puncta and induces Ca2+ entry. Sci Rep. 2017;7:42758.
Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, Kinet JP, et al. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium. 2007;41:51–61.
Shimizu T, Owsianik G, Freichel M, Flockerzi V, Nilius B, Vennekens R. TRPM4 regulates migration of mast cells in mice. Cell Calcium. 2009;45:226–32.
Arredouani MS, Lu B, Bhasin M, Eljanne M, Yue W, Mosquera JM, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin Cancer Res. 2009;15:5794–802.
Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406.
Luo JH, Yu YP, Cieply K, Lin F, Deflavia P, Dhir R, et al. Gene expression analysis of prostate cancers. Mol Carcinog. 2002;33:25–35.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–82.
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43.
Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51.
Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–36.
Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22:2790–9.
LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–506.
Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–8.
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9.
Liu P, Ramachandran S, Ali Seyed M, Scharer CD, Laycock N, Dalton WB, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66:4011–9.
Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80.
Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, et al. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001;61:5692–6.
Perrouin Verbe MA, Bruyere F, Rozet F, Vandier C, Fromont G. Expression of store-operated channel components in prostate cancer: the prognostic paradox. Hum Pathol. 2016;49:77–82.
Derouiche S, Warnier M, Mariot P, Gosset P, Mauroy B, Bonnal JL, et al. Bisphenol A stimulates human prostate cancer cell migration via remodelling of calcium signalling. Springerplus. 2013;2:54.
Flourakis M, Lehen'kyi V, Beck B, Raphael M, Vandenberghe M, Abeele FV, et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010;1:e75.
Berry PA, Birnie R, Droop AP, Maitland NJ, Collins AT. The calcium sensor STIM1 is regulated by androgens in prostate stromal cells. Prostate. 2011;71:1646–55.
Hariri W, Sudha T, Bharali DJ, Cui H, Mousa SA. Nano-targeted delivery of toremifene, an estrogen receptor-alpha blocker in prostate cancer. Pharm Res. 2015;32:2764–74.
Thompson JL, Mignen O, Shuttleworth TJ. The ARC channel—an endogenous store-independent Orai channel. Curr Top Membr. 2013;71:125–48.
Shuttleworth TJ. Orai3—the ‘exceptional’ Orai? J Physiol. 2012;590:241–57.
Bogeski I, Al-Ansary D, Qu B, Niemeyer BA, Hoth M, Peinelt C. Pharmacology of ORAI channels as a tool to understand their physiological functions. Expert Rev Clin Pharmacol. 2010;3:291–303.
Holzmann C, Kilch T, Kappel S, Dorr K, Jung V, Stockle M, et al. Differential redox regulation of Ca(2+) signaling and viability in normal and malignant prostate cells. Biophys J. 2015;109:1410–9.
Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal. 2010;3:ra24.
Alansary D, Bogeski I, Niemeyer BA (2015) Facilitation of Orai3 targeting and store-operated function by Orai1. BBA accepted for publication
Saul S, Gibhardt CS, Schmidt B, Lis A, Pasieka B, Conrad D, et al. A calcium-redox feedback loop controls human monocyte immune responses: the role of ORAI Ca2+ channels. Sci Signal. 2016;9:ra26.
Zuccolo E, Bottino C, Diofano F, Poletto V, Codazzi AC, Mannarino S, et al. Constitutive store-operated Ca(2+) entry leads to enhanced nitric oxide production and proliferation in infantile hemangioma-derived endothelial colony-forming cells. Stem Cells Dev. 2016;25:301–19.
Bhardwaj R, Hediger MA, Demaurex N. Redox modulation of STIM-ORAI signaling. Cell Calcium. 2016;60:142–52.
Barbieri CE, Chinnaiyan AM, Lerner SP, Swanton C, Rubin MA. The emergence of precision urologic oncology: a collaborative review on biomarker-driven therapeutics. Eur Urol. 2017;71:237–46.
Sarmiento D, Montorfano I, Cerda O, Caceres M, Becerra A, Cabello-Verrugio C, et al. Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc Res. 2015;98:187–96.
Barbet G, Demion M, Moura IC, Serafini N, Leger T, Vrtovsnik F, et al. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol. 2008;9:1148–56.
Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109:397–407.
Vennekens R, Olausson J, Meissner M, Bloch W, Mathar I, Philipp SE, et al. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat Immunol. 2007;8:312–20.
Weber KS, Hildner K, Murphy KM, Allen PM. Trpm4 differentially regulates Th1 and Th2 function by altering calcium signaling and NFAT localization. J Immunol. 2010;185:2836–46.
Makar TK, Gerzanich V, Nimmagadda VK, Jain R, Lam K, Mubariz F, et al. Silencing of Abcc8 or inhibition of newly upregulated Sur1-Trpm4 reduce inflammation and disease progression in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2015;12:210.
Kruse M, Schulze-Bahr E, Corfield V, Beckmann A, Stallmeyer B, Kurtbay G, et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J Clin Invest. 2009;119:2737–44.
Liu H, El Zein L, Kruse M, Guinamard R, Beckmann A, Bozio A, et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ Cardiovasc Genet. 2010;3:374–85.
Mathar I, Kecskes M, Van der Mieren G, Jacobs G, Camacho Londono JE, Uhl S, et al. Increased beta-adrenergic inotropy in ventricular myocardium from Trpm4-/- mice. Circ Res. 2014;114:283–94.
Mathar I, Vennekens R, Meissner M, Kees F, Van der Mieren G, Camacho Londono JE, et al. Increased catecholamine secretion contributes to hypertension in TRPM4-deficient mice. J Clin Invest. 2010;120:3267–79.
Mathar I, Jacobs G, Kecskes M, Menigoz A, Philippaert K, Vennekens R. Trpm4. Handb Exp Pharmacol. 2014;222:461–87.
Liu H, Chatel S, Simard C, Syam N, Salle L, Probst V, et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One. 2013;8:e54131.
Stallmeyer B, Zumhagen S, Denjoy I, Duthoit G, Hebert JL, Ferrer X, et al. Mutational spectrum in the Ca(2+)-activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum Mutat. 2012;33:109–17.
Grand T, Demion M, Norez C, Mettey Y, Launay P, Becq F, et al. 9-phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br J Pharmacol. 2008;153:1697–705.
Abriel H, Syam N, Sottas V, Amarouch MY, Rougier JS. TRPM4 channels in the cardiovascular system: physiology, pathophysiology, and pharmacology. Biochem Pharmacol. 2012;84:873–81.
Kecskes M, Jacobs G, Kerselaers S, Syam N, Menigoz A, Vangheluwe P, et al. The Ca(2+)-activated cation channel TRPM4 is a negative regulator of angiotensin II-induced cardiac hypertrophy. Basic Res Cardiol. 2015;110:43.
Jacobs G, Oosterlinck W, Dresselaers T, Geenens R, Kerselaers S, Himmelreich U, et al. Enhanced beta-adrenergic cardiac reserve in Trpm4(-)/(-) mice with ischaemic heart failure. Cardiovasc Res. 2015;105:330–9.
Piao H, Takahashi K, Yamaguchi Y, Wang C, Liu K, Naruse K. Transient receptor potential melastatin-4 is involved in hypoxia-reoxygenation injury in the cardiomyocytes. PLoS One. 2015;10:e0121703.
Armisen R, Marcelain K, Simon F, Tapia JC, Toro J, Quest AF, et al. TRPM4 enhances cell proliferation through up-regulation of the beta-catenin signaling pathway. J Cell Physiol. 2011;226:103–9.
Loo SK, Ch'ng ES, Md Salleh MS, Banham AH, Pedersen LM, Moller MB, et al. TRPM4 expression is associated with activated B-cell subtype and poor survival in diffuse large B-cell lymphoma. Histopathology. 2017;71:98–111.
• Schinke EN, Bii V, Nalla A, Rae DT, Tedrick L, Meadows GG, et al. A novel approach to identify driver genes involved in androgen-independent prostate cancer. Mol Cancer. 2014;13:120. This paper identifies TRPM4 as cancer driver gene.
• Berg KD, Soldini D, Jung M, Dietrich D, Stephan C, Jung K, et al. TRPM4 protein expression in prostate cancer: a novel tissue biomarker associated with risk of biochemical recurrence following radical prostatectomy. Virchows Arch. 2015;468:345–55. This paper demonstrates overexpression of TRPM4 in prostate cancer tissue.
• Holzmann C, Kappel S, Kilch T, Jochum MM, Urban SK, Jung V, et al. Transient receptor potential melastatin 4 channel contributes to migration of androgen-insensitive prostate cancer cells. Oncotarget. 2015;6:41783–93. This paper demonstrates a role for TRPM4 in prostate cancer cell migration.
Kilch T, Kappel S, Peinelt C. Regulation of Ca signaling in prostate cancer cells. Channels (Austin). 2016;10:170–1.
Sagredo AI, Sagredo EA, Cappelli C, Báez P, Rodrigo AM, Blanco C, et al. TRPM4 regulates Akt/GSK3-β activity and enhances β-catenin signaling and cell proliferation in prostate cancer cells. Mol Oncol. 2017. https://doi.org/10.1002/1878-0261.12100.
Vashisht A, Trebak M, Motiani RK. STIM and Orai proteins as novel targets for cancer therapy. A review in the theme: cell and molecular processes in cancer metastasis. Am J Physiol Cell Physiol. 2015;309:C457–69.
Moccia F, Zuccolo E, Poletto V, Turin I, Guerra G, Pedrazzoli P, et al. Targeting stim and Orai proteins as an alternative approach in anticancer therapy. Curr Med Chem. 2016;23:3450–80.
Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab Invest. 2010;90:234–44.
•• van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, Guzman-Ramirez N, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010;70:5163–73. This paper shows that ALDH expression levels can be used to identify MICs and TICs and analyses different in vivo mouse models.
Zoni E, Chen L, Karkampouna S, Granchi Z, Verhoef EI, La Manna F, et al. CRIPTO and its signaling partner GRP78 drive the metastatic phenotype in human osteotropic prostate cancer. Oncogene. 2017;36:4739–49.
Prevarskaya N, Skryma R, Bidaux G, Flourakis M, Shuba Y. Ion channels in death and differentiation of prostate cancer cells. Cell Death Differ. 2007;14:1295–304.
Dai J, Hensel J, Wang N, Kruithof-de Julio M, Shiozawa Y. Mouse models for studying prostate cancer bone metastasis. Bonekey Rep. 2016;5:777.
Ozdemir BC, Hensel J, Secondini C, Wetterwald A, Schwaninger R, Fleischmann A, et al. The molecular signature of the stroma response in prostate cancer-induced osteoblastic bone metastasis highlights expansion of hematopoietic and prostate epithelial stem cell niches. PLoS One. 2014;9:e114530.
Stanton MF. Diethylnitrosamine-induced hepatic degeneration and neoplasia in the aquarium fish, Brachydanio Rerio. J Natl Cancer Inst. 1965;34:117–30.
Beckwith LG, Moore JL, Tsao-Wu GS, Harshbarger JC, Cheng KC. Ethylnitrosourea induces neoplasia in zebrafish (Danio rerio). Lab Invest. 2000;80:379–85.
Langenau DM, Jette C, Berghmans S, Palomero T, Kanki JP, Kutok JL, et al. Suppression of apoptosis by bcl-2 overexpression in lymphoid cells of transgenic zebrafish. Blood. 2005;105:3278–85.
Greene HS. Heterologous transplantation of human and other mammalian tumors. Science. 1938;88:357–8.
Haldi M, Ton C, Seng WL, McGrath P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis. 2006;9:139–51.
Nicoli S, Ribatti D, Cotelli F, Presta M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007;67:2927–31.
Marques IJ, Weiss FU, Vlecken DH, Nitsche C, Bakkers J, Lagendijk AK, et al. Metastatic behaviour of primary human tumours in a zebrafish xenotransplantation model. BMC Cancer. 2009;9:128.
Pruvot B, Jacquel A, Droin N, Auberger P, Bouscary D, Tamburini J, et al. Leukemic cell xenograft in zebrafish embryo for investigating drug efficacy. Haematologica. 2011;96:612–6.
•• Bansal N, Davis S, Tereshchenko I, Budak-Alpdogan T, Zhong H, Stein MN, et al. Enrichment of human prostate cancer cells with tumor initiating properties in mouse and zebrafish xenografts by differential adhesion. Prostate. 2014;74:187–200. This paper describes zebrafish prostate cancer model.
Weiss FU, Marques IJ, Woltering JM, Vlecken DH, Aghdassi A, Partecke LI, et al. Retinoic acid receptor antagonists inhibit miR-10a expression and block metastatic behavior of pancreatic cancer. Gastroenterology. 2009;137:2136–45.e1-7.
Corkery DP, Dellaire G, Berman JN. Leukaemia xenotransplantation in zebrafish—chemotherapy response assay in vivo. Br J Haematol. 2011;153:786–9.
Ott I, Qian X, Xu Y, Vlecken DH, Marques IJ, Kubutat D, et al. A gold(I) phosphine complex containing a naphthalimide ligand functions as a TrxR inhibiting antiproliferative agent and angiogenesis inhibitor. J Med Chem. 2009;52:763–70.
Murphy AG, Casey R, Maguire A, Tosetto M, Butler CT, Conroy E, et al. Preclinical validation of the small molecule drug quininib as a novel therapeutic for colorectal cancer. Sci Rep. 2016;6:34523.
Jung DW, Oh ES, Park SH, Chang YT, Kim CH, Choi SY, et al. A novel zebrafish human tumor xenograft model validated for anti-cancer drug screening. Mol Biosyst. 2012;8:1930–9.
White RM, Sessa A, Burke C, Bowman T, LeBlanc J, Ceol C, et al. Transparent adult zebrafish as a tool for in vivo transplantation analysis. Cell Stem Cell. 2008;2:183–9.
Karlsson J, von Hofsten J, Olsson PE. Generating transparent zebrafish: a refined method to improve detection of gene expression during embryonic development. Mar Biotechnol (NY). 2001;3:522–7.
Krishnamurthy K, Wang G, Rokhfeld D, Bieberich E. Deoxycholate promotes survival of breast cancer cells by reducing the level of pro-apoptotic ceramide. Breast Cancer Res. 2008;10:R106.
Ignatius MS, Langenau DM. Fluorescent imaging of cancer in zebrafish. Methods Cell Biol. 2011;105:437–59.
Renshaw SA, Trede NS. A model 450 million years in the making: zebrafish and vertebrate immunity. Dis Model Mech. 2012;5:38–47.
Baltrunaite K, Craig MP, Palencia Desai S, Chaturvedi P, Pandey RN, Hegde RS, et al. ETS transcription factors Etv2 and Fli1b are required for tumor angiogenesis. Angiogenesis. 2017;20:307–23.
Ramachandran R, Krishnaraj C, Sivakumar AS, Prasannakumar P, Abhay Kumar VK, Shim KS, et al. Anticancer activity of biologically synthesized silver and gold nanoparticles on mouse myoblast cancer cells and their toxicity against embryonic zebrafish. Mater Sci Eng C Mater Biol Appl. 2017;73:674–83.
Chen L, Groenewoud A, Tulotta C, Zoni E, Kruithof-de Julio M, van der Horst G, et al. A zebrafish xenograft model for studying human cancer stem cells in distant metastasis and therapy response. Methods Cell Biol. 2017;138:471–96.
Fior R, Póvoa V, Mendes RV, Carvalho T, Gomes A, Figueiredo N, Ferreira MG. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc Natl Acad Sci U. S. A. 2017;114(39):E8234–E8243. https://doi.org/10.1073/pnas.1618389114.
Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503.
Drabsch Y, Snaar-Jagalska BE, Ten Dijke P. Fish tales: the use of zebrafish xenograft human cancer cell models. Histol Histopathol. 2016;32:673–86.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sven Kappel, Ines Joao Marques, Eugenio Zoni, Paulina Stokłosa, Christine Peinelt, Nadia Mercader, Marianna Kruithof-de Julio, and Anna Borgström each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article contains no studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Biology of Prostate Cancer
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Kappel, S., Marques, I.J., Zoni, E. et al. Store-Operated Ca2+ Entry as a Prostate Cancer Biomarker — a Riddle with Perspectives. Curr Mol Bio Rep 3, 208–217 (2017). https://doi.org/10.1007/s40610-017-0072-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-017-0072-8