
Abstract
Background
Recent studies have shown that gut microbiota can affect the development of Alzheimer’s disease (AD) through various mechanisms. Bile acids (BAs), which are the final byproducts of cholesterol metabolism created through both the human body and gut microbiome, appear to be influenced by gut microbiota and may impact AD pathological characteristics such as the accumulation of tau and amyloid-β. We aimed to investigate the associations between various serum BAs and CSF biomarkers (including Aβ, total tau, and p-tau). Additionally, we sought to examine the longitudinal changes in brain Aβ and tau through PET imaging in relation to BAs profile.
Methods
The data of 828 subjects including 491 diagnosed with mild cognitive impairment (MCI), 119 patients diagnosed with AD, and 267 cognitively normal (CN) participants were obtained from ADNI. The baseline and longitudinal [18F] florbetapir and [18F] flortaucipir PET standard uptake value ratios (SUVR) measures were obtained to assess the accumulation of tau and Aβ. Moreover, baseline levels of serum BAs and CSF Aβ1–42, tau, and p-tau were used.
Results
After FDR correction we observed that five BAs level and relevant calculated ratios were associated with CSF p-tau and tau, three with CSF Aβ1–42. Furthermore, three BAs level and relevant calculated ratios were associated with the tau-PET rate of change, and two with the Aβ rate of change.
Conclusion
The findings from our study suggest a correlation between altered profiles of BAs and CSF and imaging biomarkers associated with AD. These results provide supporting evidence for the link between the gut microbiome and the pathological features of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The gut-brain axis is a bidirectional communication system between the gastrointestinal tract and the central nervous system, which plays a crucial role in maintaining overall health. Recent studies have shown that the gut microbiota can affect the development of Alzheimer’s disease (AD) through various mechanisms, including neuroinflammation, amyloid-beta (Aβ) deposition, and tau protein accumulation. The gut microbiota can affect AD through several pathways, including the modulation of neurochemical and neurometabolic pathways, synthesis and secretion of neurotrophic factors, and the production of microbial molecules that influence brain function [1].
Bile acids (BAs), essential for the digestion and absorption of dietary fats, are synthesized from cholesterol in the liver. This process involves various enzymatic reactions, leading to modifications of the cholesterol molecule and the formation of bile acids with detergent-like properties. BA synthesis occurs through two major pathways: the neutral pathway, which modifies the steroid ring before side-chain cleavage, and the acidic pathway, which follows the opposite order. Enzymes located in different cellular compartments catalyze these reactions. Cholesterol 7α-hydroxylase initiates the neutral pathway and produces primary BAs, cholic acid, and chenodeoxycholic acid. Sterol 27-hydroxylase contributes to BA synthesis through the acidic pathway, particularly in specific conditions [2]. Following synthesis, most BAs are conjugated to glycine or taurine to enhance solubility and ionization. Conjugation prevents precipitation, minimizes absorption, and protects BAs from enzymatic cleavage. In the intestine, conjugated BAs are deconjugated and metabolized by gut bacteria, forming secondary BAs such as deoxycholic acid (DCA) and lithocholic acid (LCA) [3]. The enterohepatic circulation ensures the efficient recycling of BAs. BAs are secreted into bile, stored in the gallbladder, and released into the intestine during digestion. The majority of BAs are reabsorbed in the terminal ileum and transported back to the liver, maintaining a constant BA pool. BAs lost in feces are replenished by de novo synthesis in the liver [4].
BAs have been receiving growing attention in the context of AD and its potential link to the brain-gut-microbiota (BGM) axis [5]. BAs have the potential to regulate various aspects of the BGM axis, encompassing neural, immune, and neuroendocrine pathways. Within these pathways, secondary BAs, which are produced by microorganisms, can influence each of these processes. The pathologic effects of BAs may occur through multiple mechanisms, such as activating the farnesoid X receptor and inhibiting BA synthesis in the brain, blocking NMDA receptors, reducing levels of brain oxysterols, and interfering with the actions of 17β-estradiol [6].
To further address these controversies and overcome the limitations of the previous studies including small sample size and cross-sectional design, we aimed to investigate the associations between various serum BAs and cerebrospinal fluid (CSF) biomarkers (including Aβ, total tau, and p-tau). Additionally, we sought to examine the longitudinal changes in brain Aβ and tau through PET imaging in relation to BAs profile. By doing so, we aimed to provide a more comprehensive understanding of the associations between serum BAs and AD biomarkers while considering the dynamic changes over time.
Materials and methods
Data acquisition
The Alzheimer’s Disease Neuroimaging Initiative (ADNI) database provided the data for this investigation (adni.loni.usc.edu). The ADNI was founded in 2003 as a public-private partnership directed by Principal Investigator Michael W. Weiner, MD. The initial purpose of ADNI is to assess the progression of mild cogntive impairment (MCI) and early AD by combining all serial PET, MRI, biological markers, and clinical and neuropsychological measures. www.adni-info.org has the most up-to-date information.
Participants
In this study, we utilized the data collected from the baseline visits of participants in the ADNI cohort who had available serum BA levels, CSF biomarkers, and longitudinal PET. From the dataset, we included participants aged between 55 and 90 years, who had a minimum of 6 years education, fluent in either Spanish or English, without any significant neurological disorder other than AD, and had available baseline serum BA levels, baseline CSF Aβ1–42, tau, and p-tau, available longitudinal Aβ-PET and tau-PET scans. Specifically, we included 491 individuals diagnosed with MCI, 119 patients diagnosed with AD, and 267 cognitively normal (CN) participants. All individuals with MCI were identified as having amnestic MCI based on specific criteria, which included a memory complaint, an education-adjusted score on the Wechsler Memory Scale Logical Memory II test indicating objective memory loss, a Mini-Mental State Examination (MMSE) score between 24 and 30, a Clinical Dementia Rating (CDR) of 0.5, preserved activities of daily living, and no significant impairment in other cognitive domains or signs of dementia. The ADNI participants with AD were diagnosed according to the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) probable AD criteria, with MMSE scores ranging from 20 to 26 and CDR of either 0.5 or 1.
Assessment of CSF Aβ1–42, tau, and p-tau
The Luminex platform was used to obtain CSF samples, and the levels of specific CSF biomarkers, including Aβ1–42, total tau, and p-tau, were measured using Luminex’s micro-bead-based multiplex immunoassay. Additional information regarding the collection of CSF specimens and analytical measurement can be found on the ADNI website (http://adni.loni.usc.edu/methods/documents/).
PET imaging biomarkers
Aβ deposition was visualized with PET tracer [18F]AV45 [7]. The measurements of regional Aβ standard uptake value ratios (SUVR) that were calculated by the ADNI core at UC Berkeley, which they accessed through adni.loni.usc.edu were acquired. To determine global Aβ deposition, we standardized the mean uptake in several regions of interest (ROIs), including prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus, by dividing it by the cerebellum and composite reference region for cross-sectional and longitudinal analysis, respectively. We obtained Aβ SUVR values at various time points to estimate the rate of change.
Data of tau accumulation measured by tau-PET were retrieved from the ADNI server (Jagust Lab, UC Berkeley). The image processing techniques were previously described [8]. Tau-PET SUVR was defined by averaging flortaucipir (AV-1451) uptake in the Braak stage composite regions divided by a reference region such as inferior cerebellar GM (cross-sectional) or hemispheric WM (Longitudinal). The rate of change of tau-PET was estimated using available data of baseline and follow-up visits.
Serum bile acids profile
We used quantitated data of BAs in human serum samples provided by Duke University for the ADNI GO/2 and ADNI 1 projects. The samples were stored at -80 °C until preparation and analysis using a bile acid-free matrix (BAFM) prepared using a charcoal-stripping protocol to minimize analytical variations and compensate for matrix effects. Sample preparation involved spiking each 50 µL of serum with 150 µL of acetonitrile containing internal standards, followed by extraction of bile acids using a laboratory shaker and centrifugation. The supernatant was lyophilized, reconstituted, and analyzed using ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC-MS/MS). Quality control (QC) samples, including test mixtures and pooled biological samples, were used along with internal standards to monitor the stability of the large-scale analysis. Any metabolite with signal-to-noise ratio < 3.0 was rejected from statistical analysis, and the RSDs for each metabolite in the test mixtures and pooled QC samples measured were calculated. The study found that the RSDs of the lower concentration metabolites in the reference standard mixture were less than 30%, and the R.S.D. for the higher concentration metabolites shall be better than 15% for each batch of sample analysis. Overall, the study provides a reliable method for quantifying bile acids in human serum samples, which could facilitate the diagnosis and treatment of diseases associated with altered bile acid metabolism. The level of 33 serum BAs and 3 ratios were entered into the analysis.
The CA: chenodeoxycholic acid (CDCA) ratio was picked to investigate whether a change in the production of BAs occurs in the liver, moving from the primary to the alternative pathway. Additionally, the DCA:CA and glycolithocholic acid (GLCA):CDCA ratios were analyzed to observe variations in the gut microbiome’s enzymatic activity that might result in differences in the creation of secondary BAs.
ApoE genotyping
The APOE genotyping of MCI patients was performed on collected blood samples. The participants with at least one ε4 allele are considered carriers, as described by ADNI (http://adni.loni.usc.edu/methods/documents/).
Cognitive assessments
All participants underwent Mini-Mental State Examination (MMSE) which included 30 questions to measure the cognitive status at baseline. The MMSE score of patients was downloaded from ADNI.
Statistical analysis
The difference in demographical, clinical, and CSF biomarkers between groups was measured using ANOVA analysis. To investigate the association between serum Bas level and CSF and PET biomarkers, multivariable linear regression models adjusted for the effect of age, sex, APOE ε4, and BMI were used. To address the false discovery rate (FDR) due to the multiple comparisons, we used the Benjamini-Hochberg method. R software (version 3.3.3; R Foundation for Statistical Computing, Vienna, Austria) was used for statistical analysis.
Results
Participant’s characteristics
The demographical and clinical characteristics of the participants are detailed in Table 1. The level of 33 serum BA levels and 3 ratios were available for analysis (Table 2). The average time interval between the baseline and follow-up scans for Aβ-PET and tau-PET were 2.12 (± 1.88) and 1.65 (± 1.72) years respectively.
CSF biomarkers and serum BA
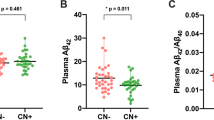
We investigated the association between cross-sectional CSF Aβ1–42, tau, and p-tau with 33 serum BA levels and 4 relevant ratios using multivariable linear regression models while adjusting for age, sex, APOE ε4, and BMI. After FDR correction we observed a significant correlation between CSF Aβ1–42 and one BA serum level and two ratios. The CSF Aβ1–42 was negatively associated with HDCA (r:-0.081, p:0.021), GLCA/CDCA ratio (r:-0.083, p:0.019), and DCA/CA ratio (r:-0.100, p:0.004) (Fig. 1).

Significant associations between CSF biomarkers and BA profile after FDR correction
Using the same model, we identified three BA serum levels and two ratios to be associated with CSF tau level (Fig. 1). We found that individuals with a higher level of CSF tau have higher serum BA levels including GCDCA (r: 0.070, p:0.046), glycodeoxycholic acid (GDCA) (r: 0.108, p:0.002), and HDCA (r: 0.085, p:0.015). Furthermore, there was a positive correlation between CSF tau with DCA/CA (r: 0.078, p:0.027) and GLCA/CDCA ratios (r: 0.080, p:0.024).
Also, we found that GDCA (r:0.106, p:0.002), GLCA (r:0.087, p:0.014), and HDCA serum level (r:0.096, p:0.006) were associated with CSF p-tau level among our participants (Fig. 1). Moreover, two BA ratios including DCA/CA (r:0.089, p:0.012) and GLCA/CDCA (r:0.082, p:0.020) were found to be correlated with CSF p-tau.
PET imaging and serum BA
We aimed to investigate whether the Aβ- or tau-PET is associated with the BA profile. Using the same regression model used for CSF biomarkers there was no significant association between baseline Aβ deposition and BA serum levels and ratios. However, when we examined the association between Aβ-PET rate of change and BA profile we found significant results (Fig. 2). The GDCA (r:0.106, p:0.049) and 7-KETOLCA (r:-0.114, p:0.026) were associated with the Aβ-PET rate of change.

Significant associations between Aβ- and tau-PET and BA profile after FDR correction
Based on our analysis there was no significant association between baseline tau-PET and BA serum levels and ratios. While cross-sectional was non-significant, the longitudinal changes in tau-PET (rate of change) were associated with GDCA (r:0.264, p:0.004), taurodeoxycholic acid (TDCA) (r:0.229, p:0.013) level, and DCA/CA ratio (r:0.183, p:0.005) after FDR correction (Fig. 2).
Discussion
In this study, we examined the BA profiles in the ADNI cohort to explore the correlation between peripheral metabolic indicators and central biomarkers related to AD pathophysiology. Additionally, we studied the connection between the rate of change in Aβ-PET and metabolic measures over time. The findings from our study indicated a significant relationship between BA profiles and three CSF biomarkers: Aβ1–42, tau, and p-tau. Our study revealed a negative association between levels of CSF Aβ1–42 and serum HDCA, as well as two ratios of secondary to primary BAs (DCA:CA and GLCA:CDCA). Additionally, a positive correlation was observed between CSF tau levels and serum GCDCA, GDCA, and HDCA levels, along with the same two secondary to primary BAs ratios. A positive correlation was also found between CSF p-tau levels and serum GLCA, GDCA, and HDCA levels, as well as the aforementioned ratios of secondary to primary BAs. Moreover, our longitudinal analysis revealed significant associations between the rate of change in Aβ-PET and levels of two BAs: GDCA and 7-KETOLCA. Additionally, we found that the rate of change in tau-PET was linked to levels of GDCA, TDCA, and the DCA:CA ratio.
Several studies investigated the association between serum BA concentrations and CSF and imaging biomarkers and symptoms of AD. Furthermore, a study on 1,464 subjects including cognitively normal, patients with MCI, and patients with AD showed that the while the serum levels of cholic acid (a primary BA) were significantly lower in AD patients compared to controls, AD patients had significantly higher serum levels of secondary bacterially-produced BAs including DCA and its glycine and taurine conjugated forms. Also, the ratios of secondary BAs to primary BAs such as DCA: CA, GDCA: CA, and TDCA: CA ratios were significantly associated with worse cognitive functions and were significantly higher in AD compared to MCI [9]. In another study, Nho et al. investigated the association between serum levels of 23 BAs and CSF and PET biomarkers of AD. They showed that GDCA: CA, TDCA: CA, and GLCA:CDCA ratios negatively correlated with CSF Aβ1–42 while there was a positive correlation between GDCA: CA ratio and amyloid deposition. Moreover, they found that higher secondary to primary BA ratios were significantly associated with higher CSF p-tau, lower hippocampal volume, and reduced brain glucose metabolism [10]. Additionally, in a longitudinal study, Varma et al. reported sex-specific associations between serum BAs and rates of brain atrophy indicating that in males, lower serum CDCA and CA levels were associated with faster rates of atrophy in total gray matter volume and various brain regions. However, in females, the results were controversial showing that in the Baltimore Longitudinal Study of Aging (BLSA) population, lower serum CA levels were associated with slower rates of total gray matter atrophy whereas in the ADNI population, lower CA levels correlated with faster rates of atrophy in only the fusiform gyrus and temporal gray matter [11].
The involvement of hepatic cholesterol metabolism is proposed to play a role in AD pathogenesis [12]. Cholesterol in the liver undergoes a process of synthesis to produce primary BAs, namely CDCA and cholic acid CA. These primary BAs are conjugated with glycine or taurine, then released into the gallbladder and subsequently transported to the intestine [13]. The liver-derived BAs are deconjugated and are further transformed into secondary BAs by intestinal anaerobic bacteria. For instance, CA is converted into DCA, while CDCA can be transformed into LCA or UDCA through either 7α-dehydroxylation or 7β-dehydroxylation, respectively [13,14,15,16]. Thus, The ratios of specific secondary to primary BAs, such as DCA:CA and GLCA:CDCA, typically indicate alterations in the gut microbial metabolism [16]. BAs are reabsorbed in the colon and terminal ileum and then released into the portal vein to be transported back to the liver, where they undergo conjugation to form their glycine and taurine derivatives [9].
Several metabolomics investigations conducted on individuals with AD or animal models have revealed substantial disparities in the composition and levels of BAs present in AD patients’ serum and brain tissue compared to healthy individuals [17,18,19,20,21,22,23]. A recent large-scale clinical study revealed that lower serum CA and CDCA concentrations were associated with faster brain atrophy in patients [11]. Moreover, the administration of supplemental UDCA, CDCA, and TUDCA has been shown to have beneficial effects in alleviating Aβ deposition, inhibiting Aβ-induced synaptic toxicity, reducing neuroinflammation, improving mitochondrial function, and ameliorating cognitive deterioration [24,25,26,27,28]. These findings indicate that altered BA profiles could contribute to cognitive dysfunction in AD and could potentially serve as a biomarker for early AD detection.
Interestingly, our study, for the first time, demonstrated that GDCA and 7-KETOLCA levels were associated with the Aβ-PET rate of change, and GDCA and TDCA levels and DCA:CA ratio were associated with the tau-PET rate of change. The novel longitudinal patterns of association between specific BA profiles and PET biomarkers observed in our study indicate a potential mechanistic connection between peripheral and central biochemical changes, highlighting the potential role of the gut-liver-brain axis in AD pathophysiology. Recent research on the gut-brain axis has revealed an emerging consensus that BAs, as metabolites intricately linked to the intestinal flora, play a crucial role as messengers within this axis [29, 30].
Previous research suggests a link between gut microbiota and brain amyloidosis. Mouse AD models lacking intestinal microbiota exhibited remarkably lower cerebral Aβ pathology than control mice with normal gut microbiota populations. Introducing microbiota from control AD mice into germ-free AD models led to a significant increase in Aβ pathology [31]. A hypothesis posits that disrupted gut microbiota and increased intestinal permeability facilitate the accumulation of amyloids derived from bacteria in the gastrointestinal tract at both systemic and brain levels, consequently promoting the buildup of Aβ42, potentially contributing to hippocampal dysfunction [32,33,34,35,36]. In addition, differences in the bacterial taxonomic composition of fecal samples were observed between cognitively impaired patients and controls. These differences included decreased levels of Firmicutes, Bifidobacterium, and Eubacterium rectale and increased levels of Bacteroidetes, Escherichia, and Shigella in the microbiome of patients. Notably, these variations were correlated with CSF biomarkers in both the patient and the control groups [37, 38]. These findings provide additional support to the growing evidence linking the gut microbiota with the development of brain amyloidosis.
Limitations
As an observational study, the ADNI and other similar cross-sectional investigations pose challenges in controlling for confounding variables and unraveling the cause-and-effect relationships. For instance, numerous factors, such as sleep patterns, medications, nutrition, and environmental influences, impact the gut microbiota population, potentially contributing to the onset of AD [39, 40]. In this research, we thoroughly investigated age, gender, APOE ε4, and BMI as possible confounding factors. However, additional factors, such as medication usage and sleep patterns, were not taken into consideration. Consequently, future experimental studies are required to unravel the intricate mechanisms through which BA influences AD progression and untangle the web of causality.
Given the lack of adequate data, we could not examine the longitudinal changes in BA profiles. While a connection has been uncovered linking altered gut microbiome, BA profiles, and AD, it’s important to note that correlation does not necessarily imply causation. The microbiome and BA metabolism might change due to AD-related factors such as weight loss, medication use, or sleep disruption [39, 41]. Up to now, there haven’t been any extensive longitudinal clinical studies that have gathered fecal samples to examine alterations in the composition of the gut microbiome thoroughly. To further elucidate the association between BA profiles, gut dysbiosis, and AD-related pathologies, future large-scale studies are required to investigate these relationships over an extended period of time.
Conclusions
To our knowledge, this is the first study examining the link between altered BA profiles and the longitudinal shifts in brain biomarkers, as identified through PET imaging. The findings from our study suggest a correlation between altered profiles of secondary BAs and CSF biomarkers associated with AD. These results provide supporting evidence for the link between the gut microbiome and the pathological features of AD. Furthermore, through our longitudinal analysis, we revealed novel connections between the pace of alteration in Aβ-PET and tau-PET and the specific BA profiles, suggesting a possible mechanistic link between peripheral and central biochemical transformations and shedding light on the potential involvement of the gut-liver-brain axis in the pathophysiology of AD. Although our findings offer additional support for the involvement of BA signaling pathways in AD, the precise cause-and-effect relationship still demands comprehensive exploration through experimental studies. Moreover, future extensive longitudinal studies are necessary to establish the connection between altered BA profiles, gut microbiome, and AD-related pathology and to elucidate intricate mechanisms through which gut bacteria and BAs contribute to the pathophysiology of AD.
Data availability
The datasets analyzed during the current study are available upon request with no restriction.
References
Yin JT, Xu XW, Jin CY, Yuan XY, Wang XG (2023) The influence of the gut microbiota on Alzheimer’s Disease: a narrative review. J Integr Neurosci 22(2):38. https://doi.org/10.31083/j.jin2202038
Šarenac TM, Mikov M (2018) Bile acid synthesis: from nature to the Chemical modification and synthesis and their applications as drugs and nutrients. Front Pharmacol 9:939. https://doi.org/10.3389/fphar.2018.00939
Chiang JY (2013) Bile acid metabolism and signaling. Compr Physiol 3(3):1191–1212. https://doi.org/10.1002/cphy.c120023
Stellaard F, Lütjohann D (2021) Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am J Physiol Gastrointest Liver Physiol 321(1):G55–g66. https://doi.org/10.1152/ajpgi.00476.2020
Mulak A (2021) Bile acids as key modulators of the Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J Alzheimers Dis 84(2):461–477. https://doi.org/10.3233/jad-210608
Ehtezazi T, Rahman K, Davies R, Leach AG (2023) The pathological effects of circulating hydrophobic bile acids in Alzheimer’s Disease. J Alzheimers Dis Rep 7(1):173–211. https://doi.org/10.3233/adr-220071
Choi SR, Golding G, Zhuang Z, Zhang W, Lim N, Hefti F et al (2009) Preclinical properties of 18F-AV-45: a PET agent for Abeta plaques in the brain. J Nucl Med 50(11):1887–1894. https://doi.org/10.2967/jnumed.109.065284
Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL et al (2011) Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32(7):1207–1218. https://doi.org/10.1016/j.neurobiolaging.2009.07.002
MahmoudianDehkordi S, Arnold M, Nho K, Ahmad S, Jia W, Xie G et al (2019) Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement 15(1):76–92. https://doi.org/10.1016/j.jalz.2018.07.217
Nho K, Kueider-Paisley A, MahmoudianDehkordi S, Arnold M, Risacher SL, Louie G et al (2019) Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: relationship to neuroimaging and CSF biomarkers. Alzheimers Dement 15(2):232–244. https://doi.org/10.1016/j.jalz.2018.08.012
Varma VR, Wang Y, An Y, Varma S, Bilgel M, Doshi J et al (2021) Bile acid synthesis, modulation, and dementia: a metabolomic, transcriptomic, and pharmacoepidemiologic study. PLoS Med 18(5):e1003615. https://doi.org/10.1371/journal.pmed.1003615
Di Paolo G, Kim T-W (2011) Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci 12(5):284–296. https://doi.org/10.1038/nrn3012
Chiang JYL, Ferrell JM (2018) Bile Acid Metabolism in Liver Pathobiology. Gene Expr 18(2):71–87. https://doi.org/10.3727/105221618X15156018385515
Ridlon JM, Kang D-J, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47(2):241–259. https://doi.org/10.1194/jlr.R500013-JLR200
Donova MV (2007) [Transformation of steroids by actinobacteria: a review]. Prikl Biokhim Mikrobiol 43(1):5–18
Guzior DV, Quinn RA (2021) Review: microbial transformations of human bile acids. Microbiome 9(1):140. https://doi.org/10.1186/s40168-021-01101-1
Weng Z-B, Chen Y-R, Lv J-T, Wang M-X, Chen Z-Y, Zhou W et al (2022) A review of bile acid metabolism and signaling in cognitive dysfunction-related diseases. Oxidative medicine and cellular longevity. 2022:4289383. https://doi.org/10.1155/2022/4289383
Pan X, Elliott CT, McGuinness B, Passmore P, Kehoe PG, Hölscher C et al (2017) Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer’s Disease. Metabolites 7(2):28. https://doi.org/10.3390/metabo7020028
Baloni P, Funk CC, Yan J, Yurkovich JT, Kueider-Paisley A, Nho K et al (2020) Metabolic Network Analysis reveals altered bile acid synthesis and metabolism in Alzheimer’s Disease. Cell Rep Med 1(8):100138. https://doi.org/10.1016/j.xcrm.2020.100138
Koike S, Miyaji Y, Sano H, Aikawa N, Kai M, Kasahara S et al (2021) Simultaneous determination of five bile acids as potential biomarkers for Alzheimer’s Disease in Mouse Brain and plasma. Anal Sci 37(8):1165–1170. https://doi.org/10.2116/analsci.20P429
Marksteiner J, Blasko I, Kemmler G, Koal T, Humpel C (2018) Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 14(1):1. https://doi.org/10.1007/s11306-017-1297-5
Shao Y, Ouyang Y, Li T, Liu X, Xu X, Li S et al (2020) Alteration of Metabolic Profile and potential biomarkers in the plasma of Alzheimer’s Disease. Aging Disease 11(6):1459. https://doi.org/10.14336/AD.2020.0217
Sun C, Gao M, Wang F, Yun Y, Sun Q, Guo R et al (2020) Serum metabolomic profiling in patients with Alzheimer disease and amnestic mild cognitive impairment by GC/MS. Biomed Chromatogr 34(9). https://doi.org/10.1002/bmc.4875
Nunes AF, Amaral JD, Lo AC, Fonseca MB, Viana RJS, Callaerts-Vegh Z et al (2012) TUDCA, a bile acid, attenuates amyloid precursor protein Processing and Amyloid-β deposition in APP/PS1 mice. Mol Neurobiol 45(3):440–454. https://doi.org/10.1007/s12035-012-8256-y
Ramalho RM, Nunes AF, Dias RB, Amaral JD, Lo AC, D’Hooge R et al (2013) Tauroursodeoxycholic acid suppresses amyloid β-induced synaptic toxicity in vitro and in APP/PS1 mice. Neurobiol Aging 34(2):551–561. https://doi.org/10.1016/j.neurobiolaging.2012.04.018
Bell SM, Barnes K, Clemmens H, Al-Rafiah AR, Al-ofi EA, Leech V et al (2018) Ursodeoxycholic acid improves mitochondrial function and redistributes Drp1 in fibroblasts from patients with either sporadic or familial Alzheimer’s Disease. J Mol Biol 430(21):3942–3953. https://doi.org/10.1016/j.jmb.2018.08.019
Zangerolamo L, Vettorazzi JF, Solon C, Bronczek GA, Engel DF, Kurauti MA et al (2021) The bile acid TUDCA improves glucose metabolism in streptozotocin-induced Alzheimer’s disease mice model. Mol Cell Endocrinol 521:111116. https://doi.org/10.1016/j.mce.2020.111116
Bazzari FH, Abdallah DM, El-Abhar HS (2019) Chenodeoxycholic Acid ameliorates AlCl3-Induced Alzheimer’s Disease neurotoxicity and cognitive deterioration via enhanced insulin signaling in rats. Molecules 24(10):1992. https://doi.org/10.3390/molecules24101992
Jia W, Xie G, Jia W (2018) Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Reviews Gastroenterol Hepatol 15(2):111–128. https://doi.org/10.1038/nrgastro.2017.119
Schoeler M, Caesar R (2019) Dietary lipids, gut microbiota and lipid metabolism. Reviews Endocr Metabolic Disorders 20(4):461–472. https://doi.org/10.1007/s11154-019-09512-0
Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G et al (2017) Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep 7:41802. https://doi.org/10.1038/srep41802
Zhao Y, Lukiw WJ (2015) Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J Nat Sci. ;1(7)
Alam MZ, Alam Q, Kamal MA, Abuzenadah AM, Haque A (2014) A possible link of gut microbiota alteration in type 2 diabetes and Alzheimer’s disease pathogenicity: an update. CNS Neurol Disord Drug Target 13(3):383–390. https://doi.org/10.2174/18715273113126660151
Daulatzai MA (2014) Chronic functional bowel syndrome enhances gut-brain axis dysfunction, neuroinflammation, cognitive impairment, and vulnerability to dementia. Neurochem Res 39(4):624–644. https://doi.org/10.1007/s11064-014-1266-6
Daulatzai MA (2014) Role of stress, depression, and aging in cognitive decline and Alzheimer’s disease. Curr Top Behav Neurosci 18:265–296. https://doi.org/10.1007/7854_2014_350
Berger AL (2016) Insulin resistance and reduced brain glucose metabolism in the aetiology of Alzheimer’s disease. J Insulin Resist 1(1). https://doi.org/10.4102/jir.v1i1.15
Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC et al (2017) Gut microbiome alterations in Alzheimer’s disease. Sci Rep 7(1):13537. https://doi.org/10.1038/s41598-017-13601-y
Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C et al (2017) Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging 49:60–68. https://doi.org/10.1016/j.neurobiolaging.2016.08.019
Morwani-Mangnani J, Giannos P, Belzer C, Beekman M, Eline Slagboom P, Prokopidis K (2022) Gut microbiome changes due to sleep disruption in older and younger individuals: a case for Sarcopenia? Sleep 45(12):zsac239. https://doi.org/10.1093/sleep/zsac239
Singh RK, Chang H-W, Yan D, Lee KM, Ucmak D, Wong K et al (2017) Influence of diet on the gut microbiome and implications for human health. J Translational Med 15(1):73. https://doi.org/10.1186/s12967-017-1175-y
Seganfredo FB, Blume CA, Moehlecke M, Giongo A, Casagrande DS, Spolidoro JVN et al (2017) Weight-loss interventions and gut microbiota changes in overweight and obese patients: a systematic review. Obes Rev 18(8):832–851. https://doi.org/10.1111/obr.12541
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding
We do not have any financial support for this study.
Author information
Authors and Affiliations
Consortia
Contributions
FN & PV: Designed the study, analyzed the data, and wrote the paper; FN, PV & MF: collected data, analyzed and interpreted the data, and wrote the draft version of the manuscript. The manuscript was revised and approved by all authors.
Corresponding author
Ethics declarations
Ethical approval
Since the data in this paper were obtained from the ADNI database (adni.loni.usc.edu), it does not include any research involving human or animal subjects.
Consent for publication
This manuscript has been approved for publication by all authors.
Conflict of interest
The author declares no conflict of interest regarding the publication of this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
**Data used to prepare this article were obtained from the Alzheimer’s disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nabizadeh, F., Valizadeh, P., Fallahi, M.S. et al. Bile acid profile associated with CSF and PET biomarkers in Alzheimer’s disease. Aging Clin Exp Res 36, 62 (2024). https://doi.org/10.1007/s40520-024-02729-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40520-024-02729-3