
Abstract
Lung aging is associated with structural remodeling, a decline of respiratory function and a higher susceptibility to acute and chronic lung diseases. Individual factors that modulate pulmonary aging include basic genetic configuration, environmental exposure, life-style and biography of systemic diseases. However, the actual aging of the lung takes place in pulmonary resident cells and is closely linked to aging of the immune system (immunosenescence). Therefore, this article reviews the current knowledge about the impact of aging on pulmonary cells and the immune system, without analyzing those factors that may accelerate the aging process in depth. Hallmarks of aging include alterations at molecular, cellular and cell–cell interaction levels. Because of the great variety of cell types in the lung, the consequences of aging display a broad spectrum of phenotypes. For example, aging is associated with more collagen and less elastin production by fibroblasts, thus increasing pulmonary stiffness and lowering compliance. Decreased sympathetic airway innervation may increase the constriction status of airway smooth muscle cells. Aging of resident and systemic immune cells leads to a pro-inflammatory milieu and reduced capacity of fighting infectious diseases. The current review provides an overview of cellular changes occurring with advancing age in general and in several cell types of the lung as well as of the immune system. Thereby, this survey not only aims at providing a better understanding of the mechanisms of pulmonary aging but also to identify gaps in knowledge that warrant further investigations.
Similar content being viewed by others
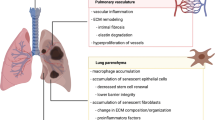


Introduction
With advancing age, the respiratory tract undergoes structural and functional changes. These changes are mostly associated with a decrease in lung function, pulmonary remodeling, limited regeneration and an enhanced susceptibility to pulmonary diseases in the elderly (>65 years of age) (Sharma and Goodwin 2006).
Even in healthy humans, lung function declines with increasing age. Breathing patterns were reported to show a smaller tidal volume and a higher respiratory rate (Janssens et al. 1999) and the maximal aerobic capacity (VO2 max) is reduced with age (McClaran et al. 1995). As a consequence, the elderly show a lower pulmonary oxygenation and exercise capacity. This age-dependent decline in pulmonary function is directly linked to structural remodeling of the lung (Miller 2010). For example, different studies in humans and rodents give evidence that the pulmonary elastic recoil capacity regresses with age due to structural remodeling of the extracellular matrix in the lung parenchyma and lowers the pulmonary forced expiratory volume (FEV1) in the elderly (Janssens et al. 1999; Huang et al. 2007; Miller 2010). Other characteristics of age-related structural remodeling include a decrease in alveolar depth and an increase in acinar airway lumen, as measured in a recent real-time morphology study in healthy elderly (Quirk et al. 2016). These changes go along with a reduction of the pulmonary gas exchange area and promote a diminished lung function with advanced age.
Pulmonary remodeling is even more pronounced in classical age-related pulmonary diseases, such as senescence-related emphysema, chronic obstructive pulmonary disease (COPD) or interstitial lung disease (ILD) (Fukuchi 2009; Meyer 2012). The likelihood of developing any of these diseases significantly increases with age. The international comparison study on the Burden of Obstructive Lung Disease (BOLD Study) shows that individuals with an age of > 70 years have a much higher chance of developing COPD (Buist et al. 2007). Another study on ILD shows that 227 out of 100,000 Americans older than 75 years of age develop ILD whereas only 4 out of 100,000 did within 18 and 34 years of age (Raghu et al. 2006). COPD or ILD show very different pathologies but are both associated with chronic structural remodeling of the extracellular matrix and impaired pulmonary function. Intrinsic factors or inhalation exposure to noxious gases, microbes or air pollutants such as cigarette smoke, asbestos or silica can promote the development of chronic pulmonary diseases (Kipen et al. 1987; Yoshida and Tuder 2007; Cohen et al. 2008); however, the underlying mechanisms are still being investigated by large scientific communities.
Besides a predisposition to develop chronic diseases, the aged lung has also been shown to be more susceptible to environmental exposures and infectious agents (Green and Pinkerton 2004). Various epidemiological studies give evidence that the elderly are less resistant to pulmonary infectious diseases and suffer from prolonged recovery times. For example, pneumonia and acute respiratory distress syndrome (ARDS) are linked to higher morbidity and mortality rates in the elderly (Suchyta et al. 1997; Meyer 2005). A survey with ARDS patients between 55 and 85 years of age showed that mortality in the >70 years old was twice as likely and survivors had more difficulties in recovery than patients < 70 years of age (Ely et al. 2002). The elderly also show a decreased capacity of fighting pulmonary viral infections such as influenza or severe acute respiratory syndrome (SARS) (Gross et al. 1995; Loeb 2004) and are less responsive to vaccination (Gross et al. 1995). These findings indicate a predisposition of the elderly to pulmonary infections and diseases and give evidence that the immune response is declining with advanced age—an effect also called immunosenescence (Weiskopf et al. 2009; Busse and Mathur 2010).
With increasing life expectance and demographic shifts towards aged societies, the research focus on mechanisms of aging and age-related diseases has been reinforced in the last decade. However, despite extensive investigations, our understanding of the mechanisms of cell and tissue aging is still limited. Different factors such as telomere shortening, stem cell exhaustion, oxidative stress or inflammation are being discussed but it is often difficult to distinguish origin, cause or effect of aging. Most likely, age-related changes originate from cumulative damage or stress occurring due to multiple incidences that living beings experience over a life-time—also called the exposome (Vrijheid 2014). The lung is in direct contact with the external environment with its vast alveolar surface area and faces constant challenges that promote the development of a senescent phenotype. Hence, the exposome supposably plays a crucial role in the development of pulmonary senescence and is strongly influenced by the individual genetic background and life style (Fig. 1). Nevertheless, despite individual histories of aging, the pathophysiology of pulmonary aging shows common characteristics and within the current review we provide an overview on the mechanisms and consequences of lung aging.

Internal and external influences on pulmonary cellular senescence and pulmonary aging
Cellular aging in the lung
Age-related changes of the lung mainly include pulmonary stem cell exhaustion and senescence of pulmonary and inflammatory cells, remodeling of the extracellular matrix, alterations in the composition of the pulmonary surfactant system, mucociliary clearance or the pulmonary nervous system. The implications of these different aspects in pulmonary aging are discussed here and summarized in Table 1.
Cellular aging and senescence
A main aspect of aging is the limitation in regeneration and repair, associated with cell senescence. Cell senescence is recognized as a state of irreversible growth cycle arrest accompanied by impaired cellular function, increased production of reactive oxygen species (ROS) and pro-inflammatory signaling and the expression of senescence-associated molecules such as β-galactosidase, p16, p53 or p21 (Campisi 2013). Different reasons can lead to senescence-associated cell cycle arrest. A well-recognized cause is telomere deficiency. Telomeres are repetitive DNA sequences with associated proteins, located at the chromosomal end to stabilize and protect the chromosomal DNA (Blackburn et al. 2006). With each cell division, the chromosomal DNA end is shortened, since the end of the linear DNA cannot be replicated by the DNA polymerase. Telomeres in regular somatic cells are therefore reduced with each replication until critical telomere shortening is leading to cell cycle arrest (Campisi 2013). The enzyme telomerase is capable of maintaining telomere regeneration but its activity is limited to embryonic and adult stem cells and only a few somatic cells. The incidence of cell senescence is thus increasing with progressing age. Additionally, different chronic diseases or environmental exposure to a noxious agent, such as cigarette smoke, promote tissue repair and cell proliferation and thereby lead to telomere shortening and premature cellular senescence (Tsuji et al. 2006; Chilosi et al. 2013).
Cell senescence can also be stress-induced, for example, due to elevated ROS production or mitochondrial dysfunction (Sahin and DePinho 2010). Cellular exposure to ROS such as superoxide anions can lead to oxidative protein or DNA damage and thereby accelerate cell senescence. Besides oxidative DNA damage, ROS have also been suggested to promote an inflammatory response and thereby potentially contribute to the progress of inflamm-aging—an age-dependent decline of the function of the immune system (Franceschi et al. 2000; El Assar et al. 2013). The free radical theory by D. Harman was an early established hypothesis (in 1954; Harman 2006), suggesting that a variety of intrinsic or extrinsic factors can contribute to induced cellular ROS levels such as environmental exposure or chronic inflammatory diseases and lead to cellular damage and a senescent phenotype. Up until now, it has been recognized that ROS play an important role in aging and contribute to a senescent phenotype. The source and action of ROS, however, are manifold and often it is not clear if their presence is the result or the cause of senescence. For example, damaged mitochondria are a source for cellular ROS but at the same time enhanced cellular ROS itself can impair mitochondrial function. More recent theories of aging therefore suggest that the hallmarks of aging can be divided into three different categories: (1) cellular dysfunction including genomic instability, epigenetic alterations, loss of proteostasis and telomere attrition, (2) antagonistic response to damage such as deregulated nutrient sensing, mitochondrial dysfunction and cell senescence and (3) a changed phenotype with altered intercellular communication and stem cell exhaustion (López-Otín et al. 2013; Meiners et al. 2015; Aunan et al. 2016). The characterization of these different aspects of aging is clearly pointing out the variety of factors involved in the phenomena of aging and their interactions at molecular, cellular and systemic levels (Fig. 2). Furthermore, it has been suggested that a critical amount of senescent cells can create a senescent environment by further promoting senescence itself via paracrine mediators (Acosta et al. 2013; Tasdemir and Lowe 2013). An accumulation of impaired or senescent cells in the lung could thereby exacerbate pulmonary senescence of certain compartments or the whole lung.

Molecular, cellular and interactive hallmarks of aging. (Figure adapted from López-Otín et al. 2013)
The occurrence of cell senescence could be tissue- or cell-type-specific and the impact or outcome depends on the specific function of the affected tissue or cells. In the human lung, there are about 40 different cell types (Ochs and Weibel 2008). Coarsely, the different pulmonary cell types can be categorized into epithelial cells (such as bronchiolar and alveolar epithelial cells), endothelial cells, fibroblasts or immune cells. Cell-type-specific senescence has been associated with different pulmonary diseases, like, for example, senescence of alveolar epithelial precursor cells with idiopathic pulmonary fibrosis (IPF) and mesenchymal stem cell exhaustion with COPD (see review by Chilosi et al. 2013). Current investigations are therefore further elaborating the impact of cell-type-specific senescence in the lung and its impact on age-related pulmonary diseases.
Epithelial cells, mucous and surfactant
Pulmonary epithelial cells form a tight barrier to protect the lung from external influences. Ciliated bronchoepithelial cells and mucous producing goblet cells are the most abundant epithelial cell types in the conducting airways, as are alveolar type 1 (AT1) and type 2 (AT2) epithelial cells in the lung parenchyma. Little is known about particular effects of senescence on bronchoepithelial cells. Given their specific functions, it is reasonable that senescence of ciliated bronchoepithelial cells would lead to an impaired performance in mucociliary transport and senescence in goblet cells and to alterations in mucous composition and production. Indeed, a few studies have shown that mucociliary clearance decreases in healthy elderly (Svartengren et al. 2005; De Oliveira-Maul et al. 2013) and ciliary beat frequency is reduced in old C57BL/6 mice (Bailey et al. 2014). Another recent study showed an increase in apoptotic bronchoepithelial cells in lungs of old mice, resulting in a lower number of epithelial cells and epithelial thinning (Ortega-Martínez et al. 2016). It is, however, not yet fully investigated whether bronchoepithelial cell senescence coincides with diminished ciliary clearance.
More information is available on pulmonary epithelial cell senescence of AT2 cells. These cells produce surfactant to lower the surface tension in respiration and serve as progenitor cells for the air–blood barrier forming AT1 cells. An accelerated cell senescence in AT2 cells has been observed in COPD (Tsuji et al. 2006) as well as in different animal models of fibrosis (Aoshiba et al. 2003; Torres-González et al. 2012). Alveolar stem cell exhaustion has been discussed as a key factor in age-related pulmonary diseases (Faner et al. 2012; Chilosi et al. 2013). Since AT2 cells also serve as progenitors for AT1 cells, AT2 cell senescence can lead to a critical decline in alveolar epithelial stem cell renewal (Alder et al. 2015; Chen et al. 2015). Murine models to study telomerase-related senescence in stem cell regeneration include TERT and TERC KO mice. These mice are either lacking the telomere reverse transcriptase (TERT) or the telomerase RNA component (TERC)—hence disabled telomerase function—and show many signs of premature aging, also including elevated apoptosis in AT2 cells (Amsellem et al. 2011; Chen et al. 2015). A study by Alder and colleagues specifically investigated AT2 cell-targeted telomere dysfunction and the effects of AT2 cell senescence. They found an enhanced pro-inflammatory response with increased numbers of macrophages in the bronchoalveolar fluid (BALF) and an enhanced susceptibility to injury such as bleomycin-induced ARDS in the animals with induced AT2 cell senescence (Alder et al. 2015). This finding gives evidence that telomere shortening is not only a result of chronic diseases but also a potential trigger.
Implications of aging on AT2 cell-mediated surfactant production and compositions have also been investigated. A surfactant is crucial for lowering the alveolar surface tension and surfactant protein A and D (SP-A and SP-D) are part of the complement system. The surfactant plays not only an essential role in regular breathing but also in pulmonary inflammation. For instance, in acute lung injury, the surfactant is qualitatively and quantitatively reduced and injury of AT2 cells worsens the prognosis in acute lung injury (Ingenito et al. 2001). However, very little is still known about age-related changes in surfactant composition. Early investigations on the surfactant and age, including studies in rats and dogs, show varying outcomes (Orgeig and Daniels 2004), which might be due to interspecies variability and varying interpretations of the definition “old”. A more recent study addressed changes in the ultra-structure of the pulmonary surfactant in 2- to 3-month and 26-month-old rats (Walski et al. 2009), showing that the structures of lamellar bodies and tubular myelin in older rats resemble those of young animals that were treated with a protein synthesis inhibitor. Another study in mice and humans found that levels of pro-inflammatory cytokines, SP-A and SP-D and activity of myeloid peroxidase increase with advanced age and lipid composition of surfactant changes in the alveolar lining fluid (ALF) (Moliva et al. 2014). The authors suggest a more pro-inflammatory and more oxidative milieu of the ALF with advanced age, partly due to AT2 cell dysfunction, which could also be a result of AT2 cell senescence. However, more research addressing the composition and secretion of the surfactant is needed to understand the impact of senescence-related surfactant dysfunction and its impact in pulmonary health and disease.
Endothelial cells
Senescence in vascular endothelial cells has been well studied, particularly in regard to arteriosclerosis, angiogenesis or arterial stiffening and remodeling. A detailed report on the current research on vascular endothelial senescence would be beyond the scope of this review, hence we summarize here only the most relevant findings, in particular in regard to pulmonary vasculature and refer to other reviews on this subject for further information (Erusalimsky 2009; El Assar et al. 2013; Regina et al. 2016). In brief, senescence in endothelial cells has a direct impact on angiogenesis by limiting vascular growth (Zaccagnini et al. 2005; Falchetti et al. 2008). Furthermore, endothelial vasodilatation has been shown to be compromised with advanced age, as senescent endothelial cells show less endothelial nitric oxide synthase (eNOS) activity and NO expression (Tschudi et al. 1996; Van der Loo et al. 2000). It is suggested that this effect is mostly governed by increased ROS levels in senescent cells that directly compromise NO signaling (Bhayadia et al. 2016). Besides ROS, pro-atherogenic and pro-thrombogenic effects have also been reported in senescence, including an increased expression of intercellular adhesion molecule 1 (ICAM-1) and plasminogen activator inhibitor-1 (PAI-1) (Maier et al. 1993; Comi et al. 1995). Results of investigations in pulmonary endothelial cells are mostly in line with these findings. For example, a study with primary pulmonary microvascular endothelial cells demonstrated that cells isolated from old rats have a decreased migration and proliferation ability, suggesting cell senescence and limited regeneration capacity to injury (Lu et al. 2012). Ex-vivo experiments with pulmonary endothelial arteries of young and old rats have furthermore shown that pulmonary arteries of old rats have impaired endothelial function with higher levels of ROS and decreased catalase expression (Podlutsky et al. 2010). Accordingly, a review by Jane-Wit and Chun also summarized the three main features of pulmonary endothelial senescence as the vulnerability to oxidative stress, impaired NO signaling and deficits in repair (Jane-Wit and Chun 2012). These features make the elderly more susceptible to diseases such as pulmonary sepsis or pulmonary hypertension.
Fibroblasts and ECM
Pulmonary fibroblasts are involved in tissue repair and the construction and/or remodeling of the extracellular matrix (ECM). The composition of the ECM has, particularly in the lung parenchyma, a direct impact on lung function and most senescence-related lung diseases, including COPD, ILD or lung cancer, show remodeling of the ECM (reviewed in Meiners et al. 2015). It is therefore not surprising that fibroblast senescence has been associated with pulmonary remodeling and age-related diseases such as emphysema and COPD or fibrosis and has been addressed in various studies (Calhoun et al. 2015; Yanai et al. 2015; Hashimoto and Sugiura 2016). Investigations on fibroblast senescence were mostly performed in vitro with cell lines. In these models, cell senescence was induced by exhaustive replication or chemical stressors and measured by specific senescence markers such as β-galactosidase or the upregulation of cell cycle proteins p21, p53 and p16 that control and arrest cell division (Tigges et al. 2014; Maciel-Barón et al. 2016). The expression of certain microRNAs has also been attributed to senescence (Lee et al. 2014; Holly et al. 2015). Fibroblast senescence has furthermore been investigated in regard to the development of fibrosis. For instance, primary fibroblasts isolated from IPF patients show signs of cell senescence, i.e., decreased cell growth, β-galactosidase increase after induction of oxidative stress, altered cell morphology and expression of α-smooth muscle actin (α-SMA) in replicative stress-induced senescence (Yanai et al. 2015). Studies with murine models suggest that pulmonary fibroblasts from old mice are more resistant to bleomycin-induced apoptosis compared to fibroblasts from young mice and seem to have a greater fibrotic response to transforming growth factor β (TGF-β) (Huang et al. 2015), whereas investigations in the senescence-accelerated prone or resistant mouse model (SAM P/R mice) with bleomycin, induced lung injury show more fibrotic lesions and higher levels of TGF-β expression in SAMP compared to SAMR animals (Xu et al. 2009). Hence, these data support the hypothesis of senescence-related mechanisms in fibrosis.
Alterations in the composition of the ECM with age have been associated with changes in fibroblast function and have received increasing attention. Proteomics and microarray analysis in young and senescent human fibroblasts show that transcription and expression of ECM proteins is altered in senescent cells (Yang et al. 2015). The impact of altered ECM composition on the lung parenchyma with age has mostly been investigated in rodents, with particular focus on elastin and collagen. Generally, it was shown that the percentage of elastic fibers is reduced with progressing age and collagen is increased, with a direct impact on lung function such as reduced elasticity and tissue dampening (Huang et al. 2007; Calhoun et al. 2015). Changes in the ECM correlated with an increase in cell senescence markers (p16 and p53) in old mice along with an activation of mTOR signaling, suggesting that cell senescence is responsible for physiological changes with advanced age (Calhoun et al. 2015). A recent study with decellularized murine lung scaffolds of various ages also confirmed changes in ECM, showing that collagen increases and elastin and laminin decrease with age (Godin et al. 2016). Interestingly, the authors also demonstrated that the repopulation of the scaffolds with primary human bronchoepithelial cells and lung fibroblasts is affected by age-related changes of the ECM. These studies emphasize the role of the ECM in pulmonary senescence and certainly more investigations will follow on this subject in the future. Furthermore, investigations addressing the three-dimensional organization of the pulmonary ECM would improve our understanding on ECM remodeling with advancing age.
Alveolar macrophages
Immune cells are also affected by senescence, a condition which is collectively termed immunosenescence. Different cells of the innate and adaptive immunity are present in the lung and their senescence has an impact on the pulmonary immunity. However, most of them, including non-tissue-resident macrophages and other leukocytes, are recruited from the circulation/bone marrow and transiently present in the lungs. The immune cell that is resident to the lung is the alveolar macrophage. The alveolar macrophages belong to the tissue-resident macrophages. They represent a population of specialized, long-lived resident macrophages in different tissues that evolve during embryogenesis (Gomez Perdiguero et al. 2014). The alveolar macrophages act as the first immune defense of the lung by clearing airborne and microbial particles in the lung and activating the innate immune system via a pathogen-associated molecular pattern (PAMP) and danger-associated molecular pattern (DAMP) molecule recognition. Studies on age-related effects of alveolar and pulmonary macrophages provide evidence that the phagocytic capacity declines with age (Higashimoto et al. 1993; Hearps et al. 2012), hence the clearance of pathogens in infection is potentially impaired or delayed in the elderly. The capability of PAMP recognition via toll-like receptor (TLR) signaling was also investigated in this context but the results are controversial and probably differ with pathogen stimulus and TLR signaling pathways (Murciano et al. 2008; Shaw et al. 2011; Boyd et al. 2012). Different experimental studies furthermore give evidence that macrophages from old animals have higher ROS levels or a decline in the ROS defense system. For example, BALF macrophages from old mice or blood monocytes from the elderly show a decreased anti-oxidative response via the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway, whereas oxidized glutathione and carboxylated albumin levels increase after cigarette smoke exposure (Suzuki et al. 2008) compared to those of young individuals. Similarly, the expression of heme oxigenase-1 (HO-1) was reduced in BALF macrophages from old compared to young mice after LPS exposure (Ito et al. 2009). The increased ROS levels are likely a sign of cell senescence and also attributed to pro-inflammatory signaling. However, reports on inflammatory signaling and cytokine expression with advanced age in alveolar macrophages are controversial. Some studies report that cytokine expression by alveolar macrophages from old animals is enhanced after inflammatory stimulation (Canan et al. 2014), whereas others show the contrary (Higashimoto et al. 1993; Lakshman et al. 2006). It is furthermore not clear if alterations in macrophage response with advanced age are related to cell senescence or to other age-related changes. In addition, non-tissue-resident macrophages and monocytes are also recruited to the lung in inflammation and influence inflammatory signaling. Macrophages show high plasticity and form heterogenic subpopulations, distinguished by different cellular markers (Misharin et al. 2013). Depending on the environment and stimulus, their phenotypic profile tends more to a pro-inflammatory, also called M1 profile or to an M2 or alternative macrophage profile, which has been considered as anti-inflammatory but is also associated with the development of fibrosis or allergies (Johnston et al. 2012; Lech and Anders 2013). It has been discussed how the macrophage plasticity changes with age and how different sub-populations are affected by aging (Stout and Suttles 2005; Mahbub et al. 2012). However, no conclusive answer to that question is available so far. Besides, a new nomenclature for macrophage activation and polarization has recently been suggested (Murray et al. 2014). Hence, proper characterizations of macrophage populations in the aging lung are required in healthy and diseased individuals to better understand the impact of macrophage senescence in pulmonary aging.
Autonomous and sensory nervous system
The pulmonary nervous system consists in afferent and efferent nerve fibers that help maintain organ homeostasis. Afferent fibers play important roles in sensing mechanical (e.g., inflation status) or chemical (e.g., capsaicin) stimuli and in being involved in protective reflexes such as cough reflex. The efferent fibers mainly belong to the sympathetic and parasympathetic nervous system and affect the constriction status of airways and blood vessels as well as the secretion of mucus. In general, the innervation of the proximal airways is far more abundant than the innervation of the alveolar region (reviewed in Belvisi 2002). The functional characteristics of the autonomous innervation make it an important pharmaceutical target in pulmonary diseases such as asthma and COPD. Besides the innervation of the lower airways, the passage from the upper to the lower airways at the larynx requires a complex interplay between sensory and motor nerve fibers regulating the swallowing reflex. The disturbance of this reflex may lead to aspiration of contents from the oropharynx or the stomach and thereby cause pulmonary syndromes of which aspiration pneumonia is most frequent (Ebihara et al. 2012). In addition, the patency of the upper airways relies on the function of the genioglossus muscle that is impaired in obstructive sleep apnea.
As with COPD, aspiration pneumonia and obstructive sleep apnea become more frequent in later life (Petroianni et al. 2006; Edwards et al. 2010; Meiners et al. 2015) and therefore it is of importance to know whether changes of the upper or lower airway innervation may contribute to them. Currently, studies on the nervous system of the upper and lower airways during aging are scarce and far from delivering a comprehensive picture. However, those studies that are available clearly indicate that the nervous system of the respiratory tract and related structures do change during aging. For example, experimental evidence suggests that in male rats—in contrast to female rats—serotonin decreases in the hypoglossal nucleus but is not compensated for by an increase in receptor density (Seebart et al. 2007), which may be related to the higher collapsibility of the upper airways in 30-month-old than in 6-month-old rats (Ray et al. 2008). Several recent studies have indicated that aging affects the sensory side of reflexes rather than the efferent part (Ebihara et al. 2011; Malandraki et al. 2011) and that stimulation of the transient receptor potential channel TRPV1 decreases the delay in the swallowing reflex (Ebihara et al. 2006). In the lower airways, aging is associated with a reduction of the noradrenergic innervation of the bronchial tree (Ricci et al. 1997) as well as with changes in neuropeptide levels, namely a decrease in vasoactive intestinal peptide concentration and nerve fiber density (Geppetti et al. 1988). The latter changes may be related to a higher susceptibility of bronchoconstriction in the elderly.
Alterations of the immune system and the microbiome with impact on the lung
Immunosenescence
The poor prognosis and recovery in pulmonary inflammatory diseases with advanced age goes along with various age-related changes in innate and adaptive immunity (Frasca and Blomberg 2015). In general, this includes decreased phagocytotic function of macrophages and neutrophils, reduced activity of natural killer (NK) cells, altered serum levels of pro-inflammatory cytokines greater numbers of airway neutrophils, as well as decreased T-cell stimulation by dendritic cells. These changes support the development of chronic pulmonary diseases and favor a poor prognosis in infectious and inflammatory diseases in the elderly (Opal et al. 2005; Boyd and Orihuela 2011).
Changes in innate immunity with impact on the respiratory tract
The first response to pathogens is regulated by the innate immunity, including DAMP/ PAMP recognition by phagocyte and other cells, cytokine and chemokine secretion, recruitment of leukocytes such as neutrophils and macrophages to the site of infection/injury and elimination of pathogens. Several reports have shown that systemic and pulmonary levels of pro-inflammatory cytokines interleukin (IL)-1β, IL-6 and tumor necrosis factor α (TNFα) are enhanced in the elderly (Meyer et al. 1996; Ogawa et al. 2008). This effect is also called inflamm-aging and associated with a greater chronic pro-inflammatory milieu (Franceschi et al. 2000). The elevated levels of pro-inflammatory cytokines are potentially the result of cell senescence, as senescent cells show increased ROS and NF-kB signaling (Salminen et al. 2008). As a consequence, the elderly with enhanced IL-6 and TNFα serum levels are more likely to develop pneumonia, as shown in a longitudinal study over the course of 6.5 years by Yende et al. (2005). It is furthermore suggested that the pro-inflammatory response in the elderly is more severe and prolonged in comparison to younger individuals (Inoue et al. 2014). Pathogen elimination by phagocytic cells is delayed with advanced age, due to a decline in phagocytic capability by macrophages and neutrophils (Higashimoto et al. 1993; Fulop et al. 2012) and DAMP/PAMP signaling potentially impaired (Murciano et al. 2008; Boyd et al. 2012). Murine studies on acute inflammation furthermore give evidence that levels of pro-inflammatory cytokines are higher in old animals, particularly the neutrophil chemoattractant keratinocyte-derived chemokine (KC, CXCL1), a murine homologue of IL-8 (Gomez et al. 2007; Chen et al. 2014). This goes along with increased numbers of neutrophils as, for example, in pneumonia or acute lung injury of old rodents (Gomez et al. 2007; Linge et al. 2015). Neutrophils secrete a variety of enzymes and radicals in order to defeat pathogens; however, if present in excess, they can also induce tissue injury in the host. Although it has been reported that pathogen elimination by neutrophils declines with age (Fortin et al. 2008), increasing numbers of neutrophils could still lead to greater tissue injury (Starr et al. 2012). Particularly in acute lung injury, elevated neutrophil numbers can promote the development of edema and enhance the severity of the disease (Zemans et al. 2009).
Another cell type of the innate immunity that has the ability to recognize and eliminate virally infected and damaged cells are the NK cells. It has been described that both function and phenotype of NK cells are altered with advanced age (Hayhoe et al. 2010; Gayoso et al. 2011; Campos et al. 2014) and that NK cell senescence impairs the antiviral immune response (Beli et al. 2011). Other functions of NK cells have been recognized, such as resolution of inflammation and elimination of senescent or stressed cells as well as stimulating the adaptive immune response (reviewed in Hazeldine and Lord 2013). With respect to the lung, it has been shown that impaired NK function is leading to greater incidence in mycobacterium tuberculosis or staphylococcus aureus infection as well as the development of lung cancer (Small et al. 2008; Hazeldine and Lord 2013; Hodge et al. 2014). Hence, NK senescence might have further implications besides declined elimination of virally infected cells, which have not yet been fully explored.
In summary, it has been shown that certain aspects of the innate immune response decline with age, such as the elimination of pathogens and infected or damaged cells. But other aspects of pro-inflammatory signaling remain intact or are even chronically enhanced with advanced age and potentially support the development of chronic inflammatory diseases.
Changes in adaptive immunity with impact on lung
The adaptive or humoral immunity regulates the specific pathogen defense. In brief, antigen-presenting cells, such as dendritic cells, collect foreign particles, process antigens, migrate to lymph nodes and activate T-cells and B-cells for specific pathogen elimination, antibody production and elimination of infected cells. The dendritic cells act here as mediators between the innate and the adaptive immunity and senescence of dendritic cells has a direct impact on the activation of the adaptive immunity. Different investigations have shown a decline in efficacy of dendritic cells in antigen presentation, including impaired DAMP recognition via nod-like receptor signaling (Stout-Delgado et al. 2012), reduced migration (Zhao et al. 2011), antigen processing (Chougnet et al. 2015) or T-cell activation (Zacca et al. 2015). The pool of available naïve T-cells was furthermore found to degenerate with age, whereas the number of memory T-cells increases (Brandenberger et al. 2014), which diminishes the efficiency for T-cell activation (Haynes and Swain 2006). This effect is accompanied by impaired B-cell-mediated antibody production and specificity (Meyer 2010) and by a reduced IL-2 release—a cytokine that promotes the stimulation of the adaptive immunity (Haynes and Swain 2006). Together, these incidences lead to an enhanced susceptibility of the elderly to infections and a declined response to vaccines (Toapanta and Ross 2009; Meyer 2010; Stout-Delgado et al. 2012) in pulmonary infection and disease.
In addition, a shift in T-helper cell subpopulations has been proposed with advanced age. T-helper cells are classified into T-helper type I (Th1) cells that secrete interferon γ (IFNγ) and IL-2 and stimulate the adaptive immune response, Th2 cells that secrete IL-5 and IL-13 and are involved in parasitic defense or allergic reaction and Th17 cells that secrete IL-17, IL-21 and IL-23 that activate neutrophilic granulocytes and are predominantly present in chronic inflammation (Lee et al. 2012; Hirahara and Nakayama 2016). Reports on alterations of the Th-subpopulation with age are not consistent and vary with subject age and model of investigation (Lee et al. 2012). In chronic pulmonary diseases such as COPD or in allergic airway disease with advanced age, Th17 cells have been shown to play a predominant role (Doe et al. 2010; Vanaudenaerde et al. 2011; Brandenberger et al. 2014). Th17 cells were described as developing from the same lineage as the anti-inflammatory regulatory T-cells (T-regs) (Diller et al. 2016). Hence, a bias toward Th17 response may abate the T-reg response and possibly support a pro-inflammatory milieu as reported in the elderly (Schmitt et al. 2013).
Most aspects of the adaptive immune response have been reported to decline with advancing age, with the consequence that pulmonary infections are prolonged and more severe in the elderly. If or how alterations in Th-cell subpopulations contribute to the pro-inflammatory status in the elderly, however, still needs further investigation.
Microbiome
Current research has focused on investigations on the commensal human microbiome in health and disease. However, little is still known about the microbiome in the lung and only lately has it been found that the lungs are not as sterile as presumed (Dickson et al. 2015). Alterations in the pulmonary microbiome with advancing age have not yet been analyzed but investigations have shown that the pulmonary microbiome is altered in patients with COPD (Pragman et al. 2012; Garcia-Nunez et al. 2014; Sze et al. 2015). To what extent the changes correlate with age is still unknown. In contrast to the lung, the microbiome in the gut has been well investigated and a close relationship between the composition of the gut microbiome, diet and healthy aging has been suggested (Claesson et al. 2011, 2012; Odamaki et al. 2016). The gut microbiome not only plays an important role in maintaining intestinal homeostasis by supporting colonic energy balance but also in modulating the intestinal immune response (Hooper et al. 2012). The immune response influenced by the gut microbiome, however, is not restricted just to the gut. Other organs, including the lung, can also be affected (Chen et al. 2011). Alterations in immune function due to changes in the gut microbiome have been related to a number of pulmonary diseases. For instance, the susceptibility to pneumonia was increased by disturbance of the commensal gut bacterial community using antibiotics (Tsay et al. 2011; Chen et al. 2011) and an enhanced susceptibility for the development of allergic airway disease with a decreased immunological tolerance has been shown in germ-free mice (Herbst et al. 2011; Hörmannsperger et al. 2012). A correlation between the severity of allergic airway disease and changes in gut microbiome has also been found with advanced age in a house dust mite model of young and old mice, showing a more pronounced pathology with advanced age along with a shift in the microbial community (Vital et al. 2015). With respect to aging, it has been reported that the integrity of the gut epithelium declines and the secretions of mucus and defensins are reduced (Larbi et al. 2008; Biagi et al. 2013). This can facilitate the entry of pathogens and generate a low-grade inflammation (Biagi et al. 2013). An enhanced level of bacterial LPS has been measured in feces and serum of old mice and related to greater levels of p16 and NF-kB activation, thereby potentially accelerating inflamm-aging (Kim et al. 2016). Certainly, these findings indicate that changes of the immune status as well as the host microbiome with progressing age have an impact on health and disease. However, little is still known about how individual microbial species or the composition of entire communities interact with and shape the immune system with impact on the lung.
Concluding remarks
The present review article aimed at providing an overview on the changes of pulmonary resident cells and the immune system associated with aging. Despite a growing knowledge about general mechanisms of cellular and molecular aging, the contribution of the different cellular systems of the lung to the impact of aging has been addressed to a much lesser extent. In particular, the interaction between various types of cell and how this is affected by aging needs to be further addressed. We are convinced that a better understanding of pulmonary aging (i.e., degeneration as the physiological opposite of regeneration) may help to develop new strategies for modern regenerative approaches.
References
Acosta JC, Banito A, Wuestefeld T et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–90
Alder JK, Barkauskas CE, Limjunyawong N et al (2015) Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A 112:5099–5104
Amsellem V, Gary-Bobo G, Marcos E et al (2011) Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 184:1358–1366
Aoshiba K, Tsuji T, Nagai A (2003) Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J 22:436–443
Aunan JR, Watson MM, Hagland HR, Søreide K (2016) Molecular and biological hallmarks of ageing. Br J Surg 103:e29–e46
Bailey KL, Bonasera SJ, Wilderdyke M et al (2014) Aging causes a slowing in ciliary beat frequency, mediated by PKCε. Am J Physiol Lung Cell Mol Physiol 306:L584–9
Beli E, Clinthorne JF, Duriancik DM et al (2011) Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech Ageing Dev 132:503–510. doi:10.1016/j.mad.2011.08.005
Belvisi MG (2002) Overview of the innervation of the lung. Curr Opin Pharmacol 2:211–215
Bhayadia R, Schmidt BMW, Melk A, Hömme M (2016) Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A 71:161–9
Biagi E, Candela M, Turroni S et al (2013) Ageing and gut microbes: perspectives for health maintenance and longevity. Pharm Res 69:11–20
Blackburn EH, Greider CW, Szostak JW (2006) Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 12:1133–1138
Boyd AR, Orihuela CJ (2011) Dysregulated inflammation as a risk factor for pneumonia in the elderly. Aging Dis 2:487–500
Boyd AR, Shivshankar P, Jiang S et al (2012) Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Exp Gerontol 47:507–18
Brandenberger C, Li N, Jackson-Humbles DN et al (2014) Enhanced allergic airway disease in old mice is associated with a Th17 response. Clin Exp Allergy 44:1282–1292
Buist AS, McBurnie MA, Vollmer WM et al (2007) International variation in the prevalence of COPD (The BOLD Study): a population-based prevalence study. Lancet 370:741–750
Busse PJ, Mathur SK (2010) Age-related changes in immune function: effect on airway inflammation. J Allergy Clin Immunol 126:690–9
Calhoun C, Shivshankar P, Saker M et al (2015) Senescent cells contribute to the physiological remodeling of aged lungs. J Gerontol A 1:1–9
Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75:685–705
Campos C, Pera A, Lopez-Fernandez I et al (2014) Proinflammatory status influences NK cells subsets in the elderly. Immunol Lett 162:10–14. doi:10.1016/j.imlet.2014.06.015
Canan CH, Gokhale NS, Carruthers B et al (2014) Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol 96:473–80
Chen L-W, Chen P-H, Hsu C-M (2011) Commensal microflora contribute to host defense against Escherichia coli pneumonia through Toll-like receptors. Shock 36:67–75
Chen MM, Palmer JL, Plackett TP et al (2014) Age-related differences in the neutrophil response to pulmonary pseudomonas infection. Exp Gerontol 54:42–46
Chen R, Zhang K, Chen H et al (2015) Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J Biol Chem 290:30813–30829
Chilosi M, Carloni A, Rossi A, Poletti V (2013) Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res 162:156–173
Chougnet CA, Thacker RI, Shehata HM et al (2015) Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol 195:2624–32
Claesson MJ, Cusack S, O’Sullivan O et al (2011) Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108:4586–91
Claesson MJ, Jeffery IB, Conde S et al (2012) Gut microbiota composition correlates with diet and health in the elderly. Nature 488:178–84
Cohen RAC, Patel A, Green FHY (2008) Lung disease caused by exposure to coal mine and silica dust. Semin Respir Crit Care Med 29:651–661
Comi P, Chiaramonte R, Maier JAM (1995) Senescence-dependent regulation of type 1 plasminogen activator inhibitor in human vascular endothelial cells. Exp Cell Res 219:304–308
De Oliveira-Maul JP, De Carvalho HB, Goto DM et al (2013) Aging, diabetes, and hypertension are associated with decreased nasal mucociliary clearance. Chest 143:1091–1097
Dickson RP, Erb-Downward JR, Huffnagle GB (2015) Homeostasis and its Disruption in the Lung Microbiome. Am J Physiol Lung Cell Mol Physiol 309:L1047–55
Diller ML, Kudchadkar RR, Delman KA et al (2016) Balancing inflammation: The link between Th17 and regulatory T cells. Mediat Inflamm. doi:10.1017/CBO9781107415324.004
Doe C, Bafadhel M, Siddiqui S et al (2010) Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 138:1140–1147
Ebihara T, Ebihara S, Watando A et al (2006) Effects of menthol on the triggering of the swallowing reflex in elderly patients with dysphagia. Br J Clin Pharmacol 62:369–371
Ebihara S, Ebihara T, Kanezaki M et al (2011) Aging deteriorated perception of urge-to-cough without changing cough reflex threshold to citric acid in female never-smokers. Cough. doi:10.1186/1745-9974-7-3
Ebihara S, Ebihara T, Kohzuki M (2012) Effect of aging on cough and swallowing reflexes: Implications for preventing aspiration pneumonia. Lung 190:29–33
Edwards BA, O’Driscoll DM, Ali A et al (2010) Aging and sleep: physiology and pathophysiology. Semin Respir Crit Care Med 31:618–633
El Assar M, Angulo J, Rodriguez-Manas L (2013) Oxidative stress and vascular inflammation in aging. Free Radic Biol Med 65:380–401. doi:10.1016/j.freeradbiomed.2013.07.003
Ely EW, Wheeler AP, Thompson BT et al (2002) Recovery rate and prognosis in older persons who develop acute lung injury and the acute respiratory distress syndrome. Ann Intern Med 136:25–36
Erusalimsky JD (2009) Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol 106:326–32
Falchetti ML, Mongiardi MP, Fiorenzo P et al (2008) Inhibition of telomerase in the endothelial cells disrupts tumor angiogenesis in glioblastoma xenografts. Int J Cancer 122:1236–1242
Faner R, Rojas M, Macnee W, Agustí A (2012) Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 186:306–13
Fortin CF, McDonald PP, Lesur O, Fülöp T (2008) Aging and neutrophils: there is still much to do. Rejuvenation Res 11:873–882
Franceschi C, Bonafè M, Valensin S et al (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254
Frasca D, Blomberg BB (2015) Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology. doi:10.1007/s10522-015-9578-8
Fukuchi Y (2009) The aging lung and chronic obstructive pulmonary disease: similarity and difference. Proc Am Thorac Soc 6:570–572
Fulop T, Fortin C, Lesur O (2012) The innate immune system and ageing: what is the contribution to immunosenescence. Bentham Open 6:121–131. doi:10.2174/1876326X01206010121
Garcia-Nunez M, Millares L, Pomares X et al (2014) Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol 52:4217–4223
Gayoso I, Sanchez-Correa B, Campos C et al (2011) Immunosenescence of human natural killer cells. J Innate Immun 3:337–343
Geppetti P, De Rossi M, Mione MC et al (1988) Age-related changes in vasoactive intestinal polypeptide levels and distribution in the rat. J Neural Transm 74:1–10
Godin LM, Sandri BJ, Wagner DE et al (2016) Decreased laminin expression by human lung epithelial cells and fibroblasts cultured in acellular lung scaffolds from aged mice. PLoS ONE. doi:10.1371/journal.pone.0150966
Gomez Perdiguero E, Klapproth K, Schulz C et al (2014) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551
Gomez CR, Hirano S, Cutro BT et al (2007) Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit Care Med 35:246–51
Green FHY, Pinkerton KE (2004) Environmental determinants of lung aging. In: Harding R, Pinkerton KE, Plopper CG (eds) The lung: development, aging and the environment. Elsevier, San Diego, pp 377–395
Gross P, Hermogenes A, Sacks H et al (1995) The efficacy of influenza vaccine in elderly persons. A meta-analysis and review of the literature. Ann Intern Med 123:518–527
Harman D (2006) Free radical theory of aging: An update - Increasing the functional life span. Ann N Y Acad Sci 1067:10–21
Hashimoto Y, Sugiura H (2016) 27-Hydroxycholesterol Accelerates Cellular Senescence in Human Lung Resident Cells. Am J Physiol Lung Cell Mol Physiol 310:L1028–1041
Hayhoe RPG, Henson SM, Akbar AN, Palmer DB (2010) Variation of human natural killer cell phenotypes with age: Identification of a unique KLRG1-negative subset. Hum Immunol 71:676–681
Haynes L, Swain SL (2006) Why aging T cells fail: implications for vaccination. Immunity 24:663–6
Hazeldine J, Lord JM (2013) The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Res Rev 12:1069–1078
Hearps AC, Martin GE, Angelovich TA et al (2012) Aging is associated with chronic innate immune activation and dysregulation of monocyte phenotype and function. Aging Cell 11:867–875
Herbst T, Sichelstiel A, Schär C et al (2011) Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am J Respir Crit Care Med 184:198–205
Higashimoto Y, Fukuchi Y, Shimada Y et al (1993) The effects of aging on the function of alveolar macrophages in mice. Mech Ageing Dev 69:207–217
Hirahara K, Nakayama T (2016) CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int Immunol 28:163–171
Hodge G, Barnawi J, Jurisevic C et al (2014) Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin Exp Immunol 178:79–85
Holly AC, Grellscheid S, van de Walle P et al (2015) Comparison of senescence-associated miRNAs in primary skin and lung fibroblasts. Biogerontology 16:423–434
Hooper LV, Littman DR, Macpherson AJ (2012) Interactions between the microbiota and the immune system. Science 336:1268–73
Hörmannsperger G, Clavel T, Haller D (2012) Gut matters: microbe-host interactions in allergic diseases. J Allergy Clin Immunol 129:1452–9
Huang K, Rabold R, Schofield B et al (2007) Age-dependent changes of airway and lung parenchyma in C57BL/6J mice. J Appl Physiol 102:200–206
Huang WT, Akhter H, Jiang C et al (2015) Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp Gerontol 61:62–75
Ingenito EP, Mora R, Cullivan M et al (2001) Decreased surfactant protein-B expression and surfactant dysfunction in a murine model of acute lung injury. Am J Respir Cell Mol Biol 25:35–44
Inoue S, Suzuki K, Komori Y et al (2014) Persistent inflammation and T cell exhaustion in severe sepsis in the elderly. Crit Care. doi:10.1186/cc13941
Ito Y, Betsuyaku T, Moriyama C et al (2009) Aging affects lipopolysaccharide-induced upregulation of heme oxygenase-1 in the lungs and alveolar macrophages. Biogerontology 10:173–80
Jane-Wit D, Chun HJ (2012) Mechanisms of dysfunction in senescent pulmonary endothelium. J Gerontol A 67:236–241
Janssens JP, Pache JC, Nicod LP (1999) Physiological changes in respiratory function associated with ageing. Eur Respir J 13:197–205
Johnston LK, Rims CR, Gill SE et al (2012) Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol 47:417–426
Kim K-A, Jeong J-J, Yoo S-Y, Kim D-H (2016) Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. doi:10.1186/s12866-016-0625-7
Kipen HM, Lilis R, Suzuki Y et al (1987) Pulmonary fibrosis in asbestos insulation workers with lung cancer: a radiological and histopathological evaluation. Br J Ind Med 44:96–100
Lakshman CR, Liu Y, Popa D et al (2006) Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leucoc Biol 79:1314–1327
Larbi A, Franceschi C, Mazzatti D et al (2008) Aging of the immune system as a prognostic factor for human longevity. Physiology 23:64–74
Lech M, Anders HJ (2013) Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta Mol Basis Dis 1832:989–997
Lee N, Shin MS, Kang I (2012) T-cell biology in aging, with a focus on lung disease. J Gerontol A 67:254–63
Lee Y, Kim SY, Bae Y (2014) Upregulation of miR-760 and miR-186 is associated with replicative senescence in human lung fibroblast cells. Mol Cells 37:620–627
Linge HM, Ochani K, Lin K et al (2015) Age-dependent alterations in the inflammatory response to pulmonary challenge. Immunol Res 63:209–215
Loeb M (2004) Pneumonia in the elderly. Curr Opin Infect Dis 17:127–130
López-Otín C, Blasco MA, Partridge L et al (2013) The hallmarks of aging. Cell 153:1194–217
Lu H, Yuan H, Chen S et al (2012) Senescent endothelial dysfunction is attributed to the up-regulation of sphingosine-1-phosphate receptor-2 in aged rats. Mol Cell Biochem 363:217–224
Maciel-Barón LA, Morales-Rosales SL, Aquino-Cruz AA et al (2016) Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age. doi:10.1007/s11357-016-9886-1
Mahbub S, Deburghgraeve CR, Kovacs EJ (2012) Advanced age impairs macrophage polarization. J Interf Cytokine Res 32:18–26
Maier JA, Statuto M, Ragnotti G (1993) Senescence stimulates U937-endothelial cell interactions. Exp Cell Res 208:270–274
Malandraki GA, Perlman AL, Karampinos DC, Sutton BP (2011) Reduced somatosensory activations in swallowing with age. Hum Brain Mapp 32:730–743
McClaran SR, Babcock MA, Pegelow DF et al (1995) Longitudinal effects of aging on lung function at rest and exercise in healthy active fit elderly adults. J Appl Physiol 78:1957–1968
Meiners S, Eickelberg O, Königshoff M (2015) Hallmarks of the ageing lung. Eur Respir J 45:807–827
Meyer KC (2005) Aging. Proc Natl Acad Sci U S A 2:433–9
Meyer K (2010) The role of immunity and inflammation in lung senescence and susceptibility to infection in the elderly. Sem Respir Crit Care Med 31:561–574
Meyer KC (2012) Management of interstitial lung disease in elderly patients. Curr Opin Pulm Med 18:483–92
Meyer KC, Ershler W, Rosenthal NS et al (1996) Immune dysregulation in the aging human lung. Am J Respir Crit Care Med 153:1072–1079
Miller MR (2010) Structural and physiological age-associated changes in aging lungs. Semin Respir Crit Care Med 31:521–527
Misharin AV, Morales-Nebreda L, Mutlu GM et al (2013) Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49:503–10
Moliva JI, Rajaram MVS, Sidiki S et al (2014) Molecular composition of the alveolar lining fluid in the aging lung. Age 36:1187–1199
Murciano C, Yáñez A, O’Connor JE et al (2008) Influence of aging on murine neutrophil and macrophage function against Candida albicans. FEMS Immunol Med Microbiol 53:214–221
Murray PJ, Allen JE, Biswas SK et al (2014) Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 41:14–20
Ochs M, Weibel ER (2008) Functional design of the human lung for gas exchange. In: Fishman AP (ed) Fishman’s Pulmonary Disease and Disorders, 4th edn. McGraw Hill, New York, pp 23–70
Odamaki T, Kato K, Sugahara H et al (2016) Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. doi:10.1186/s12866-016-0708-5
Ogawa K, Suzuki K, Okutsu M et al (2008) The association of elevated reactive oxygen species levels from neutrophils with low-grade inflammation in the elderly. Immun Ageing. doi:10.1186/1742-4933-5-13
Opal SM, Girard TD, Ely EW (2005) The immunopathogenesis of sepsis in elderly patients. Clin Infect Dis 41:S504–12
Orgeig S, Daniels CB (2004) Effects of aging, disease and the environment on the surfactant system. In: Harding R, Pinkerton KE, Plopper CG (eds) The lung: development, aging and the environment. Elsevier, San Diego, pp 363–375
Ortega-Martínez M, Rodríguez-Flores LE, Ancer-Arellano A et al (2016) Analysis of cell turnover in the bronchiolar epithelium through the normal aging process. Lung. doi:10.1007/s00408-016-9890-3
Petroianni A, Ceccarelli D, Conti V, Terzano C (2006) Aspiration pneumonia Pathophysiological aspects, prevention and management: A review. Panminerva Med 48:231–239
Podlutsky A, Ballabh P, Csiszar A (2010) Oxidative stress and endothelial dysfunction in pulmonary arteries of aged rats. AJP Heart Circ Physiol 298:H346–51
Pragman AA, Kim HB, Reilly CS et al (2012) The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS ONE. doi:10.1371/journal.pone.0047305
Quirk JD, Sukstanskii AL, Woods JC et al (2016) Experimental evidence of age-related adaptive changes in human acinar airways. J Appl Physiol 120:159–165
Raghu G, Weycker D, Edelsberg J et al (2006) Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174:810–816
Ray AD, Ogasa T, Magalang UJ et al (2008) Aging increases upper airway collapsibility in Fischer 344 rats. J Appl Physiol 105:1471–1476
Regina C, Panatta E, Candi E et al (2016) Vascular ageing and endothelial cell senescence: Molecular mechanisms of physiology and diseases. Mech Ageing Dev. doi:10.1016/j.mad.2016.05.003
Ricci A, Mariotta S, Greco S et al (1997) Age-related changes of the noradrenergic innervation of rat tracheo-bronchial tree and pulmonary vasculature. Mech Ageing Dev 99:245–255
Sahin E, DePinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464:520–8
Salminen A, Huuskonen J, Ojala J et al (2008) Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7:83–105
Schmitt V, Rink L, Uciechowski P (2013) The Th17/Treg balance is disturbed during aging. Exp Gerontol 48:1379–1386
Seebart BR, Stoffel RT, Behan M (2007) Age-related changes in the serotonin 2A receptor in the hypoglossal nucleus of male and female rats. Respir Physiol Neurobiol 158:14–21
Sharma G, Goodwin J (2006) Effect of aging on respiratory system physiology and immunology. Clin Interv Aging 1:253–260
Shaw AC, Panda A, Joshi SR et al (2011) Dysregulation of human Toll-like receptor function in aging. Ageing Res Rev 10:346–53
Small C-L, Mccormick S, Gill N et al (2008) NK cells play a critical protective role in host defense against acute extracellular. J Immunol 180:5558–68
Starr ME, Ueda J, Yamamoto S et al (2012) The effects of aging on pulmonary oxidative damage, protein nitration and extracellular dismutase down-regulation during systemic inflammation. Free Radic Biol Med 50:371–380. doi:10.1016/j.freeradbiomed.2010.11.013.The
Stout RD, Suttles J (2005) Immunosenescence and macrophage functional plasticity: dysregulation of macrophage function by age-associated microenvironmental changes. Immunol Rev 205:60–71
Stout-Delgado HW, Vaughan SE, Shirali AC et al (2012) Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol 188:2815–24
Suchyta MR, Clemmer TP, Elliott CG et al (1997) Increased mortality of older patients with acute respiratory distress syndrome. Chest 111:1334–1339
Suzuki M, Betsuyaku T, Ito Y et al (2008) Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 39:673–82
Svartengren M, Falk R, Philipson K (2005) Long-term clearance from small airways decreases with age. Eur Respir J 26:609–615
Sze MA, Dimitriu PA, Suzuki M et al (2015) The host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 192:438–445
Tasdemir N, Lowe SW (2013) Senescent cells spread the word: non-cell autonomous propagation of cellular senescence. EMBO J 32:1975–6
Tigges J, Krutmann J, Fritsche E et al (2014) The hallmarks of fibroblast ageing. Mech Ageing Dev 138:26–44
Toapanta FR, Ross TM (2009) Impaired immune responses in the lungs of aged mice following influenza infection. Respir Res. doi:10.1186/1465-9921-10-112
Torres-González E, Bueno M, Tanaka A et al (2012) Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis. Am J Respir Cell Mol Biol 46:748–56
Tsay T-B, Yang M-C, Chen P-H et al (2011) Gut flora enhance bacterial clearance in lung through toll-like receptors 4. J Biomed Sci. doi:10.1186/1423-0127-18-68
Tschudi MR, Barton M, Bersinger NA et al (1996) Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest 98:899–905
Tsuji T, Aoshiba K, Nagai A (2006) Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med 174:886–893
Van der Loo B, Labugger R, Skepper JN et al (2000) Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med 192:1731–44
Vanaudenaerde BM, Verleden SE, Vos R et al (2011) Innate and adaptive interleukin-17-producing lymphocytes in chronic inflammatory lung disorders. Am J Respir Crit Care Med 183:977–86
Vital M, Harkema JR, Rizzo M et al (2015) Alterations of the Murine Gut Microbiome with Age and Allergic Airway Disease. J Immunol Res. doi:10.1155/2015/892568
Vrijheid M (2014) The exposome: a new paradigm to study the impact of environment on health. Thorax 69:876–8
Walski M, Pokorski M, Antosiewicz J et al (2009) Pulmonary surfactant: ultrastructural features and putative mechanisms of aging. J Physiol Pharmacol 60:121–125
Weiskopf D, Weinberger B, Grubeck-Loebenstein B (2009) The aging of the immune system. Transpl Int 22:1041–50
Xu J, Gonzalez ET, Iyer SS et al (2009) Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J Gerontol A 64:731–739
Yanai H, Shteinberg A, Porat Z et al (2015) Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (iIpact) 7:664–672
Yang S-R, Park J-R, Kang K-S (2015) Reactive oxygen species in mesenchymal stem cell aging: implication to lung diseases. Oxidative Med Cell Longev. doi:10.1155/2015/486263
Yende S, Tuomanen EI, Wunderink R et al (2005) Preinfection systemic inflammatory markers and risk of hospitalization due to pneumonia. Am J Respir Crit Care Med 172:1440–6
Yoshida T, Tuder R (2007) Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 87:1047–1082
Zacca ER, Crespo MI, Acland RP et al (2015) Aging Impairs the ability of conventional dendritic cells to cross-prime CD8+ T cells upon stimulation with a TLR7 Ligand. PLoS ONE. doi:10.1371/journal.pone.0140672
Zaccagnini G, Gaetano C, Della Pietra L et al (2005) Telomerase mediates vascular endothelial growth factor-dependent responsiveness in a rat model of hind limb ischemia. J Biol Chem 280:14790–14798
Zemans RL, Colgan SP, Downey GP (2009) Transepithelial migration of neutrophils: Mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol 40:519–535
Zhao J, Zhao J, Legge K, Perlman S (2011) Age-related increases in PGD2 expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J Clin Invest 121:4921–30
Acknowledgments
The authors’ work is supported by DFG via the Cluster of Excellence Rebirth, BMBF via the Deutsches Zentrum für Lungenforschung (DZL) and by the Hochschulinterne Leistungsförderung (HilF) of the Hannover Medical School.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brandenberger, C., Mühlfeld, C. Mechanisms of lung aging. Cell Tissue Res 367, 469–480 (2017). https://doi.org/10.1007/s00441-016-2511-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-016-2511-x