
Abstract
Purpose of Review
Inhaled environmental exposures cause over 12 million deaths per year worldwide. Despite localized efforts to reduce environmental exposures, tobacco smoking and air pollution remain the urgent public health challenges that are contributing to the growing prevalence of respiratory diseases. The purpose of this review is to describe the mechanisms through which inhaled environmental exposures accelerate lung aging and cause overt lung disease.
Recent Findings
Environmental exposures related to fossil fuel and tobacco combustion and occupational exposures related to silica and coal mining generate oxidative stress and inflammation in the lungs. Sustained oxidative stress causes DNA damage, epigenetic instability, mitochondrial dysfunction, and cell cycle arrest in key progenitor cells in the lung. As a result, critical repair mechanisms are impaired, leading to premature destruction of the lung parenchyma.
Summary
Inhaled environmental exposures accelerate lung aging by injuring the lungs and damaging the cells responsible for wound healing. Interventions that minimize exposure to noxious antigens are critical to improve lung health, and novel research is required to expand our knowledge of therapies that may slow or prevent premature lung aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The human lung is continuously exposed to inhaled agents and pathogens from the external environment. A combination of individual genetics and environmental exposures influence lung aging, which manifests as structural remodeling of the respiratory tract that generates declining lung function over time [1]. During normal breathing, the trachea conducts air through the bronchi, which divide into bronchioles and end in clusters of alveoli. The alveoli are lined by an epithelial layer and basement membrane that lay adjacent to a thin interstitial space, under which lies the pulmonary capillary network [2]. The interstitial space contains lung extracellular matrix, which consists of elastic and collagen fibers that maintain the structural integrity of the lung [3]. Age-related weakening of the connective tissue in the lung generates progressive dilation of the airspaces and early collapse of the small airways [4]. The surface area of lung available for gas exchange thereby decreases with age, leading to reduced oxygenation and capacity for exercise [5]. Structural changes are even more pronounced in age-related respiratory diseases including chronic obstructive pulmonary disease (COPD), which is characterized by mucus hypersecretion and alveolar wall destruction, [6] and idiopathic pulmonary fibrosis (IPF), which is defined by interstitial fibrosis [7].
Age-related structural alterations in the lung are driven in part by inhaled exposures that damage the lung epithelium and underlying tissue [8]. Efficient repair mechanisms are critical to maintain the structural integrity of the lung and prevent pathological remodeling. Specialized type II alveolar epithelial cells contribute to wound healing by generating new type I epithelial cells after injury, which cover most of the alveolar surface [9]. Wound healing is also stimulated by multipotent mesenchymal stem cells that populate subepithelial lung tissue and differentiate into epithelial cells, macrophages, and reparative fibroblasts [10, 11]. Pulmonary fibroblasts reside in the interstitial space and mend the lung extracellular matrix in order to re-establish and maintain alveolar architecture [3]. However, when lung progenitor cells and cellular repair mechanisms are inhibited, aberrant structural remodeling distorts the lung architecture and leads to premature lung function impairment [1].
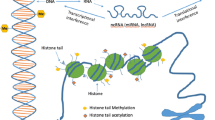
Compromised cellular repair mechanisms are one of the hallmarks of lung aging (Figure 1). Inhaled exposures diminish the lung’s regenerative potential by generating oxidative stress, DNA damage, epigenetic instability, telomere attrition, mitochondrial injury, and abnormal protein homeostasis in key progenitor and structural cells [12••]. Accumulated damage in mesenchymal stem cells leads to apoptosis and stem cell depletion, while repeated insults in type II alveolar epithelial cells and lung fibroblasts lead to cellular senescence [13]. Cellular senescence is characterized by arrested growth and diminished cellular function, and cell-specific senescence can generate different forms of lung degeneration [14]. For example, senescent alveolar epithelial cells are unable to induce re-epithelialization after lung injury, while senescent fibroblasts produce aberrant collagen in the lung extracellular matrix [15, 16]. Cumulative inhalational exposures over the lifespan introduce accumulating inflammatory-oxidative stress and act in concert to induce widespread pulmonary cellular senescence and premature lung aging [17, 18]. This review provides an overview of environmental exposures that can impact lung health and details the mechanisms and biological pathways through which environmental exposures accelerate lung aging.

Mechanisms of lung aging induced by environmental exposures141. Figure 1 enumerates the biological pathways through which environmental exposures accelerate lung aging. Inhalation of noxious antigens induces oxidative stress, inflammation, telomere shortening, DNA damage, mitochondrial dysfunction, epigenetic instability, immune dysregulation, and impaired proteostasis in multiple cell lines in the lung. As a result, pulmonary stem cells are depleted, key progenitor cells become senescent, and critical repair mechanisms are impaired, leading to premature lung aging
Environmental Exposures and Biological Impact on the Lung
Tobacco Smoke
Tobacco smoking is a combustion process that leads to the aerosolization of thousands of toxic chemicals including carbon monoxide, hydrogen cyanide, and polycyclic aromatic hydrocarbons [19]. Many of the components in tobacco smoke chemically react with oxygen to generate free radicals and inhibit protective antioxidants [20]. Through the combustion of noxious chemicals and generation of harmful reactive oxygen species (ROS), tobacco smoke induces widespread tissue damage in a manner that mimics biological aging (Table 1) [21].
At the molecular level, tobacco smoke alters DNA methylation levels in lung cells and circulating leukocytes [22, 23]. Smoking-related oxidative stress generates DNA demethylation, and nicotine downregulates DNA methyltransferase enzymes that transfer methyl groups to cytosine-phosphate-guanine (CpG) sites [24, 25]. Accordingly, prior epidemiological studies have demonstrated marked differences in total DNA methylation among smokers compared to never smokers [26]. Changes in DNA methylation in gene-coding sequences can alter gene expression and may link tobacco smoke exposure to smoking-related diseases [22]. For example, tobacco smoke has been shown in mouse models to increase methylation of the Bcl-2 promoter [27••]. Bcl-2 promoter methylation lowers Bcl-2 expression, which leads to apoptosis of cells in the alveolar walls and generates premature emphysema [27••]. Similarly, human studies have shown that smoking reduces DNA methylation at the aryl hydrocarbon receptor repressor (AHRR) gene in blood and lung tissue [28,29,30]. Smoking-induced reductions in AHRR methylation inhibit expression of detoxifying enzymes that remove harmful environmental chemicals including hydrocarbons contained in tobacco smoke [31]. Thus, smoking-induced AHRR demethylation may represent a mechanism of premature lung disease in smokers [32].
Recent studies have shown that DNA methylation levels are also a robust biomarker of biological aging [33]. The “epigenetic clock” effectively predicts biological age in all tissues based upon DNA methylation levels at a collection of CpG sites [34]. The epigenetic clock also quantifies age acceleration, which is a metric of premature aging defined as the difference between the chronological age and the biological or DNA methylation age. Tobacco smoking has been shown in human studies to accelerate DNA methylation age in airway cells and lung tissue, likely by modulating expression of genes that regulate cellular senescence and cell survival [35,36,37,38] Accordingly, in two independent population-based studies, accelerated epigenetic aging was associated with incident COPD, which is characterized by premature pulmonary senescence [39, 40]. Specifically, the odds of incident COPD increased by 1–2% per 5-year elevation in epigenetic aging, suggesting that accelerated epigenetic aging is an independent risk factor for age-related lung diseases.
Closely tied to age- and smoking-related epigenetic changes are alterations in cellular protein homeostasis. In natural aging, abnormal proteostasis leads to the accumulation of toxic misfolded protein aggregates [41]. In vitro studies have shown that tobacco smoking also precipitates impaired protein folding in the endoplasmic reticulum (ER) of human lung cells [42•]. The ER regulates protein folding and degradation and activates a stress response when misfolded proteins accumulate in the ER lumen. Smoking additionally inhibits lysosomal-mediated degradation of dysfunctional proteins, which allows abnormal proteins to accumulate in the perinuclear space [43]. Accumulation of dysfunctional proteins generates lung inflammation and dilation of the airways, which are principal components of premature emphysema [44].
In addition to stimulating inflammation in the lung, animal models have demonstrated that smoking-induced protein misfolding inhibits production of functional surfactant proteins [42•]. Pulmonary surfactant is a lipoprotein fluid secreted by type II alveolar cells that decreases surface tension in the alveoli and prevents lung collapse during normal breathing [45]. Smoking-induced inhibition of surfactant protein production induces apoptosis of airway epithelial cells [42]. While epithelial damage typically triggers airway repair mechanisms, tobacco smoke also disrupts airway progenitor cells, impeding alveolar re-epithelialization after injury [46]. Persistent epithelial damage generates alveolar wall destruction and pathological airway remodeling, which are integral components of premature lung aging [47].
Prior studies have shown that tobacco smoking induces premature cellular senescence in lung epithelial cells and basal progenitor cells [15, 48]. Smoking reduces telomere length in both cell populations in vitro, leading to cell cycle arrest and cellular dysfunction [49]. As a result, disordered cell differentiation and impaired epithelial remodeling limit effective cellular repair mechanisms. Tobacco smoke also induces cellular senescence in lung fibroblasts, which are critical for maintaining normal lung architecture [50]. Inhibited wound healing generates architectural disturbances in the alveoli and airway epithelium, which precipitates premature lung disease.
In addition to inducing cellular senescence in airway fibroblasts and progenitor cells, tobacco smoking stimulates an airway epithelial-mesenchymal transition [51]. Human studies comparing smokers to non-smokers demonstrated that some airway epithelial cells undergo biochemical transformations and adopt a mesenchymal cell phenotype following smoking-induced epithelial damage [52]. Transformed mesenchymal cells secrete components of the extracellular matrix and contribute to lung regeneration and healing. However, in the setting of prolonged inflammation and recurrent smoking-related injury, the transformed cells produce a disorganized extracellular matrix and generate fibrosis [53]. Interstitial fibrosis can lead to IPF, which is an irreversible aging-associated lung disease [40].
While a subset of epithelial cells adopt a mesenchymal cell phenotype in response to tobacco smoke exposure, other epithelial cells transform into mucus-secreting cells [54••]. In vitro studies have shown that smoking-related inflammation alters energy production and protein translation in exposed epithelial cells, inducing widespread mucus cell metaplasia. The resulting increase in mucus production contributes to airflow obstruction and premature functional impairment and is a defining feature of many inflammatory lung diseases [55].
Mucus hypersecretion facilitates bacterial colonization of the airways, which leads to recurrent airway infections when coupled with the immunosuppressing effect of chronic smoke exposure [56]. Tobacco smoking suppresses macrophage phagocytosis of bacteria in the lungs and impairs maturation of pulmonary dendritic cells, which are critical activators of the adaptive immune system [57, 58]. Tobacco smoke also inhibits B and T cell immune responses and is associated with decreased immunoglobulin production in animal models [59•]. Dysregulated immunity in the lung can lead to repeated and prolonged respiratory tract infections, which can induce structural changes in the lung and generate premature lung aging [60].
Particulate Matter
Particulate matter (PM) is a mixture of aerosolized microscopic particles that can be inhaled into the lungs [61]. PM is primarily generated by emissions from motor vehicles and industrial facilities, and fine PM (diameter < 2.5 μm [PM2.5]) is small enough to traverse the tracheobronchial tree and deposit in the small airways [62]. PM2.5 contains transition metals and organic aerosols that generate ROS and inhibit antioxidant enzyme activity in the lung [63]. As a result, oxidative stress builds in the respiratory tract and accelerates lung aging through multiple pathways (Table 2) [64].
PM-induced oxidative stress modulates enzymes that regulate DNA methylation, leading to differential methylation at thousands of CpG sites in the lungs and circulating leukocytes [65, 66]. DNA methylation regulates gene expression and may provide a link between particulate air pollution exposure and premature lung aging [67••]. For example, PM2.5 exposure has been shown in population-based studies to alter DNA methylation in the interleukin-6 (IL-6) and tissue factor (F3) genes, suggesting PM2.5-induced changes in DNA methylation upregulate expression of pro-inflammatory cytokines and acute phase reactants [68]. A corresponding mediation analysis suggested gene-specific methylation mediated the relationship between air pollution and inflammatory biomarkers in plasma. Accordingly, in vitro studies have shown that PM2.5 exposure triggers increased epithelial cell production of inflammatory cytokines (IL-6, IL-1β, tumor necrosis factor alpha [TNF-α]) and chemotactic molecules (IL-8, monocyte chemoattractant protein 1 [MCP1]). The resulting pro-inflammatory milieu triggers architectural changes that are characteristic of premature lung aging. PM2.5-induced upregulation of IL-1β stimulates mucus hypersecretion in the airway epithelial cells, which generates airflow obstruction [69]. Pro-inflammatory cytokines also stimulate infiltration of neutrophils and macrophages in the lung, which release proteases that degrade the airway epithelial barrier [70, 71]. PM2.5 exposure has also been independently associated with AHRR demethylation, [72] suggesting that PM and tobacco smoke may have shared mechanisms of DNA demethylation and may accelerate lung aging through similar biological pathways [73].
The airway epithelial barrier provides both physical and immunological protection against inhaled foreign antigens, and damage to the barrier drives pathogenesis of age-related lung diseases [74]. In addition to generating inflammatory injury to the epithelial barrier, PM2.5-induced ROS generate DNA strand breaks and suppress DNA repair enzymes in airway epithelial cells [75, 76]. DNA damage stimulates mitochondrial dysfunction, which can induce epithelial cell death [77]. In vitro studies have demonstrated that oxidative stress also damages the proteins that form the tight junctions in the epithelial cell barrier, thereby diminishing the barrier function of the airway epithelial layer [78, 79]. In response, alveolar progenitor cells are activated and recruited to repair the injured alveolar barrier [80]. However, when epithelial repair mechanisms are impaired, pathological airway remodeling ensues.
Prior in vitro studies have shown that inhaled PM decreases the viability of epithelial progenitor cells. PM2.5 exposure decreases expression of human telomerase reverse transcriptase in lung epithelial cells [81]. As a result, epithelial cell telomeres are shortened and cell cycle arrest ensues [82••]. Cellular senescence in type II alveolar epithelial cells limits the regenerative capacity of the lung epithelium, leading to impaired wound healing and increased inflammation [83]. Unrepaired epithelial injury also induces proliferation of lung fibroblasts [84, 85]. Activated fibroblasts increase collagen deposition, and excessive production of extracellular matrix leads to aberrant alveolar remodeling and fibrosis [86]. Accordingly, epidemiological studies have shown that PM exposure is a known risk factor for IPF, which is a progressive age-related lung disease [87].
Ground-Level Ozone
Tropospheric ozone forms when emissions from industrial plants and motor vehicles chemically react in the presence of UV light [88]. Ozone is not filtered by the upper airways, which allows inhaled ozone to deposit in the lower respiratory tract [89]. When ozone comes into contact with lung epithelial cells in vitro, it alters expression of tight junction proteins in the epithelial barrier [90••]. Disintegration of tight junctions increases permeability of the epithelial barrier, which stimulates release of inflammatory cytokines and ROS [91]. In turn, ozone-induced ROS generate mitochondrial dysfunction [92•]. Acute ozone exposure diminishes mitochondrial energy storage and decreases mitochondrial oxygen consumption in the lung, leading to release of mitochondrial ROS. Mitochondrial ROS activate the NLRP3 inflammasome, which is a protein complex that induces inflammation-mediated cell death and generates alveolar wall destruction [93]. Accordingly, in prior epidemiological studies, chronic ozone exposure was associated with lung function impairment and emphysema independent of smoking [94••]. Long-term ozone exposure was also associated with shortness of breath and impaired functional status resulting from respiratory symptoms, suggesting ambient air pollution may contribute significantly to respiratory symptoms that characterize premature lung aging.
Sulfur Dioxide
Fossil fuel combustion generates sulfur dioxide, which is a toxic gas that contributes heavily to air pollution in industrialized countries [95]. Human studies have demonstrated that sulfur dioxide converts to sulfuric acid after inhalation and increases bronchial reactivity and bronchoconstriction, which are hallmarks of reactive airway disease and asthma [96]. Sulfur dioxide also decreases mucociliary clearance, which increases the viscosity of airway mucus and promotes pathogen colonization and reproduction [97]. Pathogen colonization in the airways promotes infiltration by immune and inflammatory cells, which in turn promotes airway remodeling and premature lung aging [98].
Nitrogen Dioxide
Fossil fuel combustion generates nitrogen dioxide, which is a primary source of urban air pollution [99]. Nitrogen dioxide is a water-soluble gas that deposits in the small airways where it is converted to nitrous and nitric acids [100••]. Nitric acids directly damage airway epithelial cells, leading to a chemical pneumonitis that manifests as pulmonary edema [101]. Nitrogen dioxide exposure also suppresses alveolar macrophage-mediated production of inflammatory cytokines in response to bacterial infection, which dampens the immune response. The pollutant further diminishes respiratory immunity by reducing mucociliary clearance in the lower respiratory tract of animal models, leading to impaired clearance of respiratory pathogens [102]. Immune dysregulation in the lungs increases susceptibility to respiratory infections, which can lead to cellular and structural damage in the airways [103].
Silica Dust
Silica is a common mineral that is a large component of granite and sandstone rocks [104]. Silica exposure can occur after any activity that requires breaking ground or handling silica-containing stone. Inhaled crystalline silica particles deposit in the distal airways of the lungs and are phagocytosed by resident macrophages, which release ROS and inflammatory cytokines [105]. Animal models have demonstrated that silica-induced ROS disrupt a telomere-binding protein complex that preserves telomere length in progenitor lung cells [106]. The resulting telomere attrition generates DNA damage in type II alveolar epithelial cells, leading to cellular senescence and apoptosis. Increased alveolar cell loss triggers aberrant healing mechanisms in lung fibroblasts, which proliferate and increase collagen production around silica particles [107]. The resulting interstitial fibrosis leads to decreased lung compliance and impaired gas exchange, which are hallmarks of premature lung aging.
Coal Dust
Coal mining, transport, and processing generate airborne respirable dust that can deposit in the small airways of the lungs [108]. Coal dust cannot be eliminated from the lungs but rather is engulfed by macrophages that reside in the alveolar space. Activated macrophages release TNF-α and IL-6 in vitro, which stimulate infiltration of neutrophils and lymphocytes in the lungs. Neutrophils secrete elastases that break down elastic fibers, leading to dilation and destruction of the alveolar walls [109]. Alveolar macrophages also secrete insulin-like growth factor-1 (IGF-1) and platelet-derived growth factor (PDGF), which recruit fibroblasts to sites of coal dust deposition and stimulate fibroblast proliferation [110] Activated fibroblasts upregulate collagen production, leading to collagen accumulation in pneumoconiotic lesions in the lungs. Areas of focal emphysema and pneumoconiotic nodules distort the lung architecture and generate premature lung function impairment.
Asbestos
Asbestos fibers are durable minerals that were historically used in construction and insulation because of their resilience and affordability [111]. Deteriorating buildings and asbestos-containing products can generate airborne asbestos fibers that deposit in the distal airways [112]. Alveolar macrophages are too small to completely engulf larger asbestos fibers, and incomplete phagocytosis generates biomineralization and formation of iron-rich envelopes around asbestos fibers [111]. Animal models have shown that asbestos-iron complexes trigger mitochondrial ROS production in alveolar epithelial cells, mesothelial cells, and macrophages [113]. Persistent oxidative stress induces mitochondrial DNA damage, triggering apoptosis of mesothelial cells and type II alveolar epithelial cells. Asbestos exposure also activates p53 expression in key progenitor cells, leading to cellular senescence in alveolar epithelial and mesothelial cells [114]. Cellular senescence in progenitor cells impairs physiologic repair mechanisms and triggers an exaggerated fibroblast response characterized by pathological collagen deposition. Proliferating fibroblasts generate interstitial fibrosis, which is a hallmark of asbestosis and premature lung aging [115]. Notably, tobacco smoking hinders clearance of asbestos bodies from the human lung, leading to exaggerated pulmonary toxicity in smokers with asbestos exposures [116].
Bioaerosols
Indoor air contains ubiquitous biological contaminants including bacteria, viruses, and fungi [117]. Indoor dust also contains bacterial extracellular vesicles (EVs), which are membrane-bound nanoparticles produced from Gram-negative bacteria that can reach and accumulate in the lung alveoli [118]. Inhalation of bacterial EVs activates the innate immune response and triggers production of inflammatory cytokines including TNF-α and IL-6 in vitro [119]. Cytokine release triggers an influx of neutrophils in the lung, which release protease and elastase enzymes that destroy structural components of the alveolar septa [109]. Repeated exposure to bacterial EVs alters the lung architecture by damaging alveolar walls and generating alveolar enlargement, as well as by increasing collagen deposition in the airways. The prolonged inflammation that follows EV exposure can generate mucus gland hyperplasia and emphysema and indicates that dust EVs may contribute significantly to premature lung aging [120••].
Strategies for Improving Environmental and Respiratory Health
Environmental exposures accelerate lung aging and contribute to the development of age-related respiratory diseases (Figure 2). Inhaled antigens generate oxidative stress and inflammation that lead to destruction and fibrosis of the lung parenchyma, which are irreversible distortions of the lung architecture [121]. Thus, evaluation and minimization of noxious environmental exposures are critical to improve lung health and prevent age-related lung diseases.

Environmental exposures and associated age-related changes in the lung142. Environmental exposures impair cellular repair mechanisms in the lung, leading to structural alterations including emphysema, mucus hypersecretion, and pulmonary fibrosis that generate functional impairment and characterize premature lung aging
Several strategies can help reduce exposure to ambient air pollution. Portable or centralized air filters reduce concentrations of indoor air pollutants, which reflect a combination of pollutants from cooking and organic dust as well as outdoor source pollutants that infiltrate indoors [122, 123]. Clinical trials have shown that high efficiency particulate air (HEPA) in-duct filtration systems reduce indoor particulate concentrations and improve microvascular health, which is a determinant of lung health [124, 125]. Similarly, individuals who commute via personal vehicles or public transportation can reduce in-vehicle particulate exposure through use of cabin filters and air conditioners to stimulate air recirculation [126].
Avoiding rigorous outdoor activity when air pollutant concentrations are high can also help minimize harmful exposures. Air pollution monitoring networks such as the Environmental Protection Agency’s Air Quality Index measure concentrations of harmful air pollutants and forecast air pollutant levels using mathematical models [127]. When air pollution levels are elevated, minimizing outdoor activity and reducing rigorous outdoor exercise can decrease inhalation of harmful pollutants [128, 129]. Reducing exposure to high-pollution microenvironments can also reduce noxious exposures. For example, avoiding physical activity near high-traffic roads can help optimize respiratory health [130]. Similarly, smoking cessation and complete removal of secondhand smoke exposure are essential to preserve lung health among smokers and their family members. There are multiple proven interventions to aid with smoking cessation and removal of residual smoke pollution [131].
With regard to occupational exposures including silica dust, coal dust, and asbestos, minimizing dust exposure is the most effective prevention strategy. While engineering controls including dust extraction systems are the primary method for reducing exposures, individual prevention strategies including wearing a mask and removing dust from skin and clothing can help reduce harmful exposures [132]. Secondary prevention strategies including monitoring for lung function impairment and early radiographic signs of disease in high-risk individuals can help prevent severe disease, as miners with radiographic evidence of lung disease are legally entitled to work with enhanced protections against dust exposure [133].
In addition to minimizing inhaled environmental exposures, primary prevention strategies in the form of influenza, pneumococcal, and Covid-19 vaccinations can minimize risk of respiratory infections and promote lung health among individuals with high-risk environmental exposures [134]. Individuals over the age of 65, and particularly those with chronic medical conditions, are recommended to stay up to date on vaccinations in order to prevent severe respiratory infections.
Prior research has suggested that pharmacological and dietary interventions may reduce susceptibility to inhaled exposures. An epidemiological study showed that nonsteroidal anti-inflammatory drugs (NSAIDs) partially attenuated lung function decline after PM exposure, [135] and a small trial demonstrated that NSAID use attenuated lung function decline in adults exposed to ozone [136]. Similarly, B vitamin supplementation prevented PM2.5-induced alterations in DNA methylation levels in a small trial of adults, suggesting B vitamins can neutralize PM-related oxidative stress [137]. Dietary antioxidants including vitamin C also preserved lung function in a study of young adults exposed to ozone [138]. Finally, stem cell therapies are emerging as potential mitigators of environmental pollutants, and transplantation of adipose-derived stem cells attenuated PM2.5-induced lung inflammation in mice [139]. However, while preliminary studies are promising, further research is required before pharmaceutical, biological, and lifestyle interventions can be safely recommended to minimize or reverse damage from inhaled environmental exposures.
Conclusions
The lungs are one of few organs in the body that continuously interface with the external environment. Environmental exposures trigger oxidative and inflammatory stress that damage the lung parenchyma, impair physiologic repair mechanisms, and induce accelerated lung aging. Premature lung aging manifests as lung function impairment and overt lung disease and causes significant global morbidity and mortality. While individual interventions that minimize noxious environmental exposures and reduce risk of respiratory infections can optimize lung health, cohesive national and international policies that minimize carbon emissions are required to decrease the global burden of inhaled environmental exposures. In addition, novel research is required to expand our knowledge of therapeutic interventions that may slow or prevent premature lung aging. Future research in the following areas may expand available strategies to prevent age-related lung diseases:
-
Human trials examining pharmacologic interventions (i.e., NSAIDs) are required to determine if medical interventions protect the lungs from environmental pollutants.
-
Larger human trials are required to determine whether dietary interventions including antioxidant supplements (i.e., vitamins B, C, and E) can mitigate the impact of inhaled exposures on lung health.
-
While stem cell therapies represent an emerging and promising field, additional studies in animal models and eventually humans are required to validate the effectiveness of stem cells in preserving lung health.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Brandenberger C, Muhlfeld C. Mechanisms of lung aging. Cell Tissue Res. 2017;367(3):469–80.
Suki B, Stamenovic D, Hubmayr R. Lung parenchymal mechanics. Compr Physiol. 2011;1(3):1317–51.
White ES. Lung extracellular matrix and fibroblast function. Ann Am Thorac Soc. 2015;12(Suppl 1):S30–3.
Gillooly M, Lamb D. Airspace size in lungs of lifelong non-smokers: effect of age and sex. Thorax. 1993;48(1):39–43.
McClaran SR, Babcock MA, Pegelow DF, Reddan WG, Dempsey JA. Longitudinal effects of aging on lung function at rest and exercise in healthy active fit elderly adults. J Appl Physiol (1985). 1995;78(5):1957-68.
Rennard SI. COPD: overview of definitions, epidemiology, and factors influencing its development. Chest. 1998;113(4 Suppl):235S–41S.
Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013;41(5):1207–18.
Cooper DM, Loxham M. Particulate matter and the airway epithelium: the special case of the underground? Eur Respir Rev 2019;28(153):190066.
Jacob A, Vedaie M, Roberts DA, Thomas DC, Villacorta-Martin C, Alysandratos KD, et al. Derivation of self-renewing lung alveolar epithelial type II cells from human pluripotent stem cells. Nat Protoc. 2019;14(12):3303–32.
Spees JL, Pociask DA, Sullivan DE, Whitney MJ, Lasky JA, Prockop DJ, et al. Engraftment of bone marrow progenitor cells in a rat model of asbestos-induced pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(4):385–94.
Fang S, Zhang S, Dai H, Hu X, Li C, Xing Y. The role of pulmonary mesenchymal cells in airway epithelium regeneration during injury repair. Stem Cell Res Ther. 2019;10(1):366.
•• Peters A, Nawrot TS, Baccarelli AA. Hallmarks of environmental insults. Cell. 2021;184(6):1455-68. This manuscript outlines mechanisms through which common environmental exposures damage bodily tissues and cause premature death and disease.
Mora AL, Rojas M. Adult stem cells for chronic lung diseases. Respirology. 2013;18(7):1041–6.
Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705.
Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2004;31(6):643–9.
• Lin Y, Xu Z. Fibroblast senescence in idiopathic pulmonary fibrosis. Front Cell Dev Biol. 2020;8:593283. This manuscript highlights pathways that generate lung fibroblast senescence and describes mechanisms that connect fibroblast senescence to pulmonary fibrosis.
•• Vij N, Chandramani-Shivalingappa P, Van Westphal C, Hole R, Bodas M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am J Physiol Cell Physiol. 2018;314(1):C73-C87. This study demonstrates the impact of tobacco smoke exposure on autophagy and connects impaired autophagy to premature emphysema and lung aging.
Vrijheid M. The exposome: a new paradigm to study the impact of environment on health. Thorax. 2014;69(9):876–8.
Kapp R. Encyclopedia of Toxicology (Second Edition). In: Wexler P, editor. Tobacco Smoke. London: Elsevier; 2005. p. 200–2.
Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6(2):445–62.
Choukrallah MA, Hoeng J, Peitsch MC, Martin F. Lung transcriptomic clock predicts premature aging in cigarette smoke-exposed mice. BMC Genomics. 2020;21(1):291.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–47.
• Ringh MV, Hagemann-Jensen M, Needhamsen M, Kular L, Breeze CE, Sjoholm LK, et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine. 2019;46:290-304. This study demonstrates that tobacco smoke exposure alters DNA methylation levels in lung cells.
Satta R, Maloku E, Zhubi A, Pibiri F, Hajos M, Costa E, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. Proc Natl Acad Sci U S A. 2008;105(42):16356–61.
Zhou X, Zhuang Z, Wang W, He L, Wu H, Cao Y, et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell Signal. 2016;28(9):1163–71.
Philibert RA, Sears RA, Powers LS, Nash E, Bair T, Gerke AK, et al. Coordinated DNA methylation and gene expression changes in smoker alveolar macrophages: specific effects on VEGF receptor 1 expression. J Leukoc Biol. 2012;92(3):621–31.
•• Zeng H, Li T, He X, Cai S, Luo H, Chen P, et al. Oxidative stress mediates the apoptosis and epigenetic modification of the Bcl-2 promoter via DNMT1 in a cigarette smoke-induced emphysema model. Respir Res. 2020;21(1):229. This study connects smoking-induced changes in DNA methylation to premature emphysema.
Guida F, Sandanger TM, Castagne R, Campanella G, Polidoro S, Palli D, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet. 2015;24(8):2349–59.
Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7(11):1331–8.
Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22(5):843–51.
Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478(7368):197–203.
Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115.
Hsu CC, Leu YW, Tseng MJ, Lee KD, Kuo TY, Yen JY, et al. Functional characterization of Trip10 in cancer cell growth and survival. J Biomed Sci. 2011;18:12.
McLoughlin KJ, Pedrini E, MacMahon M, Guduric-Fuchs J, Medina RJ. Selection of a real-time PCR housekeeping gene panel in human endothelial colony forming cells for cellular senescence studies. Front Med (Lausanne). 2019;6:33.
Donlon TA, Morris BJ, Chen R, Masaki KH, Allsopp RC, Willcox DC, et al. Analysis of polymorphisms in 59 potential candidate genes for association with human longevity. J Gerontol A Biol Sci Med Sci. 2018;73(11):1459–64.
•• Wu X, Huang Q, Javed R, Zhong J, Gao H, Liang H. Effect of tobacco smoking on the epigenetic age of human respiratory organs. Clin Epigenetics. 2019;11(1):183. This study demonstrates that tobacco smoking accelerates the epigenetic aging of lung tissue.
•• Breen M, Nwanaji-Enwerem JC, Karrasch S, Flexeder C, Schulz H, Waldenberger M, et al. Accelerated epigenetic aging as a risk factor for chronic obstructive pulmonary disease and decreased lung function in two prospective cohort studies. Aging (Albany NY). 2020;12(16):16539-54. This study demonstrates that accelerated epigenetic aging is associated with increased risk of incident COPD.
Chilosi M, Carloni A, Rossi A, Poletti V. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013;162(3):156–73.
Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421–35.
• Bradley KL, Stokes CA, Marciniak SJ, Parker LC, Condliffe AM. Role of unfolded proteins in lung disease. Thorax. 2021;76(1):92-9. This manuscript outlines how environmental toxins induce ER stress and impaired protein folding, which describes how impaired proteostasis contributes to the development of respiratory diseases.
Bodas M, Vij N. Augmenting autophagy for prognosis based intervention of COPD-pathophysiology. Respir Res. 2017;18(1):83.
Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047–82.
Han S, Mallampalli RK. The role of surfactant in lung disease and host defense against pulmonary infections. Ann Am Thorac Soc. 2015;12(5):765–74.
Deeb RS, Walters MS, Strulovici-Barel Y, Chen Q, Gross SS, Crystal RG. Smoking-associated disordering of the airway basal stem/progenitor cell metabotype. Am J Respir Cell Mol Biol. 2016;54(2):231–40.
Hadzic S, Wu CY, Avdeev S, Weissmann N, Schermuly RT, Kosanovic D. Lung epithelium damage in COPD - an unstoppable pathological event? Cell Signal. 2020;68:109540.
Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3(9-10):545–56.
Walters MS, De BP, Salit J, Buro-Auriemma LJ, Wilson T, Rogalski AM, et al. Smoking accelerates aging of the small airway epithelium. Respir Res. 2014;15:94.
Nyunoya T, Monick MM, Klingelhutz AL, Glaser H, Cagley JR, Brown CO, et al. Cigarette smoke induces cellular senescence via Werner's syndrome protein down-regulation. Am J Respir Crit Care Med. 2009;179(4):279–87.
Kalluri RW, RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Milara JPT, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–20.
Shaykhiev RZWL, Chao I, Fukui T, Witover B, Brekman A, Crystal RG. EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype. Proc Natl Acad Sci U S A. 2013;110:12102–7.
•• Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, Rios CL, et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat Commun. 2020;11(1):2485. This study defines how tobacco smoking impacts lung epithelial cells and induces transformation into pathological cells that contribute to the development of inflammatory lung diseases.
Reynolds CJ, Quigley K, Cheng X, Suresh A, Tahir S, Ahmed-Jushuf F, et al. Lung defense through IL-8 carries a cost of chronic lung remodeling and impaired function. Am J Respir Cell Mol Biol. 2018;59(5):557–71.
Shen Y, Huang S, Kang J, Lin J, Lai K, Sun Y, et al. Management of airway mucus hypersecretion in chronic airway inflammatory disease: Chinese expert consensus (English edition). Int J Chron Obstruct Pulmon Dis. 2018;13:399–407.
Givi ME, Folkerts G, Wagenaar GT, Redegeld FA, Mortaz E. Cigarette smoke differentially modulates dendritic cell maturation and function in time. Respir Res. 2015;16:131.
O'Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette smoking impairs human pulmonary immunity to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2014;190(12):1430–6.
• Bhat TA, Kalathil SG, Bogner PN, Miller A, Lehmann PV, Thatcher TH, et al. Secondhand smoke induces inflammation and impairs immunity to respiratory infections. J Immunol. 2018;200(8):2927-40. This study describes how chronic tobacco smoke exposure induces inflammation and impaired immunity in the lungs.
Jiang C, Chen Q, Xie M. Smoking increases the risk of infectious diseases: a narrative review. Tob Induc Dis. 2020;18:60.
Nogueira JB. Air pollution and cardiovascular disease. Rev Port Cardiol. 2009;28(6):715–33.
Kodros JK, Volckens J, Jathar SH, Pierce JR. Ambient particulate matter size distributions drive regional and global variability in particle deposition in the respiratory tract. Geohealth. 2018;2(10):298–312.
de Oliveira AAF, de Oliveira TF, Dias MF, Medeiros MHG, Di Mascio P, Veras M, et al. Genotoxic and epigenotoxic effects in mice exposed to concentrated ambient fine particulate matter (PM2.5) from Sao Paulo city, Brazil. Part Fibre Toxicol. 2018;15(1):40.
Lin CI, Tsai CH, Sun YL, Hsieh WY, Lin YC, Chen CY, et al. Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice. Int J Biol Sci. 2018;14(3):253–65.
Mostafavi N, Vermeulen R, Ghantous A, Hoek G, Probst-Hensch N, Herceg Z, et al. Acute changes in DNA methylation in relation to 24h personal air pollution exposure measurements: a panel study in four European countries. Environ Int. 2018;120:11–21.
Somineni HK, Zhang X, Biagini Myers JM, Kovacic MB, Ulm A, Jurcak N, et al. Ten-eleven translocation 1 (TET1) methylation is associated with childhood asthma and traffic-related air pollution. J Allergy Clin Immunol. 2016;137(3):797–805 e5.
•• Huang SK, Tripathi P, Koneva LA, Cavalcante RG, Craig N, Scruggs AM, et al. Effect of concentration and duration of particulate matter exposure on the transcriptome and DNA methylome of bronchial epithelial cells. Environ Epigenet. 2021;7(1):dvaa022. This study demonstrates how particulate matter exposure induces changes in DNA methylation in lung epithelial cells
Bind MA, Lepeule J, Zanobetti A, Gasparrini A, Baccarelli A, Coull BA, et al. Air pollution and gene-specific methylation in the Normative Aging Study: association, effect modification, and mediation analysis. Epigenetics. 2014;9(3):448–58.
Wang J, Zhu M, Wang L, Chen C, Song Y. Amphiregulin potentiates airway inflammation and mucus hypersecretion induced by urban particulate matter via the EGFR-PI3Kalpha-AKT/ERK pathway. Cell Signal. 2019;53:122–31.
Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol. 2010;298(6):L715–31.
Manicone AM. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev Clin Immunol. 2009;5(1):63–75.
Tantoh DM, Lee KJ, Nfor ON, Liaw YC, Lin C, Chu HW, et al. Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations. Clin Epigenetics. 2019;11(1):69.
Tantoh DM, Wu MC, Chuang CC, Chen PH, Tyan YS, Nfor ON, et al. AHRR cg05575921 methylation in relation to smoking and PM2.5 exposure among Taiwanese men and women. Clin Epigenetics. 2020;12(1):117.
Watson JK, Sanders P, Dunmore R, Rosignoli G, Jule Y, Rawlins EL, et al. Distal lung epithelial progenitor cell function declines with age. Sci Rep. 2020;10(1):10490.
Prahalad AK, Inmon J, Dailey LA, Madden MC, Ghio AJ, Gallagher JE. Air pollution particles mediated oxidative DNA base damage in a cell free system and in human airway epithelial cells in relation to particulate metal content and bioreactivity. Chem Res Toxicol. 2001;14(7):879–87.
Meng Z, Zhang Q. Damage effects of dust storm PM2.5 on DNA in alveolar macrophages and lung cells of rats. Food Chem Toxicol. 2007;45(8):1368–74.
Upadhyay D, Panduri V, Ghio A, Kamp DW. Particulate matter induces alveolar epithelial cell DNA damage and apoptosis: role of free radicals and the mitochondria. Am J Respir Cell Mol Biol. 2003;29(2):180–7.
• Zhao R, Guo Z, Zhang R, Deng C, Xu J, Dong W, et al. Nasal epithelial barrier disruption by particulate matter </=2.5 mum via tight junction protein degradation. J Appl Toxicol. 2018;38(5):678-87. This study demonstrates how particulate matter exposure disrupts tight junction proteins in the respiratory epithelial barrier.
Yan J, Lai CH, Lung SC, Chen C, Wang WC, Huang PI, et al. Industrial PM2.5 cause pulmonary adverse effect through RhoA/ROCK pathway. Sci Total Environ. 2017;599-600:1658-66.
Olajuyin AM, Zhang X, Ji HL. Alveolar type 2 progenitor cells for lung injury repair. Cell Death Discov. 2019;5:63.
Leao R, Apolonio JD, Lee D, Figueiredo A, Tabori U, Castelo-Branco P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: clinical impacts in cancer. J Biomed Sci. 2018;25(1):22.
•• Chang-Chien J, Huang JL, Tsai HJ, Wang SL, Kuo ML, Yao TC. Particulate matter causes telomere shortening and increase in cellular senescence markers in human lung epithelial cells. Ecotoxicol Environ Saf. 2021;222:112484. This study demonstrates how particulate matter exposure generates telomere attrition and cellular senescence in lung epithelial cells.
Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174(8):886–93.
Bove H, Devoght J, Rasking L, Peters M, Slenders E, Roeffaers M, et al. Combustion-derived particles inhibit in vitro human lung fibroblast-mediated matrix remodeling. J Nanobiotechnology. 2018;16(1):82.
Alley D, Langley-Turnbaugh S, Gordon N, Wise J, Van Epps G, Jalbert A. The effect of PM10 on human lung fibroblasts. Toxicol Ind Health. 2009;25(2):111–20.
Pardo A, Selman M. Lung fibroblasts, aging, and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2016;13(Suppl 5):S417–S21.
Makela K, Ollila H, Sutinen E, Vuorinen V, Peltola E, Kaarteenaho R, et al. Inorganic particulate matter in the lung tissue of idiopathic pulmonary fibrosis patients reflects population density and fine particle levels. Ann Diagn Pathol. 2019;40:136–42.
Guenther A, Geron C, Pierce T, Lamb B, Harley P, Fall R. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmospheric Environment. 2000;34(12-14):2205–30.
Zhang JJ, Wei Y, Fang Z. Ozone pollution: a major health hazard worldwide. Front Immunol. 2019;10:2518.
•• Kim BG, Lee PH, Lee SH, Park CS, Jang AS. Impact of ozone on claudins and tight junctions in the lungs. Environ Toxicol. 2018;33(7):798-806. Thus study elucidates mechanisms through which ozone exposure damages tight junctions in the airway epithelial barrier.
Michaudel C, Fauconnier L, Jule Y, Ryffel B. Functional and morphological differences of the lung upon acute and chronic ozone exposure in mice. Sci Rep. 2018;8(1):10611.
• Wiegman CH, Li F, Ryffel B, Togbe D, Chung KF. Oxidative stress in ozone-induced chronic lung inflammation and emphysema: a facet of chronic obstructive pulmonary disease. Front Immunol. 2020;11:1957. This manuscript outlines mechanisms through which ozone exposure damages the lungs and accelerates lung aging.
Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128.
•• Paulin LM, Gassett AJ, Alexis NE, Kirwa K, Kanner RE, Peters S, et al. Association of long-term ambient ozone exposure with respiratory morbidity in smokers. JAMA Intern Med. 2020;180(1):106-15. This study elucidated the association between long-term ozone exposure and adverse respiratory outcomes.
National Research Council (US) Committee on Toxicology. Emergency and Continuous Exposure Limits for Selected Airborne Contaminants: Volume 2. Washington (DC): National Academies Press (US); 1984. SULFUR DIOXIDE.
Utell MJ, Morrow PE, Hyde RW. Airway reactivity to sulfate and sulfuric acid aerosols in normal and asthmatic subjects. J Air Pollut Control Assoc. 1984;34(9):931–5.
Lin VY, Kaza N, Birket SE, Kim H, Edwards LJ, LaFontaine J, et al. Excess mucus viscosity and airway dehydration impact COPD airway clearance. Eur Respir J. 2020;55(1):1900419.
Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–8.
Ji X, Meng X, Liu C, Chen R, Ge Y, Kan L, et al. Nitrogen dioxide air pollution and preterm birth in Shanghai. China. Environ Res. 2019;169:79–85.
•• Zhang Z, Wang J, Lu W. Exposure to nitrogen dioxide and chronic obstructive pulmonary disease (COPD) in adults: a systematic review and meta-analysis. Environ Sci Pollut Res Int. 2018;25(15):15133-45. This review evaluates and establishes the association between nitrogen dioxide and incident COPD.
Amaducci A, Downs JW. Nitrogen Dioxide Toxicity. 2021 Jul 27. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021.
Cao Y, Chen M, Dong D, Xie S, Liu M. Environmental pollutants damage airway epithelial cell cilia: Implications for the prevention of obstructive lung diseases. Thorac Cancer. 2020;11(3):505–10.
Globinska A, Kowalski ML. Innate lymphoid cells: the role in respiratory infections and lung tissue damage. Expert Rev Clin Immunol. 2017;13(10):991–9.
Lapp NL. Lung disease secondary to inhalation of nonfibrous minerals. Clin Chest Med. 1981;2(2):219–33.
Leso V, Fontana L, Romano R, Gervetti P, Iavicoli I. Artificial stone associated silicosis: a systematic review. Int J Environ Res Public Health. 2019;16(4):568.
Shoeb M, Mustafa GM, Joseph P, Umbright C, Kodali V, Roach KA, et al. Initiation of pulmonary fibrosis after silica inhalation in rats is linked with dysfunctional shelterin complex and DNA damage response. Sci Rep. 2019;9(1):471.
Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015;12(2):286–99.
Perret JL, Plush B, Lachapelle P, Hinks TS, Walter C, Clarke P, et al. Coal mine dust lung disease in the modern era. Respirology. 2017;22(4):662–70.
Demkow U, van Overveld FJ. Role of elastases in the pathogenesis of chronic obstructive pulmonary disease: implications for treatment. Eur J Med Res. 2010;15(Suppl 2):27–35.
Vanhee D, Gosset P, Boitelle A, Wallaert B, Tonnel AB. Cytokines and cytokine network in silicosis and coal workers' pneumoconiosis. Eur Respir J. 1995;8(5):834–42.
Bardelli F, Brun F, De Panfilis S, Cloetens P, Capella S, Belluso E, et al. Chemo-physical properties of asbestos bodies in human lung tissues studied at the nano-scale by non-invasive, label free x-ray imaging and spectroscopic techniques. Toxicol Lett. 2021;348:18–27.
Giacobbe C, Di Giuseppe D, Zoboli A, Lassinantti Gualtieri M, Bonasoni P, Moliterni A, et al. Crystal structure determination of a lifelong biopersistent asbestos fibre using single-crystal synchrotron X-ray micro-diffraction. IUCrJ. 2021;8(Pt 1):76–86.
Liu G, Beri R, Mueller A, Kamp DW. Molecular mechanisms of asbestos-induced lung epithelial cell apoptosis. Chem Biol Interact. 2010;188(2):309–18.
Kopnin PB, Kravchenko IV, Furalyov VA, Pylev LN, Kopnin BP. Cell type-specific effects of asbestos on intracellular ROS levels, DNA oxidation and G1 cell cycle checkpoint. Oncogene. 2004;23(54):8834–40.
Kim SJ, Cheresh P, Jablonski RP, Rachek L, Yeldandi A, Piseaux-Aillon R, et al. Mitochondrial 8-oxoguanine DNA glycosylase mitigates alveolar epithelial cell PINK1 deficiency, mitochondrial DNA damage, apoptosis, and lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2020;318(5):L1084–L96.
Churg A, Stevens B. Enhanced retention of asbestos fibers in the airways of human smokers. Am J Respir Crit Care Med. 1995;151(5):1409–13.
Ferguson RMW, Garcia-Alcega S, Coulon F, Dumbrell AJ, Whitby C, Colbeck I. Bioaerosol biomonitoring: sampling optimization for molecular microbial ecology. Mol Ecol Resour. 2019;19(3):672–90.
Lee EY, Bang JY, Park GW, Choi DS, Kang JS, Kim HJ, et al. Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics. 2007;7(17):3143–53.
Kim YLW, Choi E, Choi J, Heo Y, Gho Y, Jee Y, Oh Y, Kim Y. Extracellular vesicles derived from gram-negative bacteria, such as Escherichia coli, induce emphysema mainly via IL-17A–mediated neutrophilic inflammation. Journal of Immunology. 2015;194(7):3361–8.
•• Meganathan V, Moyana R, Natarajan K, Kujur W, Kusampudi S, Mulik S, et al. Bacterial extracellular vesicles isolated from organic dust induce neutrophilic inflammation in the lung. Am J Physiol Lung Cell Mol Physiol. 2020;319(6):L893-L907. This study demonstrates that inhalation of bacterial extracellular vesicles generates inflammatory damage in the lungs.
Wang X, Zhao Y, Li D, Feng Y, Xie Y, Zhou Y, et al. Intrapulmonary distal airway stem cell transplantation repairs lung injury in chronic obstructive pulmonary disease. Cell Prolif. 2021;54(6):e13046.
Macintosh DL, Myatt TA, Ludwig JF, Baker BJ, Suh HH, Spengler JD. Whole house particle removal and clean air delivery rates for in-duct and portable ventilation systems. J Air Waste Manag Assoc. 2008;58(11):1474–82.
Macintosh DL, Minegishi T, Kaufman M, Baker BJ, Allen JG, Levy JI, et al. The benefits of whole-house in-duct air cleaning in reducing exposures to fine particulate matter of outdoor origin: a modeling analysis. J Expo Sci Environ Epidemiol. 2010;20(2):213–24.
Brauner EV, Forchhammer L, Moller P, Barregard L, Gunnarsen L, Afshari A, et al. Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med. 2008;177(4):419–25.
Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104(22):2673–8.
Xu BZ, Y. Investigation on lowering commuters’ in-cabin exposure to ultrafine particles. Transportation Research. 2013;18:122–30.
Laumbach R, Meng Q, Kipen H. What can individuals do to reduce personal health risks from air pollution? J Thorac Dis. 2015;7(1):96–107.
Daigle CC, Chalupa DC, Gibb FR, Morrow PE, Oberdorster G, Utell MJ, et al. Ultrafine particle deposition in humans during rest and exercise. Inhal Toxicol. 2003;15(6):539–52.
•• Carlsten C, Salvi S, Wong GWK, Chung KF. Personal strategies to minimise effects of air pollution on respiratory health: advice for providers, patients and the public. Eur Respir J. 2020;55(6). This manuscript provides personal-level recommendations for minimizing air pollution exposures.
Burns J, Boogaard H, Polus S, Pfadenhauer LM, Rohwer AC, van Erp AM, et al. Interventions to reduce ambient particulate matter air pollution and their effect on health. Cochrane Database Syst Rev. 2019;5(5):CD010919.
Guo K, Li J, Li J, Chen N, Li Y, Yang K, et al. The effects of pharmacological interventions on smoking cessation in people with alcohol dependence: a systematic review and meta-analysis of nine randomized controlled trials. Int J Clin Pract. 2021;6:e14594.
Cullinan P, Munoz X, Suojalehto H, Agius R, Jindal S, Sigsgaard T, et al. Occupational lung diseases: from old and novel exposures to effective preventive strategies. Lancet Respir Med. 2017;5(5):445–55.
Hall NB, Blackley DJ, Halldin CN, Laney AS. Current review of pneumoconiosis among US coal miners. Curr Environ Health Rep. 2019.
Bekkat-Berkani R, Wilkinson T, Buchy P, Dos Santos G, Stefanidis D, Devaster JM, et al. Seasonal influenza vaccination in patients with COPD: a systematic literature review. BMC Pulm Med. 2017;17(1):79.
Gao X, Coull B, Lin X, Vokonas P, Schwartz J, Baccarelli AA. Nonsteroidal antiinflammatory drugs modify the effect of short-term air pollution on lung function. Am J Respir Crit Care Med. 2020;201(3):374–8.
Alexis N, Urch B, Tarlo S, Corey P, Pengelly D, O'Byrne P, et al. Cyclooxygenase metabolites play a different role in ozone-induced pulmonary function decline in asthmatics compared to normals. Inhal Toxicol. 2000;12(12):1205–24.
Zhong J, Karlsson O, Wang G, Li J, Guo Y, Lin X, et al. B vitamins attenuate the epigenetic effects of ambient fine particles in a pilot human intervention trial. Proc Natl Acad Sci U S A. 2017;114(13):3503–8.
Samet JM, Hatch GE, Horstman D, Steck-Scott S, Arab L, Bromberg PA, et al. Effect of antioxidant supplementation on ozone-induced lung injury in human subjects. Am J Respir Crit Care Med. 2001;164(5):819–25.
Gao J, Yuan J, Liu Q, Wang Y, Wang H, Chen Y, et al. Adipose-derived stem cells therapy effectively attenuates PM2.5-induced lung injury. Stem Cell Res Ther. 2021;12(1):355.
Lakey PS, Berkemeier T, Tong H, Arangio AM, Lucas K, Poschl U, et al. Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci Rep. 2016;6:32916.
Adapted from "Spheroidal cell (apoptotic blebbing)", "Immune cells (inflammation", "Chromosome (with 2 telomeres)", "Histone modifications (acetylation and DNA methylation", "Bacillus (with antibodies)", "LGI1", "Mitochondria (2D, bursting, dashed)", "DNA (double-stranded break)", "Mesenchymal stem cell", "Mitosis (3. Metaphase), "Pseudostratified epithelium (damaged)", and "Lungs (with alveoli)", by Biorender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. Accessed 16 July 2021.
Adapted from "Alveoli", "Alveoli (with mucus)", and "Emphysemic Alveoli", by Biorender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. Accessed 16 July 2021.
Funding
Christina M. Eckhardt was supported by a clinical and translational science awards grant (TL1TR001875).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environment and Aging
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eckhardt, C.M., Wu, H. Environmental Exposures and Lung Aging: Molecular Mechanisms and Implications for Improving Respiratory Health. Curr Envir Health Rpt 8, 281–293 (2021). https://doi.org/10.1007/s40572-021-00328-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-021-00328-2