
Abstract
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) that is cytogenetically characterized by a balanced reciprocal translocation between chromosomes 15 and 17, which results in the fusion of the promyelocytic leukemia (PML) and retinoic acid receptor alpha (RARα) genes. Because patients with APL present a tendency for severe bleeding, often resulting in an early fatal course, APL was historically considered to be one of the most fatal forms of acute leukemia. However, therapeutic advances, including anthracycline- and cytarabine-based chemotherapy, have significantly improved the outcomes of APL patients. Due to the further introduction of all-trans retinoic acid (ATRA) and—more recently—the development of arsenic trioxide (ATO)-containing regimens, APL is currently the most curable form of AML in adults. Treatment with these new agents has introduced the concept of cure through targeted therapy. With the advent of revolutionary ATRA-ATO combination therapies, chemotherapy can now be safely omitted from the treatment of low-risk APL patients. In this article, we review the six-decade history of APL, from its initial characterization to the era of chemotherapy-free ATRA-ATO, a model of cancer-targeted therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute promyelocytic leukemia (APL) is a rare subtype of acute myeloid leukemia (AML) that accounts for < 10% of all AMLs, with an estimated incidence of 0.1/100,000 in Western countries [1]. APL is characterized by a distinctive blast morphology, the presence of coagulopathy at diagnosis, and a specific balanced reciprocal translocation t(15;17), which fuses the promyelocyte (PML) gene on chromosome 15 to the retinoic acid receptor alpha (RARα) gene on chromosome 17. APL is a medical emergency requiring prompt diagnosis and management. Indeed, the disease frequently presents in combination with a consumptive life-threatening coagulopathy. The treatment of APL has undergone a revolution over the past two decades due to the advent of therapy based on all-trans retinoic acid (ATRA) and arsenic trioxide (ATO), which are known to target RARα and PML, the two distinct moieties of the disease-specific oncoprotein PML/RARα [2]. The results of large trials of various combinations of ATRA, ATO, and chemotherapy in a number of countries have transformed the prognosis for this type of leukemia, with long-term remission rates now exceeding 80% [3,4,5,6]. Recently, large randomized trials comparing the standard ATRA plus chemotherapy approach with an ATRA-ATO chemotherapy-free combination have shown a benefit of the chemotherapy-free approach, leading to new treatment guidelines for APL patients [7,8,9]. Thus, the story of APL can serve as a model for the development of targeted therapies and curative approaches for malignant diseases.
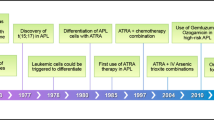
In this brief review, we examine the major advances made in the history of APL therapy, from the initial description of the disease to its present status (Fig. 1). Targeted more specifically at the young hematologist and the general educated reader, this review provides accounts of scientific and medical discoveries and practices in this domain. It utilizes chronology as an organizing framework while stressing the importance of themes.

Highlights in the history of APL treatment
The article is based on previously conducted studies and does not contain any studies with human participants or animals performed by the author.
First Descriptions and Treatments
APL was first described in 1957 by the Norwegian hematologist Leif Hillestad, who detailed three cases of a special type of AML characterized by “a very rapid fatal course of only a few weeks’ duration, a white blood cell picture dominated by promyelocytes, a severe bleeding tendency due to fibrinolysis and thrombocytopenia, a normal erythrocyte sedimentation rate, probably caused by the reduced fibrinogen concentration in the plasma.” He suggested denoting this type of AML acute promyelocytic leukemia, and concluded that “it seems to be the most malignant form of acute leukemia” [10] (Fig. 2). One of those three patients was previously described by Stormorken [11], and in his paper Hillestad mentioned three other previously reported [12,13,14] patients with identical symptoms to his cases, who may therefore also have been APL patients.

APL was first described in 1957 by Leif Hillestad, who published three cases of a special type of AML in Acta Medica Scandinavica
More detailed features of APL were described by Bernard in 1959, who reported a series of 20 patients [15]. Severe hemorrhagic diathesis was recognized as the most prominent clinical feature of APL, as it accounted for most of the fatal events at presentation and during initial cytotoxic treatment. This has been ascribed to disseminated intravascular coagulation (DIC) or hyperfibrinolysis triggered by APL blasts [16].
The predominant malignant cells were described as abnormal promyelocytes with an immature nucleus and a cytoplasm filled with azurophilic granules. In 1976, the French-American-British (FAB) Nomenclature Committee categorized them as M3 cells (hypergranular promyelocytic leukemia), with the following characteristics: “The great majority of cells are abnormal promyelocytes, with a characteristic pattern of heavy granulation. The nucleus varies greatly in size and shape and is often reniform or bilobed. The cytoplasm of most of the cells is completely occupied by closely packed or even coalescent large granules. In some cells the cytoplasm is filled with fine dust-like granules. Characteristic cells containing bundles of Auer rods (‘faggots’) randomly distributed in the cytoplasm are almost invariably present in the bone marrow and sometimes in the peripheral blood. A variable but often high proportion of the hypergranular promyelocytes and the faggot cells are disrupted” [17]. Four years later, a microgranular variant (M3v) form of the disease with usually bilobed cell nuclei, no granules visible on light microscopy, and a positive myeloperoxidase reaction was identified in approximately 20% of the cases and recognized in an updated FAB classification [18] (Fig. 3). A very rare third variant form—basophilic microgranular APL—was described in 1982 [19], and is characterized by cells with a high nucleus/cytoplasm ratio and strongly basophilic cytoplasm with sparse or no granules.

Morphologic variants of APL
Early studies with induction including 6-mercaptopurine (6-MP) alone or in combination with steroids, methyl-glyoxalguanyl hydrazine, and/or methotrexate led to poor results with complete remission (CR) rates ranging from 5 to 14% and survival periods of 3–16 weeks (median 3.5 weeks) among all patients and 4 months to more than 6 years among responders. Furthermore, the best way of managing the bleeding diathesis remained unclear [15, 20,21,22].
The first important success in APL therapy was reported by Bernard et al. [23], who showed that the disease was particularly sensitive to treatment with anthracyclines, and obtained high rates of CR (19 of 34 APL patients; 55%) with daunorubicin given as monotherapy. A significant proportion of these patients (approximately 30%) experienced long-term remission. Consequently, anthracycline-based chemotherapy became one of the mainstays of APL treatment.
By the end of this period, APL had been morphologically and clinically characterized but kept a dismal prognosis, especially because of fatal coagulation disorders.
Chemotherapy During the Pre-All-Trans Retinoic Acid Era
Between 1980 and 1988, studies focused on the management of APL treatment and on the identification of prognostic factors. The beneficial effect of anthracycline treatment was confirmed [24,25,26,27]. Following the demonstration that APL cells are relatively sensitive to daunorubicin [23] and that AML cells respond to cytarabine [28], chemotherapy composed of an anthracycline and cytosine arabinoside became the frontline treatment for APL. With this combination, CR rates reached 80% in newly diagnosed patients [29], but were not very different from those seen for treatment with anthracycline alone [25, 29,30,31]. However, the frequently observed aggravation of hemorrhagic diathesis by chemotherapy during the first days of treatment, which leads to a high early death rate, necessitated intensive management based on platelet and fibrinogen support [31,32,33]. There were contentious discussions regarding the origins of the fibrinogenopenia (either secondary or primary fibrinolysis) as well as the optimal treatment for it: low-dose heparin, antifibrinolytic agents, or platelet transfusions alone. The most consistent coagulation abnormalities were prolongation of the prothrombin and thrombin times, elevation of fibrinogen degradation products, and decreased levels of factor V and fibrinogen. Gralnick and Abrell [34] compared the procoagulant (tissue factor) and fibrinolytic activities of malignant promyelocytes to those of other AML cells, and found markedly increased tissue factor activity in promyelocytes. Normal levels of protein C and antithrombin III combined with acquired reduced alpha-2 antiplasmin levels implied a fibrinolytic process rather than DIC [35]. Regarding therapy, low-dose heparin (50 U/kg) given every 4–6 h or in continuous perfusion (100–200 U/kg/day) was shown to result in clinical and laboratory improvement [36]. Platelet transfusions were also shown to be useful for controlling the hemorrhagic diathesis, with the aim being to keep the platelet count close to 50 × 109/L. Fresh frozen plasma, plasma fractions, or fibrinogen did not prove useful for preventing hemorrhage [36].
Despite a significant improvement in outcome, the median duration of remission ranged from 11 to 24 months with a 5-year disease-free survival (DFS) of only 35–45% in patients treated by chemotherapy alone (Table 1) [25, 29, 37,38,39,40,41,42,43]. The early mortality rate was approximately 15% and the resistant rate was 10%. Resistance could be reduced by applying a more aggressive anthracycline treatment [25, 29, 44]. To increase survival, maintenance therapy with 6-MP and methotrexate was suggested [25, 29, 38].
Several prognostic factors were rapidly identified, such as severe fibrinopenia and a high white blood cell (WBC) count at diagnosis [23]. Other prognostic factors included age, fever, high blast count, serum creatinine, hyperuricemia, lactate dehydrogenase level, and albumin level [38, 40, 43]. Factors predictive of hemorrhagic deaths were age, high blast cell count, thrombocytopenia, prothrombin time, fibrinogen level, and anemia [31, 38, 40, 43].
At 2 years, the relapse rate was approximately 35%. A variety of reinduction methods were attempted in the pre-ATRA era. A second CR was reported in 53% of cases [38]. Allogeneic and autologous hematopoietic stem cell transplantation (HSCT) resulted in a longer duration of second CR [38, 42, 45].
At the biological level, a major breakthrough during this period was the confirmation that APL is a distinct entity based on the presence of an abnormal cytogenetic feature. First considered by Golomb et al. to be a partial deletion of chromosome 17 [46], the abnormality was identified a year later as a balanced reciprocal translocation between the long arms of chromosomes 15 and 17: t(15;17) (q21;q22) [47], which was consistently associated with APL [48] (Fig. 4). Variant chromosomal translocations [t(11;17) or t(5;17)] can be detected in no more than 2% of all APL patients. The presence of additional chromosomal abnormalities was further shown to have no affect on the clinical outcomes, including 17p or other adverse karyotypes known to confer poor outcome in AML [49].

APL is characterized by a specific balanced reciprocal translocation t(15;17) that fuses the PML gene on chromosome 15 to the RARα gene on chromosome 17
The HL60 cell line, established in 1977 from a patient with AML [50], was long considered to be a promyelocytic cell line. HL60 cultures comprise maturation-arrested cells with many properties that are similar to those of promyelocytes. The cells can be induced to differentiate terminally in vitro, and some reagents such as retinoic acid cause HL60 cells to differentiate to granulocyte-like cells. The HL60 cell genome contains an amplified c-myc proto-oncogene, which declines following induction of differentiation. However, the use of the HL60 cell line as a biological model has actually confused the field for many years, and HL60 cells differ from promyelocytes in many respects [51]. A re-evaluation of the original specimens indicated that FAB class M2 would have been a more accurate description [52]. Cytogenetic analysis showed the occurrence of many karyotypic abnormalities, but HL60 cells do not express PML-RARA.
During this period, anthracycline-based chemotherapies cured some patients with APL, raising hopes. At the genetic level, APL was found to correspond to a unique t(15;17) chromosomal translocation.
All-Trans Retinoic Acid Era
The Beginnings of ATRA Therapy
Some evidence emerged in the 1960s that led to the development of cancer differentiation therapy [53]. A major breakthrough was made by Sachs, who discovered in 1978 that leukemia cells could undergo differentiation upon the influence of various agents [54]. In the early 1980s, a wide variety of compounds, including retinoic acid, were shown to induce morphologic and functional maturation of HL-60 cells [55], and a specific response of APL specimens [56]. The first cases relating to the clinical effects of retinoic acid as a differentiating inducer of APL were reported [57, 58]. Flynn et al. induced in vitro maturation of leukemic promyelocytes from a patient with APL refractory to chemotherapy, and showed that similar maturation occurred in vivo after oral treatment with 13-cis-retinoic acid [57]. Nilsson reported that blood and marrow were normalized through differentiation induction by oral 13-cis-retinoic acid in one relapsing APL patient who was resistant to further lines of chemotherapy [58]. Daenen et al. administered 13-cis-retinoic acid to an APL patient who could not be treated with intensive chemotherapy because of invasive aspergillosis. The patient achieved CR with the disappearance of signs of coagulopathy [59]. During the same period, given the results achieved in Western medicine using cancer differentiation therapy and noting one of Confucius’s famous analects (“If you use laws to direct the people, and punishments to control them, they will merely try to evade the laws, and will have no sense of shame. But if by virtue you guide them, and by the rites you control them, there will be a sense of shame and of right.”), in 1980 physicians at the Shanghai Rui Jin Hospital started to screen APL patients for differentiation inducers [60]. ATRA, an isomer of retinoic acid, was shown to be superior to 13-cis-retinoic acid in both in vitro and in vivo settings and to strongly induce terminal differentiation in HL-60 and fresh APL cells. In 1985, a meeting held in Paris on differentiating agents discussed the specific activity of ATRA in APL, and resulted in a close collaboration between the Shanghai Institute of Hematology and the Saint-Louis Hospital in Paris focusing on the use of ATRA manufactured by Pharmaceutical Unit Number 6 in Shanghai. ATRA was first employed to treat APL patients in 1985 at the Rui Jin Hospital in Shanghai. The first patient treated with ATRA was a 5-year-old girl treated in 1985 at the Shanghai Children’s Hospital after the failure of anthracycline-based chemotherapy. She received ATRA at 45 mg/m2/day and achieved CR 3 weeks later [60]. After isolated case reports (4 newly diagnosed and 2 refractory to chemotherapy) were published in 1987 [61], Huang et al. reported that treatment with ATRA alone was successful in 24 APL patients (16 newly diagnosed and 8 refractory cases) with more than 90% CR and no exacerbation of coagulopathy or early death due to hemorrhage [62]. Despite these positive results, Western pharmaceutical companies refused to manufacture ATRA, which was instead obtained from the Chinese for the treatment of French APL patients. Finally, following the presentation of the results of ATRA treatment at the Memorial Sloan Kettering Cancer Center in 1989 and the intervention of Raymond Warrell, Roche Nutley began to manufacture ATRA tablets [63]. The efficacy of ATRA was confirmed in Paris for APL patients experiencing a first or subsequent relapse [64, 65]. CR was achieved in 95% of cases with no aplasia, no alopecia, and few infection episodes. The most striking feature was the gradual terminal differentiation of leukemic cells in the bone marrow, combined with the presence of Auer rods in mature granulocytes [65, 66]. In the US, the first report of an APL patient responding to ATRA was published in 1991 [67]. However, ATRA alone could not induce prolonged remission. Despite being associated with a high rate of CR, the use of ATRA alone caused progressive resistance to the drug, leading to relapse with 3 to 6 months. This was suggested to be caused by a reduction in the plasma concentration of ATRA due to accelerated clearance [64, 68, 69].
At that time, this new approach regarding APL treatment was not unanimously accepted, and divergent opinions were expressed—notably in a provocative editorial in which the idea that APL was a pseudoleukemia was explored [70].
Advances in Molecular Biology
The molecular basis for the response to ATRA was elucidated in the early 1990s [71,72,73,74]. The breakpoint on 17q21 was located within the locus of the retinoic acid receptor alpha (RARA) gene [75]. An unusual pattern of RARA mRNA was found in blast cells from APL patients [76]. The breakpoint on the RARA gene was cloned and sequenced using the NB4 cell line (established by Lanotte [77]) and fresh APL cells [71,72,73]. Its partner gene located in the breakpoint region on chromosome 15q22 was the promyelocytic leukemia (PML) gene (originally named MYL), sequenced 1 year later [78, 79]. This gene was subsequently shown to be involved in the control of cell proliferation, apoptosis, and senescence [80]. Meanwhile, the cloning of the t(15;17) translocation enabled reverse transcription polymerase chain reaction (RT PCR)-based strategies for the rapid diagnosis and sensitive detection of minimal residual disease (MRD) [81,82,83,84,85]. However, the initial interlaboratory sensitivity variations constituted a major limitation. Persistence or reappearance of PCR positivity for PML/RARA in patients with morphological remission was correlated with impending hematologic relapse [84]. This allowed the identification of patients requiring treatment intensification [86, 87] and permitted the kinetics of response and disease relapse to be determined with the development of real-time quantitative PCR of PML/RARA detection [88]. The biochemical mechanisms of transcriptional repression induced by PML/RARA protein and the molecular basis for the response to ATRA were later identified [89,90,91]. Variant translocations were subsequently discovered [92]. These variant fusion genes are clinically significant due to their differential responses to ATRA. Some of these variants are relatively or completely resistant to ATRA, such as t(11;17) (q23;q21) (leading to the PLZF-RARA fusion gene [93, 94]) and the fusion gene between STAT5b and RARA [95]. Other types are sensitive to ATRA, such as t(11;17) (q13;q21) [96], t(5;17) (q35;q21) [97], and t(4;17) (q12;q21) [98], leading to the fusion genes NuMA-RARA, NPM1-RARA, and FIP1L1-RARA, respectively. Approximately 10 diverse translocation partner genes of RARA have now been reported [99]. The nature of the fusion partner has a significant bearing upon the disease characteristics, including sensitivity to therapy, demanding a highly specific treatment approach.
Intensive Anthracycline-Based Chemotherapy in Combination with ATRA
Based on the first results with ATRA alone, French clinicians initiated a combination therapy consisting of ATRA alone until CR was achieved, followed by three courses of daunorubicin and cytarabine [100]. Twenty-six patients entered this nonrandomized study. Results, which were compared to a historical control group, showed a reduction in the early relapse rate as compared with ATRA treatment alone but a similarly low number of late relapses. In 1991, the European APL group launched the first randomized trial comparing three courses of chemotherapy with daunorubicin plus cytarabine versus ATRA until CR achievement followed by the same three courses of chemotherapy. The trial was stopped prematurely because event-free survival (EFS) was significantly better in the ATRA arm (12-month EFS: 79% versus 50%) [101]. In the ATRA group, the 4-year EFS was 63% compared to 17% in the chemotherapy-only cohort. These results were later confirmed by those from the North American Intergroup (5-year DFS: 69% versus 29%) [5, 102]. In a second randomized study (APL93 trial), the European APL group demonstrated that concurrent ATRA plus chemotherapy (daunorubicin plus cytarabine) resulted in a significantly lower 2-year relapse rate (6% versus 16%) when compared with sequential ATRA followed by chemotherapy [103]. This was further confirmed by other multicenter trials [6, 104,105,106]. The early addition of chemotherapy also decreased the incidence of retinoic acid syndrome [107]. Two randomized studies (the APL93 trial in Europe and the US Intergroup trial) tested the effects of maintenance therapy in APL [102, 103]. The European trial showed that maintenance for 2 years with intermittent ATRA therapy (2 weeks every 3 months) combined with low-dose chemotherapy (6-mercaptopurine daily plus methotrexate weekly) reduced the risk of relapse, while the US trial demonstrated that patients who had not received ATRA during induction benefited from ATRA used as maintenance therapy. However, two subsequent studies did not show any benefit of maintenance therapy in APL patients achieving molecular CR [108, 109]. New approaches to maintenance therapy were investigated, such as the use of tamibarotene (formerly Am80), which showed a trend for improved efficacy in high-risk APL patients [110]. The incidence of central nervous system (CNS) involvement in first relapse has been estimated to be 2%, with a higher prevalence in high-risk patients (5.5%) [111]. There is a general consensus for no CNS prophylaxis in low- and intermediate-risk patients [111,112,113] and CNS prophylaxis in high-risk patients and those experiencing CNS bleeding at diagnosis or during induction [111].
Overall, it became apparent during this period that APL was a curable disease when using a combination of ATRA and chemotherapy, and autologous or allogeneic stem cell transplantation was no longer indicated for first-line therapy. Long-term outcomes in large series showed a 5-year EFS of 70%, a 6-year DFS of 68%, and the best outcomes were seen in patients receiving ATRA during induction and consolidation.
Mechanisms of Action of ATRA
Reactivation of PML-RARA-repressed transcription by retinoic acid was proposed to explain the induction of differentiation and clearance of APL. PML-RARA is able to form homodimers that repress the transcriptional expression of target genes involved in granulocytic differentiation by binding to a set of retinoic acid response elements in the regulatory regions of these genes and recruiting corepressor proteins on both PML and RARA moieties [60]. PML-RARA can also recruit methylating enzymes, leading to hypermethylation of the downstream gene promoter, resulting in aberrant epigenetic control at critical gene chromatin domains. A pharmacological ATRA concentration of 10−6–10−7 M is generally considered to cause a change in PML-RARA configuration. The corepressor complex dissociates from the receptor and a coactivator complex is recruited, opening the chromatin structure and relieving transcriptional repression. There is modulation of a large number of genes involved in granulocytic differentiation [114]. Over the years, a number of reports have questioned this model of retinoic acid-mediated induction of differentiation [115]. Another effect of ATRA is to induce PML-RARA degradation by triggering caspase-mediated cleavage of the PML-RARA chimeric protein [116] and through a ubiquitin/proteasome system-mediated degradation [117].
Differentiation Syndrome
Differentiation syndrome is a clinical syndrome caused by ATRA-induced differentiation. During treatment, patients presented an increased WBC count, potentially associated with unexplained fever, hypotension, weight gain, respiratory distress, pulmonary infiltrates, pleural and/or pericardial effusions, and renal failure [65]. These disorders were defined as ATRA syndrome [118], and involved one-third of the patients treated in Western countries [119] but a smaller proportion of those treated in China [120]. The risk is greatest in patients with already high WBC counts. The pathogenesis is not well defined. However, a systemic inflammatory response driven by the release of pro-inflammatory cytokines, capillary leakage, and alterations in cellular adhesion molecule expression leading to tissue infiltration by leukemic cells and microcirculation occlusion was hypothesized [121, 122]. The standard management consists of dexamethasone 10 mg twice daily intravenously until resolution and for at least 3 days [112]. Prophylactic steroid therapy during induction can reduce the incidence of differentiation syndromes [123,124,125].
Risk-Adapted Treatment
Based on their initial studies, the Italian GIMEMA group (Gruppo Italiano per le Malattie Ematologiche dell’Adulto) and the Spanish PETHEMA group (Programa Espanol para el Tratamiento de las Hemopatias Malignas del Adulto) questioned the role of cytarabine in the treatment of APL and used only an anthracycline for chemotherapy in combination with ATRA for remission induction followed by chemotherapy consolidation and maintenance therapy [6, 105, 126]. The GIMEMA group proposed an ATRA/idarubicin (AIDA) induction and added three cycles of consolidation including idarubicin, cytarabine, etoposide, mitoxantrone, and thioguanine. The AIDA0493 protocol showed that 95% of patients achieved CR, a 2-year EFS of 79%, and a 2-year overall survival (OS) of 87% [105, 126]. The PETHEMA LPA96 trial demonstrated that removing all non-anthracycline drugs from the consolidation phase after an AIDA induction did not significantly affect patient outcomes [6]. This was confirmed by the Australasian Leukaemia and Lymphoma Group (ALLG) APML3 trial, which used an AIDA induction and an additional second cycle of idarubicin and demonstrated the importance of maintenance therapy in this setting [127]. The AIDA regimen then became one of the most popular standard treatments for newly diagnosed APL, and formed the basis for a meta-analysis leading to a proposed score for relapse risk assessment (Table 2) that was used in further trials [128]. The LPA99 PETHEMA trial evaluated consolidation therapy with ATRA plus three chemotherapy courses based on intensified idarubicin doses for patients with intermediate- and high-risk APL. Compared to the previous LPA96 trial, this schedule reduced the 3-year cumulative incidence of relapse (9% versus 20%) and significantly improved disease-free survival and overall survival [129]. In the LPA2005 trial, treatment of high-risk patients with cytarabine significantly reduced the 3-year relapse rate from 26% to 11% when compared to the results from the LPA99 trial [129, 130]. Similarly, the AIDA2000 trial showed that a risk-adapted strategy in consolidation resulted in better long-term outcomes by avoiding toxicity and infection in low- and intermediate-risk APL patients [4]. The French APL2000 trial showed a benefit of cytarabine in consolidation for high-risk patients and an increased risk of relapse when omitting cytarabine from induction and consolidation courses [131, 132]. A review of these results in combination with those from the LPA99 trial confirmed a benefit for an ATRA-idarubicin-based protocol in low- and intermediate-risk patients and better survival with the combination anthracycline-cytarabine in high-risk patients [3]. In contrast, the British Medical Research Council (MRC) AML15 study reported an increased number of deaths in CR when cytarabine was administered, but this did not translate into significant differences in terms of survival rates [133]. No studies have directly compared the type of anthracycline used. A matched-pair analysis comparing idarubicin and daunorubicin showed no significant difference [134]. However, anthracycline-related cardiotoxicity is a major concern [135], especially in patients treated with an AIDA-based protocol, who showed diastolic dysfunction and regional wall motion abnormalities [136].
Overall, these results led the European LeukemiaNet to recommend that APL be treated with an ATRA/anthracycline combination for induction, two or three cycles of anthracycline-based chemotherapy as consolidation, intermediate- or high-dose cytarabine in younger patients with WBC count > 10 × 109/L, and maintenance therapy for protocols in which it has been shown to be beneficial [112].
Relapse After Treatment with ATRA
About 20% of patients have been reported to relapse after risk-adapted ATRA and chemotherapy regimens [137]. Several studies have shown the clinical benefit of early identification of disease recurrence and pre-emptive therapy at the time of molecular relapse [87, 138, 139]. Molecular relapse is conventionally defined as two consecutive RT-PCR positive tests in marrow samples collected 2–4 weeks apart after first-line therapy. Treatment of molecular relapse was associated with a better treatment tolerance, fewer days of hospitalization, fewer early deaths, and a reduced rate of differentiation syndrome. In the case of a relapse after first-line therapy using ATRA, a second remission can be achieved in approximately 90% of cases with the use of ATRA plus chemotherapy. A subsequent allogeneic or autologous HSCT was then recommended to avoid another relapse [140,141,142]. In 2009, an expert panel on behalf of the European LeukemiaNet published recommendations for the management of relapsed APL based on literature data and the recent introduction of ATO in this situation [112]. Two cycles of ATO ± ATRA therapy were considered the best option for patients who relapsed after ATRA and chemotherapy regimens. In patients achieving molecular remission, intensification with autologous SCT was recommended, or prolonged ATO-ATRA therapy. Allogeneic HSCT was recommended for patients who failed to achieve molecular remission or for those who relapsed after less than 1 year of first CR. Central nervous system prophylaxis is usually recommended at relapse.
Mutation Landscape and Other Prognostic Factors
FLT3 emerged as the most frequently altered gene in primary APL; 27% of cases had FLT3-ITD and 16% of cases had FLT3-TKD mutations [143]. While AML with FLT3-ITD is associated with a high risk of relapse, its clinical impact in APL is still controversial [144,145,146]. FLT3-ITD was associated with high-risk groups and therefore independently associated with inferior OS. A meta-analysis of patients treated predominantly with ATRA and chemotherapy identified lower rates of DFS and OS in patients with a FLT3-ITD mutation [147]. The presence of a FLT3 mutation also appeared to increase the risk of induction death [148, 149]. However, this was not confirmed in patients who received ATO or ATRA-ATO [146]. Other commonly observed mutated genes included WT1 (14%), NRAS (10%), KRAS (4%), ETV6 (1%), and EZH2 (1%). DNMT3A, NPM1, TET2, IDH1, IDH2, and ASXL1 mutations were absent or rarely observed.
Immunophenotyping might also play a role in prognostication. CD56 overexpression (10% of APL cases) is associated with a high WBC count and an increased risk of relapse [150]. CD2, found in 24% of APL patients, was associated with CD34 positivity, a higher WBC count, Bcr3, and a microgranular subtype [151].
The mechanism of PML-RARA-induced oncogenic transformation has been studied using various APL mouse models [152, 153]. PML-RARA is the only genetic event capable of initiating typical APL disease when expressed in transgenic mice. Additional genetic/epigenetic changes play a role in progression to full transformation in the APL phenotype. The recent identification of crucial PML-RARA features has resulted in a complex image of APL pathogenesis. The PML-RARA fusion protein appears to be multifaceted, deregulating the differentiation and self-renewal of myeloid progenitors and conferring resistance to apoptosis. Heterotetramers of PML-RARA bound to RXRA allow specific binding to a large variety of DNA sites, including sites recognized by nuclear receptors controlling myeloid differentiation or stem cell self-renewal [154, 155]. PML-RARA undergoes post-translational modifications, such as PML sumoylation and RARA phosphorylation, which are important for APL initiation [156, 157]. Furthermore, RARA fusion proteins interact with key chromatin remodeling complexes, which play a crucial role in stem cell maintenance [158]. While translocations of RARA, typically PML-RARA, are a genetic hallmark of APL, a small fraction of APL cases lack translocations of RARA. Retinoic acid receptors (RARs) are nuclear hormone receptors which function as ligand-activated transcription factors that interact specifically to modulate transcription DNA elements [159]. RARs include alpha (RARA), beta (RARB), and gamma (RARG) receptor types. They function as heterodimers with retinoid X receptors (RXRs). Translocations involving RARB and TBLXR1-RARB have been identified [160]. TBLXR1-RARB homodimerizes and diminishes transcriptional activity for the RAR pathway in a dominant-negative manner. However, the response of APL with RARB translocation to retinoids was attenuated compared with that of PML-RARA. The involvement of RARG in a chimeric fusion transcript (NUP98) has also been reported [161]. Heterodimers formed by RXRA-RARG are necessary for growth arrest and visceral and primitive endodermal differentiation [162]. Artificial PML-RARG fusion proteins are able to produce the same biological effects as those mediated in the cells by PML-RARA and to confer responsiveness to differentiation treatment [162]. Inoculated transduced cells expressing PML-RARG were able to trigger leukemia in murine models [159].
Overall, the introduction of ATRA in 1985 to improve the response of APL patients to chemotherapy has led to a revolution in the prognosis of this disease. PML-RARA fusion was shown to be the only constant genomic abnormality in APL cells.
Arsenic Trioxide
Arsenic, An Old Remedy
Arsenic is one of the oldest drugs, and has been used to treat a variety of diseases. It was mentioned by Hippocrates (460–370 BC), who used realgar and orpiment pastes to treat ulcers. Arsenic was also used to treat diseases in ancient China (“Fight poison with poison”). It is mentioned in the Chinese Nei Jing treaty (263 BC) as a treatment for periodic fever; Hong Ge (284–364 AD used it as a disinfectant; Si-Miao Sun (581–682 AD) used it to treat malaria, and Shi-Zhen Li (1518–1593 AD) administered it for a variety of diseases [163, 164]. In Western countries, it was used by Avicenna (980–1037 AD) and Paracelsus (1493–1541 AD). In 1774, Lefébure proposed an arsenic-containing paste as a “remedy to radically cure all cancers” [165]. Fowler’s solution containing 1% potassium arsenite, first described in 1845, was one of the first chemotherapeutic agents used in the treatment of leukemia [166,167,168]. It was rediscovered by Forkner and Scott in 1931 for the treatment of chronic myeloid leukemia (CML) [168]. In China, the use of arsenic to treat leukemia started in the early 1970s. Tai Yun Han, a pharmacist from Harbin Medical University, made solutions (named Ailing solutions) using the same components as those in an old anticancer remedy: ATO, mercury chloride, and toad venom. His colleague, Ting Dong Zhang, explored the effect of the solution on myeloid leukemia patients and observed some promising effects [164].
First Trials with ATO
There are three inorganic forms of arsenic: red arsenic (AS4S4, also known as realgar), yellow arsenic (AS2S3, also known as orpiment), and white arsenic or ATO (AS2O3), which is made by burning realgar or orpiment [60]. In 1992, Sun et al. reported that CR was achieved in 21 of 32 APL patients treated with a solution of ATO [169]. The survival rate was 30% after 10 years. Results obtained using pure ATO were reported in the late 1990s: CR rates of 73% and 52% were obtained in 30 newly diagnosed and 42 relapsed APL cases, respectively [170]; CR was achieved in 9 of 10 patients treated with ATO alone and in 5 patients treated with a combination of ATO and low-dose chemotherapy or ATRA [171, 172]. These results were further confirmed by a group of 47 relapsed and 11 newly diagnosed APL cases with CR rates of 85 and 73%, respectively [173]. In a retrospective study of 242 cases from Harbin Medical University Hospital, the CR rates in newly diagnosed patients and relapsed patients were 88% and 60%, respectively, with a 5-year OS of 92% and a 7-year OS of 77% [174]. Furthermore, molecular remission was reported in a high proportion of patients (72–91%) who received ATO alone [175, 176]. Based on those results, ATO was introduced by many groups worldwide for the treatment of relapsed APL. ATO-based salvage therapy has been able to induce long-lasting remission. The main studies reported in Table 3 [175,176,177,178,179,180,181,182,183,184,185,186] were reviewed by Lengfelder et al. [187].
Mechanisms of Action
At the time that the efficiency of arsenic for the treatment of APL was first reported, the prevalent concept was that terminal blast differentiation drove the response to ATRA. ATO was proposed to initiate apoptosis or partial differentiation [171]. ATO was shown to exert dose-dependent effects on APL cells [171]. At high concentrations (0.5–2.0 μmol/L), ATO induced apoptosis, mainly by activating the mitochondria-mediated intrinsic apoptotic pathway. At lower concentrations (0.1–0.5 μmol/L), ATO promoted the differentiation of APL cells. This was supported by animal models and the examination of bone marrow in APL patients treated with ATO [188]. The pro-apoptotic activity of ATO was then scrutinized at the gene and protein levels, and various involvements were noted, including a pathway composed of ATR, PML, Chk2, and p53 [189]. Critically, arsenic does not activate RARA-dependent transcription [165], although it may induce differentiation syndrome in APL patients [190]. Subsequent research has shown that ATO’s therapeutic efficacy is due to its effect on the PML moiety of the PML-RARA fusion protein [2]. The PML protein is important for the formation of nuclear bodies (first described as electron-dense shells with electron-light cores in 1960 [191]), which allow the recruitment and interaction of proteins through the posttranslational modification processes of sumoylation and ubiquitination. PML-RARA expression disrupts PML nuclear bodies. Disruption of nuclear bodies leads to the loss of their tumor-suppressive activity, possibly contributing to leukemogenesis. The binding of arsenic to the PML component through two cysteine residues induces their oxidation and the formation of a disulfite bond [192, 193]. In the final step, ATO mediates the recruitment of 11S proteasome, a process essential for the degradation of PML and PML-RARA proteins [188, 194, 195]. PML-RARA destruction also abrogates self-renewal through the re-formation of PML nuclear bodies that were initially disassembled by PML-RARA and subsequent p53 activation [196]. The engagement of prosenescent responses explains why single-agent arsenic is definitively curative in up to 70% of patients, while ATRA alone only yields transient responses [197, 198]. In mouse models, the presence of PML and p53 is absolutely required for a long-term therapy response [196]. Failure of therapy in vivo is likely due to the lack of p53 activation, which occurs only when PML-RARA is efficiently degraded. Mutations in the unrearranged PML allele that affect arsenic binding were found in patients resistant to therapy [199]. Additionally, arsenic induces the degradation of PML/PML-RARA through the production of reactive oxygen species (ROS). Anthracyclines, which can stimulate the production of ROS, may also contribute to PML-RARA degradation [200]. As opposed to ATRA, there is evidence for the eradication of leukemia-initiating cells after ATO therapy [115]. There is also evidence that ATO crosses the blood–brain barrier. The concentration of arsenic in the cerebrospinal fluid has been shown to correlate with the plasma level [201]. Resistance to ATO is uncommon, but can be due to various reasons. Missense mutations in the PML-RARA transcripts have been identified in patients resistant to ATO [202].
ATO Toxicities
Despite the quick resolution of significant coagulation disturbances, severe hemorrhage was the most frequent cause (70%) of early death, with most of the fatal bleeding events located in the CNS [175]. Differentiation syndrome (retinoic-acid-like syndrome) is relatively common (reported in 7–35% of cases) and potentially life-threatening complication occurring during the first days or weeks of treatment. Preventive treatment is particularly important in high-risk patients (patients with a WBC count > 10 × 109/L). Treatment includes the prompt use of intravenous dexamethasone 10 mg twice daily until resolution or for a minimum of 3 days [122]. Hyperleukocytosis can be managed using hydroxyurea [8]. In the AML17 study, patients with a peripheral WBC count > 10 × 109/L at baseline received gemtuzumab ozogamicin (GO) 3 mg/m2 on day 1 and again on day 4 if the WBC count had not fallen below this value [7]. QTc prolongation is a common side effect of ATO. It can lead to torsade de pointes-type ventricular arrhythmia, which is potentially fatal [8]. Patients with a prolonged QTc interval above 500 ms are recommended to maintain adequate potassium and magnesium concentrations. QTc prolongation has recently been the subject of a study that aimed to identify the optimal correction formula to be employed in QTc calculations [203]. Hepatic toxicity has frequently been reported in studies employing ATO, especially in terms of an increase in liver enzymes [197]. Although frequent, this complication is usually reversible and successfully managed with the temporary discontinuation of ATO. Neurological toxicity, mainly consisting of peripheral neuropathy, has also been reported. This mild side effect is usually managed with temporary discontinuation. An unsolved issue is whether or not CNS prophylaxis is needed. Pharmacodynamic studies indicated that one-third of the ATO crosses the blood–brain barrier, which could suggest that a sufficient amount of the drug is available on site to prevent disease recurrence [204]. No arsenic accumulation or delayed toxicity was observed in patients followed up for over 10 years [205, 206].
ATO in Newly Diagnosed APL
Two therapeutic approaches were used with ATO in newly diagnosed APL: (1) minimizing or eliminating cytotoxic chemotherapy from induction and consolidation phases; (2) reinforcing the antileukemic efficacy of the ATRA plus chemotherapy regimen.
The main studies that have evaluated ATO-based induction and consolidation phases are shown in Table 4. In noncomparative prospective studies, ATO was used as a monotherapy for induction and prolonged consolidation or as maintenance therapy [197, 198]. The CR rate was 86% and the early death rate (mostly due to fatal hemorrhages) was 14%. In one study, the 5-year OS estimate and the 5-year DFS estimate were 74% and 80%, respectively [198]. In the other, the OS and DFS estimates after a median follow-up of 38 months were 64% and 67%, respectively [197]. In relapsing patients, a second CR was achieved in 52% of cases when treated with ATO again, albeit with a 2-year survival rate of only 35% [197]. Another trial combined ATO and ATRA, extended with GO, and idarubicin when the WBC count was above 10 × 109/L [207, 208]. A randomized study comparing ATO monotherapy, ATO plus ATRA, and ATRA monotherapy as the induction regimen was conducted in China [205]. Induction was followed by consolidation and maintenance therapy. The CR rates ranged from 90 to 95% among the three treatment arms, and a shorter time was needed to reach CR and PML-RARA negativity with combination therapy. The 5-year OS and the 5-year EFS were 92% and 89%, respectively.
To reduce chemotherapy exposure, the role of ATO in consolidation has been investigated. A significant benefit of the ATO-ATRA-chemotherapy arm was demonstrated in a randomized study from the US Intergroup in comparison with the ATRA-chemotherapy arm [209]. Patients who achieved a CR after ATRA and chemotherapy were randomized to receive either additional ATO consolidation (two cycles at 0.15 mg/kg/day for 5 days for 5 weeks) or two cycles of a common ATRA/daunorubicin consolidation. The results demonstrated that for all risk groups, ATO in consolidation significantly improved 3-year DFS (90% vs 70%; P < 0.0001), and there was a nonstatistically significant improvement in terms of OS. In another American study, some of the chemotherapy cycles were replaced with ATO cycles [210]. Comparable outcomes were reported, with a DFS of 90% and an OS of 88%. The APL 2006 study showed that cytarabine can be replaced by ATO without altering the relapse rate in the low/intermediate-risk group, while the combination increased myelosuppression in the high-risk group [211].
The use of ATO improved the clinical outcomes of refractory or relapsed as well as newly diagnosed APL. ATO appears to be the most effective single agent for APL. ATO cures APL by initiating PML-RARA oncoprotein degradation by targeting its PML moiety.
Chemotherapy-Free Treatment in APL
ATRA-ATO in Frontline Therapy
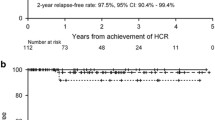
Following studies in a mouse model by the group led by de Thé [212] and that led by Waxman [213], which showed that the ATRA-ATO combination could dramatically prolong survival and even eradicate disease in animals, the group from the Shanghai Institute of Hematology was the first to report on the use of the ATRA-ATO combination in a cohort of 85 newly diagnosed APL patients [205, 214]. CR was achieved in 94% of cases and the 5-year EFS and OS were 89% and 92%, respectively. Subsequently, several groups reported the use of ATRA/ATO in newly diagnosed patients [7, 8, 207, 208, 215, 216] (Table 5). The MD Anderson group treated 187 patients with this combination in a nonrandomized trial [215]. Patients with high-risk APL as well as those in low- or intermediate-risk groups who developed leukocytosis during ATRA/ATO therapy were given a dose of GO. CR was achieved in 96% of cases with a 5-year OS of 89 and 86% in low-risk and high-risk APL, respectively. The final report of the randomized Italian-German APL0406 trial confirmed the efficacy and safety of the ATRA/ATO combination, especially in the low-risk APL group [216]. In this study, 263 patients with newly diagnosed non-high-risk APL were randomly assigned to ATRA/ATO and ATRA/chemotherapy arms [8]. CR rates of 100% and 97% were reported in the ATRA/ATO and ATRA/chemotherapy arms, respectively. Significant results were observed in terms of EFS, cumulative incidence of relapse, and OS at 50 months in the ATRA/ATO arm [97% vs 80%, P = 0.001; 1.9% vs 13.9%, P = 0.001; 99% vs 93%, P = 0.007, respectively], while induction death occurred only in the ATRA/chemotherapy arm (3%). The British group enrolled 235 patients in a randomized study of newly diagnosed patients comparing ATRA plus idarubicin as induction followed by ATRA/chemotherapy consolidation versus ATRA/ATO [7]. Ninety-three percent of the 30 high-risk patients and 7 low-risk patients who received the ATRA/ATO arm also received GO for their high WBC count. The CR rate was 94% in the ATRA/ATO arm vs 89% in the ATRA/chemotherapy arm. The 4-year EFS was significantly better in the ATRA/ATO arm (91% vs 70%; P = 0.002). This significant benefit was apparent in the low-risk group but not in the high-risk group. It therefore appears that ATRA/ATO alone was not enough for the high-risk group, so the optimal therapy for high-risk patients remains unclear. Iland et al. showed that ATRA/ATO plus chemotherapy is superior to ATRA plus chemotherapy in all-risk APL patients [9, 217]. Burnett et al. found that ATRA/ATO treatment is feasible for high-risk patients who receive GO for their high WBC count [7]. In the APL4 trial from the Australia–New Zealand group, idarubicin was incorporated into the ATRA/ATO induction regimen, followed by ATRA/ATO consolidation therapy [127]. This is also the case in the current multinational phase III APOLLO trial that randomizes ATRA/ATO plus idarubicin versus the AIDA regimen in newly diagnosed high-risk APL patients. In arm A, induction with ATRA is supplemented by two administrations of idarubicin on days 1 and 3, respectively, and ATO starting on day 5 after the start of idarubicin. The induction therapy is followed by 4 courses of ATO/ATRA-based consolidation. In arm B, standard AIDA-based induction comprises idarubicin on days 1, 3, 5, and 7 combined with daily ATRA therapy until CR is achieved. Upon achieving hematological CR, patients start three cycles of consolidation including ATRA combined with Ara-C, idarubicin, and mitoxantrone, followed by 2 years of maintenance therapy. Patients with leukocytosis may have a high risk of CNS relapse [111]. The first prevention measure is to decrease the WBC count using hydroxyurea or chemotherapy. The second could be to increase the blood–brain barrier permeability of ATO by mannitol infusion [218, 219]. While maintenance therapy was originally part of the design of the ATRA/chemotherapy regimen, the role of maintenance has come into question in the era of ATRA/ATO therapy. In the low/intermediate-risk group, maintenance therapy can be safely omitted after the ATRA/ATO regimen. In the high-risk group, it could also be omitted if the ATRA/ATO regimen is used in conjunction with a cytotoxic agent such as idarubicin, and if molecular remission has been achieved at the end of consolidation. Recently, prolonged therapy with ATRA and ATO was tested in the relapse setting and the results confirmed the potentially curative effect of this regimen in relapsed APL, especially in patients with a long first CR [220].
Mechanisms of the Synergistic Effect of ATRA-ATO
Distinct APL mouse models highlighted a synergy between ATRA and ATO [212, 221]. This combination enhanced apoptosis but not differentiation and accelerated APL clearance. The two drugs exert synergistic effects in regard to the loss of self-renewal, most likely through cooperative PML-RARA degradation. PML/RARA disrupts PML nuclear bodies and deregulates transcriptional control through the recruitment of corepressor complexes. PML/RARA ultimately induces enhanced self-renewal and a differentiation block [200, 222]. Nuclear bodies are stress-sensitive nuclear domains involved in p53 activation and the control of senescence [223, 224]. Other functions have been attributed to PML/RARA, which is also influenced by important genetic modulators [225,226,227,228,229]. ATRA and ATO bind to the PML-RARA fusion oncoprotein and initiate proteolytic degradation of PML-RARA [222]. The binding of ATRA to RARA activates PML/RARA target gene transcription and initiates fusion protein degradation. The binding of ATO to the PML part triggers PML/RARA conjugation by SUMO, resulting in its degradation [230]. The key role of oncoprotein catabolism in leukemia eradication was shown by exploring the response to ATRA or ATO in mice. Degradation of PML-RARA reverses transcriptional repression and triggers a PML/p53-dependent senescence checkpoint. In mouse models, arsenic was shown to act on both the PML-RARA oncoprotein and the remaining normal PML protein, which drives p53 activation and senescence [196]. PML-RAR degradation is required to stop APL cell self-renewal, and it predicts long-term APL clearance. A contribution of the immune system to the response to therapy has also been suggested because of the diminished antileukemic activity observed in immune-compromised mice [231]. The immune system seems to be important for eliminating senescent cells involved in APL clearance [232, 233]. Partner-mediated RARA dimerization is believed to be essential for arresting differentiation [234, 235], and transformation requires the PML coiled-coil domain [236]. Mutations at the arsenic-binding site of PML explain therapy resistance and why arsenic only cures 70% of APL patients when used as a single agent [199]. Similarly, ATRA acts on not only PML-RARA but also the Pin-1 enzyme, the overexpression of which favors transformation in multiple cell types, or PHF8-mediated transcriptional activation [225, 237].
APL is a hematological malignancy driven by the PML-RARA oncogene. ATRA and ATO were both shown to target PML-RARA, explaining their strong specificity for APL. Standard-risk APL can now be cured in virtually all patients with a combination of ATRA and ATO.
Conclusions and Perspectives
Constant progress in the treatment of APL has caused the status of APL to evolve from highly fatal to highly curable. The current results show high CR rates and high 5-year DFS rates. The superiority of ATRA-ATO compared to the conventional ATRA-chemotherapy regimen is indisputable in the low- and intermediate-risk groups, which no longer require chemotherapy, but not in the high-risk group. Results obtained in the Italian-German APL0406 trial [8] led the NCCN and a Canadian expert panel to indicate this regimen as a new standard of care in this setting [113, 238]. However, there are still open issues in this area, notably regarding the type of ATO schedule. Regimens for high-risk APL have not been compared sufficiently to be able to draw final conclusions. Preliminary evidence from trials combining ATRA-ATO with chemotherapy indicate a high efficacy of ATO in this subset of patients and suggest that this triple combination may be curative in high-risk patients [7, 9]. However, the type and dose of initial chemotherapy needed during induction in combination with ATRA-ATO remain to be determined. ATRA-ATO therapy may also serve as an attractive alternative for patients who are considered unfit for conventional therapy. The impressive improvement in APL outcomes has recently been challenged by registry-based studies exploring APL outcomes in the real world [239,240,241]. These studies indicate that early mortality is currently an underestimated phenomenon in clinical trials. There is an ongoing need to decrease the early death rate (within the first 30 days from diagnosis), which is still the primary cause of treatment failure rather than the resistant disease that is so common for all other subtypes of AML [242]. Large clinical trials have reported an early death rate of 3–10% [8, 130, 217]. However, the death rate has been reported to be approximately 20% in patients who were ineligible or excluded from trials [243]. In large population-based analyses, and among patients treated in single institutions, the rate of mortality is even higher and can range from 9.6% to 61.5% [244]. An explanation for this high rate of early death is the rarity of APL coupled with the fact that the majority of APL patients in some countries are treated outside clinical trials and in smaller inexperienced centers. Some studies have identified prognostic factors that are capable of predicting early deaths. In multicenter studies, the most frequent causes of death were identified as bleeding, differentiation syndrome, infection, and multiorgan failure. Cerebral hemorrhage and severe infection are generally involved in early deaths, which primarily occur in patients with hyperleukocytosis [245]. Delays in ATRA therapy have been suggested as a contributing factor to early deaths [246], as has initial hospitalization in nonspecialized primary care institutions [241, 247]. A risk classification system was recently proposed to identify the subgroup at a high risk of early death among APL patients in not only de novo but also relapse cohorts [248]. Overall, the rapid identification and treatment of newly diagnosed APL patients, particularly older patients, remain challenging. This emphasizes the need for rapid supportive care together with immediate access to ATRA-based therapy [249]. Better-trained hematologists and better health insurance coverage in developing countries could further reduce early deaths [250]. Other current concerns involve APL treatment in children, post-ATO relapses, refinements to strategies for high-risk APL, and the introduction of a new drug formula with an attempt to give only oral ambulatory therapy. Despite the dramatic improvement achieved in frontline therapy of childhood APL with the combination of ATRA and anthracycline-based regimens, relapse and chemotherapy-associated side effects still represent non-negligible causes of treatment failure and morbidity [251]. A frontline treatment approach based on the ATRA-ATO combination has been explored in a limited number of pediatric patients [252,253,254]. All patients achieved molecular CR after a median time of 10 weeks but experienced hyperleukocytosis, which could require the administration of chemotherapy or hydroxycarbamine [253, 254]. A prospective multicenter European trial from the International Consortium for Childhood APL is ongoing. Current literature on the treatment of relapsed APL focuses on the period after ATRA and chemotherapy. It is likely that the current scenario of APL relapse will change, and the clinical and biological features of post-ATO relapses as well as their optimal management should be examined in future investigations. The addition of a few days of idarubicin or new therapeutic agents to the ATRA-ATO regimen has been suggested for the high-risk group. GO is an anti-CD33 (a differentiation antigen detectable in almost 100% of APL patients) antibody calicheamicin conjugate that can yield a high molecular remission rate when used as a single agent in APL [255]. The combination of GO with ATRA-ATO therapy was tested by the MD Anderson Cancer Center [208]. SNG-CD33A, a next-generation anti-CD33 antibody, has demonstrated antileukemic activity in a phase 1 study [256]. Another anti-CD33, HuM195, has demonstrated efficacy on MRD [257]. Because of mutations detected in 25–45% of cases, FLT3 inhibitors could find application in the treatment of APL. FLT3 inhibitor SU11657 in combination with ATRA induced rapid regression of leukemia in an animal model [258]. Tamibarotene (formerly called Am80), a synthetic retinoic acid with more potent in vitro activity than ATRA, has demonstrated a favorable pharmacokinetic profile, as the plasma level does not decline after daily administration [259]. It produced interesting results when used as a single agent for induction in relapsed/refractory patients who previously received ATRA and ATO [260]. In a study reported by the Japan Adult Leukaemia Study Group, tamibarotene led to an improvement in relapse-free survival (RFS) when used as a maintenance therapy in high-risk APL [110]. When applied as a maintenance therapy, tamibarotene was significantly superior to ATRA in terms of decreasing relapse in high-risk patients [261]. Another arsenic compound named the Realgar-Indigo naturalis formula, a traditional Chinese oral medicine containing As4S4, has been shown to be highly effective when used to treat APL. This represents a more convenient route of administration than the standard intravenous regimen. Oral arsenic has shown favorable oral absorption, leading to a bioavailability of up to 95% of an equivalent dose of intravenous ATO [262]. It was first tested in relapsed APL cases, where it showed high efficacy and no QTc prolongation and ventricular arythmias, in contrast with intravenous ATO, likely because its peak plasma concentrations were lower [263]. The use of oral arsenic (tetra-arsenic tetra-sulfide As4S4) in combination with ATRA has shown efficacy for high-risk APL [264] and a noninferior outcome in non-high-risk APL when compared with intravenous ATO plus ATRA in trials including adults and children [265,266,267]. The rates of adverse events were similar [268], and the oral combination As4S4-ATRA has demonstrated the feasibility of out-of-hospital treatment and reducing hospital stay [267, 269]. Oral ATO was also tested in the setting of maintenance therapy after first CR, and produced similar results to the intravenous formulation [270]. However, the relative contribution of maintenance therapy remains controversial. A recent Cochrane meta-analysis could not show a benefit of maintenance for OS, despite an improvement in DFS [271]. Recent trials incorporating ATO have demonstrated that success can be achieved without any maintenance in non-high-risk patients [7, 8, 272].
APL is the best example of how targeted therapies can trigger definitive cures. ATRA-ATO combination therapy illustrates the importance of traditional medicine and represents the first model of molecular target based therapy. Several expert panels have recommended that molecular remission should be considered a therapeutic objective in APL, and molecular response has been adopted as a study endpoint in modern clinical trials [273]. APL patients around the world should soon undergo this curative treatment, perhaps based on oral formulations [274]. Oral administration raises the possibility of implementing treatment as outpatient care, thus increasing quality of life and decreasing medical costs [269, 275]. Along with the introduction of tyrosine kinase inhibitor therapy in chronic myeloid leukemia, the ATRA-ATO combination represents one of the most important advances in hematological disease therapy over the last three decades, and provides a model for the way in which cancer should be treated.
References
Sant M, Allemani C, Tereanu C, et al. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010;116:3724–34.
De Thé H, Le Bras M, Lallemand-Breitenbach V. The cell biology of disease: acute promyelocytic leukemia, arsenic, and PML bodies. J Cell Biol. 2012;198:11–21.
Adès L, Sanz MA, Chevret S, et al. Treatment of newly diagnosed acute promyelocytic leukemia (APL): a comparison of French-Belgium-Swiss and PETHEMA results. Blood. 2008;111:1078–84.
Lo-Coco F, Avvisati G, Vignetti M, et al. Front-line treatment of acute promyelocytic leukemia with AIDA induction followed by risk-adapted consolidation for adults younger than 61 years: results of the AIDA-2000 trial of the GIMEMA group. Blood. 2010;116:3171–9.
Tallman MS, Andersen JW, Schiffer CA, et al. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood. 2002;100:4298–302.
Sanz MA, Martin G, Rayon C, et al. A modified AIDA protocol with anthracycline-based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARalpha-positive acute promyelocytic leukemia. Blood. 1999;94:3015–21.
Burnett A, Russell NH, Hills RK, et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukemia in all risk groups (AML17): results of a randomized, controlled, phase 3 trial. Lancet Oncol. 2015;16:1295–305.
Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21.
Iland HJ, Collins M, Bradstock K, et al. Use of arsenic trioxide in remission induction and consolidation therapy for acute promyelocytic in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study: a non-randomized phase 2 trial. Lancet Haematol. 2015;9:e357.
Hillestad LK. Acute promyelocytic leukemia. Acta Med Scand. 1957;159:189–94.
Stormorken H. Fibrinolytic purpura associated with myelogenous leukaemia case. Tidssk Nor Laegeforen. 1956;76:754–6.
Cooperberg AA, Neiman GMA. Fibrigenopenia and fibrinolysis in acute myelogenous leukemia. Ann Intern Med. 1955;42:707–11.
Pisciotta AV, Schultz JE. Fibrinolytic purpura in acute leukaemia. Am J Med. 1955;19:824–8.
Risak E. Die fibrinopenie. Z Klin Med. 1935;128:605–29.
Bernard J, Mathe G, Boulay J, et al. Acute promyelocytic leukaemia: a study made on 20 cases. Schweiz Med Wochenschr. 1959;89:604–8.
Caen J, Larrieu MH, Bernard J. Afibrinémies acquises. Rev Fr Clin Biol. 1959;4:363–6.
Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias French-American-British (FAB) Co-operative Group. Br J Haematol. 1976;33:451–8.
Bennett JM, Catovsky D, Daniel MT, et al. A variant form of hypergranular promyelocytic leukemia (M3). Ann Intern Med. 1980;44:169–70.
McKenna RW, Parkin J, Bloomfield CD, Sandberg RD, Brunning RD. Acute promyelocytic leukemia: a study of 39 cases with identification of a hyperbasophilic microgranular variant. Br J Haematol. 1982;50:201–14.
Baker WG, Bang NU, Nachman RL, Raafat F, Horowitz HI. Hypofibrinogenemic hemorrhage in acute myelogenous leukemia treated with heparin with autopsy findings of widespread intravascular clotting. Ann Intern Med. 1964;61:116–23.
Rosenthal RL. Acute promyelocytic leukemia associated with hypofibrinogenemia. Blood. 1963;21:495–508.
Didisheim P, Trombold JS, Vandervoort LE, Mibashan RS. Acute promyelocytic leukemia with fibrinogen and factor V deficiencies. Blood. 1964;23:717–28.
Bernard J, Weil M, Boiron M, Jacquillat C, Flandrin G, Gemon MF. Acute promyelocytic leukemia: results of treatment by daunorubicin. Blood. 1973;41:489–96.
Avvisati G, Mandelli F, Petti MC, et al. Idarubicin (4-demethoxydaunorubicin) as single agent for remission induction of previously untreated acute promyelocytic leukemia: a pilot study of the Italian cooperative group GIMEMA. Eur J Haematol. 1990;44:257–60.
Marty M, Ganem G, Fisher J, et al. Leucémie aiguë promyélocytaire: étude rétrospective de 119 malades traités par daunorubicine. Nouv Rev Fr Hematol. 1984;26:371–8.
Arlin Z, Kempin S, Mertelsmann R, et al. Primary therapy of acute promyelocytic leukemia: results of amsacrine- and daunorubicin-based therapy. Blood. 1984;63:211–2.
Fenaux P, Tertian G, Castaigne S, et al. A randomized trial of amsacrine and rubidazone in 39 patients with acute promyelocytic leukemia. J Clin Oncol. 1991;9:1556–61.
Ellison RR, Holland JF, Weil M, et al. Arabinosylcytosine: a useful agent in the treatment of acute leukemias in adults. Blood. 1968;32:507–23.
Fenaux P, Pollet JP, Vandenbossche-Simon L, et al. Treatment of acute promyelocytic leukemia: a report of 70 cases. Leuk Lymphoma. 1991;4:239–48.
Petti MC, Avvisati G, Amadori S, et al. Acute promyelocytic leukemia: clinical aspects and results of treatment in 62 patients. Haematologica. 1987;72:151–5.
Rodeghiero F, Avvisati G, Castaman G, Barbui T, Mandelli F. Early deaths and anti-hemorrhagic treatments in acute promyelocytic leukemia. A GIMEMA retrospective study in 268 consecutive patients. Blood. 1990;11:2112–7.
Drapkin RL, Gee TS, Dowling MD, et al. Prophylactic heparin therapy in acute promyelocytic leukemia. Cancer. 1978;41:2484–90.
Daly PA, Schiffer CA, Wiernick PH. Acute promyelocytic leukemia—clinical management of 15 patients. Am J Hematol. 1980;8:347–59.
Gralnick HR, Abrell E. Studies of the procoagulant and fibrinolytic activity of promyelocytes in acute promyelocytic leukaemia. Br J Haematol. 1973;24:89–99.
Avvisati G, ten Cate JW, Sturk A, Lamping R, Petti MG, Mandelli F. Acquired alpha-2-antiplasmin deficiency in acute promyelocytic leukaemia. Br J Haematol. 1988;70:43–8.
Gralnick HR, Bagley J, Abrell E. Heparin treatment for the hemorrhagic diathesis of acute promyelocytic leukemia. Am J Med. 1972;52:167–74.
Cordonnier C, Vernant JP, Brun B, et al. Acute promyeocytic leukemia in 57 previously untreated patients. Cancer. 1985;55:18–25.
Kantarjian HM, Keating MJ, Walters RS, et al. Acute promyelocytic leukemia. M.D. Anderson Hospital experience. Am J Med. 1986;80:789–97.
Goldberg MA, Ginsburg D, Mayer RJ, et al. Is heparin administration necessary during induction chemotherapy for patients with acute promyelocytic leukemia? Blood. 1987;69:187–91.
Hoyle CF, Swirsky DM, Freedman L, Hayhoe FG. Beneficial effect of heparin in the management of patients with APL. Br J Haematol. 1988;68:283–9.
Sanz MA, Jarque I, Martin G, et al. Acute promyelocytic leukemia. Therapy results and prognostic factors. Cancer. 1988;61:7–13.
Cunningham I, Gee TS, Reich LM, Kempin SJ, Naval AN, Clarkson BD. Acute promyelocytic leukemia: treatment results during a decade at Memorial Hospital. Blood. 1989;73:1116–22.
Thomas X, Archimbaud E, Treille-Ritouet D, Fiere D. Prognostic factors in acute promyelocytic leukemia: a retrospective study of 67 cases. Leuk Lymphoma. 1991;4:249–56.
Head D, Kopecky KJ, Weick J, et al. Effect of aggressive daunomycin therapy on survival in acute promyelocytic leukemia. Blood. 1995;86:1717–28.
Avvisati G, Petti MC, Petrucci MT, Falconi E, Trindelli MC, Mandelli F. Treatment of recurrent promyelocytic leukemia with a combination regimen utilizing amsacrine, cytosine arabinoside and 6-thioguanine (AAT). Haematologica. 1989;74:279–82.
Golomb HM, Rowley J, Vardiman J, Baron J, Locker G, Krasnow S. Partial deletion of long arm of chromosome 17: a specific abnormality in acute promyelocytic leukemia? Arch Intern Med. 1976;136:825–8.
Rowley JD, Golomb HM, Dougherty C. The 15/17 translocation: a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;i:549–50.
Larson RA, Kondo K, Vardiman JW, Butler AE, Golomb HM, Rowley JD. Evidence for a 15;17 translocation in every patient with acute promyelocytic leukemia. Am J Med. 1984;76:827–41.
Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–65.
Collins SJ, Gallo RC, Gallagher RE. Continuous growth and differentiation of human myeloid leukemia cells in suspension culture. Nature. 1977;270:347–9.
Parmley RT, Akin DT, Barton JC, et al. Cytochemistry and ultrastructural morphometry of cultured HL60 myeloid leukemia cells. Cancer Res. 1987;47:4932–40.
Dalton WT, Ahearn MJ, McCredie KB, et al. HL-60 cell line was derived from a patient with FAB-M2 and not FAB-M3. Blood. 1988;71:242–7.
Pierce GB Jr, Verney EL. An in vitro and in vivo study of differentiation in teratocarcinomas. Cancer. 1961;14:1017–29.
Sachs L. Control of normal cell differentiation and the phenotypic reversion of malignancy in myeloid leukaemia. Nature. 1978;274:535–9.
Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci USA. 1980;77:2936–40.
Breitman TR, Collins SJ, Keene BR. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood. 1981;57:1000–4.
Flynn PJ, Miller WJ, Weisdorf DJ, et al. Retinoic acid treatment of acute promyelocytic leukemia: in vitro and in vivo observations. Blood. 1983;62:1211–7.
Nilsson B. Probable in vivo induction of differentiation by retinoic acid of promyelocytes in acute promyelocytic leukaemia. Br J Haematol. 1984;57:365–71.
Daenen S, Vellenga E, van Dobbenburgh OA, Halie MR. Retinoic acid as antileukemic therapy in a patient with acute promyelocytic leukemia and Aspergillus pneumonia. Blood. 1986;67:559–61.
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–15.
Huang ME, Ye YC, Chen SR, et al. All-trans retinoic acid with or without low dose cytosine arabinoside in acute promyelocytic leukemia: report of 6 cases. Chin Med J. 1987;100:949–53.
Huang ME, Ye YC, Chen SR, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–72.
Degos L. The history of acute promyelocytic leukaemia. Br J Haematol. 2003;122:539–53.
Degos L, Chomienne C, Daniel MT, et al. Treatment of first relapse in acute promyelocytic leukaemia with all-trans retinoic acid. Lancet. 1990;336:1440–1.
Castaigne S, Chomienne C, Daniel MT, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704–9.
Chomienne C, Ballerini P, Balitrand N, et al. Differentiation of acute promyelocytic cells by all-trans retinoic acid. II. In vitro studies: structure–function relationship. Blood. 1990;76:1710–7.
Wiernik PH, Dutcher JP, Paietta E, et al. Treatment of promyelocytic blast crisis of chronic myelogenous leukemia with all trans-retinoic acid. Leukemia. 1991;5:504–9.
Warrell RP Jr, Maslak P, Eardley A, Heller G, Miller WH Jr, Frankel SR. Treatment of acute promyelocytic leukemia with all-trans retinoic acid: an update of the New York experience. Leukemia. 1994;8:929–33.
Chen ZX, Xue YQ, Zhang R, et al. A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood. 1991;78:1413–9.
Wiernik PH. Acute promyelocytic leukemia: another pseudoleukemia? Blood. 1990;76:1675–7.
De The H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature. 1990;347:558–61.
Borrow J, Goddard AD, Sheer D, Solomon E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science. 1990;249:1577–80.
Longo L, Pandolfi P, Biondi A, et al. Rearrangement and aberrant expression of the retinoic acid receptor α in acute promyelocytic leukemia. J Exp Med. 1990;172:1571–5.
Alcalay M, Zangrilli D, Pandolfi PP, et al. Translocation breakpoint of acute promyelocytic leukemia lies within the retinoic acid receptor alpha locus. Proc Natl Acad Sci USA. 1991;88:1977–81.
Mattei MG, Petkovich M, Mattei JF, Brand N, Chambon P. Mapping of the human retinoic acid receptor to the q21 band of chromosome 17. Hum Genet. 1988;80:186–8.
Chomienne C, Ballerini P, Huang ME, et al. The retinoic acid receptor alpha gene is rearranged in retinoic acid sensitive promyelocytic leukaemias. Leukemia. 1990;4:802–7.
Lanotte M, Martin-Thouvenin V, Najman S, et al. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood. 1991;77:1080–6.
De Thé H, Lavau C, Marchio A, et al. The myl RAR alpha fusion mRNA generated by the t(15;17) translocation in APL encodes a functionally altered retinoic acid receptor. Cell. 1991;66:675–84.
Kakizuka A, Miller WH Jr, Umesono K, et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991;66:663–74.
Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–70.
Castaigne S, Balitrand N, de Thé H, et al. A PML/RAR alpha fusion transcript is constantly detected by RNA based polymerase chain reaction in acute promyelocytic leukaemia. Blood. 1992;79:3110–8.
Biondi A, Rambaldi A, Pandolfi PP, et al. Molecular monitoring of the myl/retinoic acid receptor alpha fusion gene in APL by polymerase chain reaction. Blood. 1992;80:492–7.
Miller WH Jr, Kakizuka A, Frankel SR, et al. Reverse transcription polymerase chain reaction for the rearranged retinoic acid receptor alpha clarifies diagnosis and detects minimal residual disease in acute promyelocytic leukemia. Proc Natl Acad Sci USA. 1992;89:2694–8.
Lo Coco F, Diverio D, Pandolfi PP, et al. Molecular evaluation of residual disease as a predictor of relapse in acute promyelocytic leukaemia. Lancet. 1992;340:1437–8.
Grimwade D, Howe K, Langabeer S, et al. Minimal residual disease detection in acute promyelocytic leukemia by reverse-transcriptase PCR: evaluation of PML-RAR alpha and RAR alpha-PML assessment in patients who ultimately relapse. Leukemia. 1996;10:61–6.
Diverio D, Rossi V, Avvisati G, et al. Early detection of relapse by prospective RT-PCR analysis of the PML-RARalpha fusion gene in patients with acute promyelocytic leukemia enrolled in the GIMEMA-AIEOP multicenter AIDA trial. Blood. 1998;92:784–9.
Lo Coco F, Diverio D, Avvisati G, et al. Therapy of molecular relapse in acute promyelocytic leukemia. Blood. 1999;94:2225–9.
Grimwade D, Jovanovic JV, Hills RK, et al. Prospective minimal residual disease monitoring to predict relapse of acute promyelocytic leukemia and to direct pre-emptive arsenic trioxide therapy. J Clin Oncol. 2009;27:3650–8.
He LZ, Guidez F, Tribioli C, et al. Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL. Nat Genet. 1998;18:126–35.
Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391:815–8.
Lin RJ, Evans RM. Acquisition of oncogenic potential by RAR chimeras in acute promyelocytic leukemia through formation of homodimers. Mol Cell. 2000;5:821–30.
Adams J, Nassiri M. Acute promyelocytic leukemia—a review and discussion of variant translocations. Arch Pathol Lab Med. 2015;139:1308–13.
Chen SJ, Zelent A, Tong JH, et al. Rearrangements of the retinoic acid receptor alpha and promyelocytic leukemia zinc finger genes resulting from t(11;17) (q23;q21) in a patient with acute promyelocytic leukemia. J Clin Investig. 1993;91:2260–7.
Chen Z, Brand NJ, Chen A, et al. Fusion between a novel Kruppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J. 1993;12:1161–7.
Arnould C, Philippe C, Bourdon V, et al. The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor alpha in acute promyelocytic-like leukaemia. Hum Mol Genet. 1999;8:1741–9.
Wells RA, Hummel JL, De KA, et al. A new variant translocation in acute promyelocytic leukaemia: molecular characterization and clinical correlation. Leukemia. 1996;10:735–40.
Corey SJ, Locker J, Oliveri DR, et al. A non-classical translocation involving 17q12 (retinoic acid receptor alpha) in acute promyelocytic leukemia (APML) with atypical features. Leukemia. 1994;8:1350–3.
Kondo T, Mori A, Darmanin S, et al. The seventh pathogenic fusion gene FIP1L1-RARA was isolated from a t(4;17)-positive acute promyelocytic leukemia. Haematologica. 2008;93:1414–6.
Baba SM, Pandith AA, Shah ZA, Baba RA. Pathogenetic implication of fusion genes in acute promyelocytic leukemia and their diagnostic utility. Clin Genet. 2019;95:41–52. https://doi.org/10.1111/cge.13372.
Fenaux P, Castaigne S, Dombret H, et al. All-trans retinoic acid followed by intensive chemotherapy gives a high complete remission rate and may prolong remissions in newly diagnosed acute promyelocytic leukemia: a pilot study on 26 cases. Blood. 1992;80:2176–81.
Le Deley MC, Castaigne S, et al. Effect of all trans retinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. The European APL 91 Group. Blood. 1993;82:3241–9.
Tallman MS, Andersen JW, Schiffer CA, et al. All trans retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021–8.
Fenaux P, Chastang C, Chevret S, et al. A randomized comparison of all trans retinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94:1192–200.
Lengfelder E, Reichert A, Schoch C, et al. Double induction strategy including high dose cytarabine in combination with all-trans retinoic acid: effects in patients with newly diagnosed acute promyelocytic leukemia. German AML Cooperative Group. Leukemia. 2000;14:1362–70.
Mandelli F, Diverio D, Avvisati G, et al. Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell’Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood. 1997;90:1014–21.
Asou N, Adachi K, Tamura J, et al. Analysis of prognostic factors in newly diagnosed acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. Japan Adult Leukemia Study Group. J Clin Oncol 1998; 16:78-85.
De Botton S, Chevret S, Coiteux V, et al. Early onset of chemotherapy can reduce the incidence of ATRA syndrome in newly diagnosed acute promyelocytic leukemia (APL) with low white blood cell counts: results from APL93 trial. Leukemia. 2003;17:339–42.
Avvisati G, Lo-Coco F, Paoloni FP, et al. AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood. 2011;117:4716–25.
Asou N, Kishimoto Y, Kiyoi H, et al. A randomized study with or without intensified maintenance chemotherapy in patients with acute promyelocytic leukemia who have become negative for PML/RARA transcript after consolidation therapy. Blood. 2007;110:59–66.
Shinagawa K, Yanada M, Sakura T, et al. Tamibarotene as maintenance therapy for acute promyelocytic leukemia: results from a randomized controlled trial. J Clin Oncol. 2014;32:3729–35.
Montesinos P, Diaz-Mediavilla J, Deben G, et al. Central nervous system involvement at first relapse in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline monochemotherapy without intrathecal prophylaxis. Haematologica. 2009;94:1242–9.
Sanz MA, Grimwade D, Tallman MS, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–91.
Seftel MD, Barnett MJ, Couban S, et al. A Canadian consensus on the management of newly diagnosed and relapsed acute promyelocytic leukemia in adults. Curr Oncol. 2014;21:234–50.
Zheng PZ, Wang KK, Zhang QY, et al. Systems analysis of transcriptome and proteome in retinoic acid/arsenic trioxide-induced cell differentiation/apoptosis of promyelocytic leukemia. Proc Natl Acad Sci USA. 2005;102:7653–8.
Ablain J, De Thé H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 2011;117:5795–802.
Nervi C, Ferrara FF, Fanelli M, et al. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein. Blood. 1998;92:2244–51.
Fang Y, Zhou X, Lin M, et al. The ubiquitin-proteasome pathway plays essential roles in ATRA-induced leukemia cells G0/G1 phase arrest and transition into granulocytic differentiation. Cancer Biol Ther. 2010;10:1157–67.
Frankel SR, Eardley A, Lauwers G, Weiss M, Warrell RP. The ‘retinoic acid syndrome’ in acute promyelocytic leukemia. Ann Intern Med. 1992;117:292–6.
De Botton S, Dombret H, Sanz M, et al. Incidence, clinical features, and outcome of all-trans retinoic acid syndrome in 413 cases of newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1998;92:2712–8.
Wang ZY, Sun GL, Shen ZX, et al. Differentiation therapy for acute promyelocytic leukemia with all-trans retinoic acid: 10-year experience of its clinical application. Chin Med J. 1999;112:963–7.
Luesink M, Pennings JLA, Wissink WM, et al. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: triggering the differentiation syndrome. Blood. 2009;114:5512–21.
Sanz MA, Montesinos P. How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood. 2014;123:2777–82.
Wiley JS, Firkin FC. Reduction of pulmonary toxicity by prednisolone prophylaxis during all-trans retinoic acid treatment of acute promyelocytic leukemia. Australian Leukaemia Study Group. Leukemia. 1995;9:774–8.
Kelaidi C, Chevret S, De Botton S, et al. Improved outcome of acute promyelocytic leukemia with high WBC counts over the last 15 years: The European APL Group experience. J Clin Oncol. 2009;27:2668–76.
Montesinos P, Bergua JM, Vellenga E, et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood. 2009;113:775–83.
Avvisati G, Lo-Coco F, Diverio D, et al. AIDA (all-trans retinoic acid + idarubicin) in newly diagnosed acute promyelocytic leukemia: a Gruppo Italiano Malattie Ematologiche dell’Adulta (GIMEMA) pilot study. Blood. 1996;88:1390–8.
Iland H, Bradstock K, Seymour J, et al. Results of the APML3 trial incorporating all-trans-retinoic acid and idarubicin in both induction and consolidation as initial therapy for patients with acute promyelocytic leukemia. Haematologica. 2012;97:227–34.
Sanz MA, Lo-Coco F, Martin G, et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: a joint study of the PETHEMA and GIMEMA cooperative groups. Blood. 2000;96:1247–53.
Sanz MA, Martin G, Gonzalez M, et al. Risk-adapted treatment of acute promyelocytic leukemia with all-trans retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood. 2004;103:1237–43.
Sanz MA, Montesinos P, Rayon C, et al. Risk-adapted treatment of acute promyelocytic leukemia based on all-trans retinoic acid and anthracycline with addition of cytarabine in consolidation therapy for high-risk patients: further improvements in treatment outcome. Blood. 2010;115:5135–46.
Adès L, Chevret S, Raffoux E, et al. Is cytarabine useful in the treatment of acute promyelocytic leukemia? Results of a randomized trial from the European Acute Promyelocytic Leukemia Group. J Clin Oncol. 2006;24:5703–10.
Adès L, Chevret S, Raffoux E, et al. Long-term follow-up of the European APL 2000 trial, evaluating the role of cytarabine combined with ATRA and daunorubicin in the treatment of nonelderly APL patients. Am J Hematol. 2013;88:556–9.
Burnett AK, Hills RK, Grimwade D, et al. Inclusion of chemotherapy in addition to anthracycline in the treatment of acute promyelocytic leukaemia does not improve outcomes: results of the MRC AML15 trial. Leukemia. 2013;27:843–51.
Sanz MA, Montesinos P, Kim HT, et al. All-trans retinoic acid with daunorubicin or idarubicin for risk-adapted treatment of acute promyelocytic leukemia: a matched-pair analysis of the PETHEMA LPA-2005 and IC-APL studies. Ann Hematol. 2015;94:1347–56.
Thomas X, Le QH, Fière D. Anthracycline-related toxicity requiring cardiac transplantation in long-term disease-free survivors with acute promyelocytic leukemia. Ann Hematol. 2002;81:504–7.
Pellicori P, Calicchia A, Lococo F, et al. Subclinical anthracycline cardiotoxicity in patients with acute promyelocytic leukemia in long-term remission after AIDA protocol. Congest Heart Fail. 2012;18:217–21.
Lengfelder E, Saussele S, Weisser A, Buchner T, Hehlmann R. Treatment concepts of acute promyelocytic leukemia. Crit Rev Oncol Hematol. 2005;56:261–74.
Grimwade D. The pathogenesis of acute promyelocytic leukaemia: evaluation of the role of molecular diagnosis and monitoring in the management of the disease. Br J Haematol. 1999;106:591–613.
Esteve J, Escoda L, Martin G, et al. Outcome of patients with acute promyelocytic leukemia failing to front-line treatment with all-trans retinoic acid and anthracycline-based chemotherapy (PETHEMA protocols LPA96 and LPA99): benefit of an early intervention. Leukemia. 2007;21:446–52.
Castagnola C, Lunghi M, Corso A, et al. Management of acute promyelocytic leukemia relapse in the ATRA era. Haematologica. 1998;83:714–7.
Thomas X, Dombret H, Cordonnier C, et al. Treatment of relapsing acute promyelocytic leukemia by all-trans retinoic acid therapy followed by timed sequential chemotherapy and stem cell transplantation. Leukemia. 2000;14:1006–13.
Sanz MA, Labopin M, Gorin NC, et al. Hematopoietic stem cell transplantation for adults with acute promyelocytic leukemia in the ATRA era: a survey of the European Cooperative Group for Blood and Marrow Transplantation. Bone Marrow Transpl. 2007;39:461–9.
Madan V, Shyamsunder P, Han L, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. 2016;30:2430.
Lucena-Araujo AR, Kim HT, Jacomo RH, et al. Internal tandem duplication of the FLT3 gene confers poor overall survival in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline-based chemotherapy: an International Consortium on Acute Promyelocytic Leukemia study. Ann Hematol. 2014;93:2001–10.
Poire X, Moser BK, Gallagher RE, et al. Arsenic trioxide in front-line therapy of acute promyelocytic leukemia (C9710): prognostic significance of FLT3 mutations and complex karyotype. Leuk Lymphoma. 2014;55:1523–32.
Cicconi L, Divona M, Ciardi C, et al. PML-RARα kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia. 2016;30:1987–92.
Beitinjaneh A, Jang S, Roukoz H, Majhail NS. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations in acute promyelocytic leukemia: a systematic review. Leuk Res. 2010;34:831–6.
Gale RE, Hills R, Pizzey AR, et al. NCRI Adult Leukaemia Working Party. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. 2005;106:3768–76.
Kutny MA, Moser BK, Laumann K, et al. FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2012;59:662–7.
Testa U, Lo-Coco F. Prognostic factors in acute promyelocytic leukemia: strategies to define high-risk patients. Ann Hematol. 2016;95:673–80.
Breccia M, De Propris MS, Stefanizzi C, et al. Negative prognostic value of CD34 antigen also if expressed on a small population of acute promyelocytic leukemia cells. Ann Hematol. 2014;93:1819–23.
Kogan SC. Mouse models of acute promyelocytic leukemia. Curr Top Microbiol Immunol. 2007;313:3–29.
Westervelt P, Ley TJ. Seed versus soil: the importance of the target cell for transgenic models of human leukemias. Blood. 1999;93:2143–8.
Kamashev DE, Vitoux D, de Thé H. PML/RARA-RXR oligomers mediate retinoid-/cAMP in APL cell differentiation. J Exp Med. 2004;199:1–13.
Martens JH, Brinkman AB, Simmer F, et al. PML-RARα/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell. 2010;17:173–85.
Zhu J, Zhou J, Peres L, et al. A sumoylation site in PML/RARA is essential for leukemic transformation. Cancer Cell. 2005;7:143–53.
Nasr R, Guillemin MC, Ferhi O, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML/RARA degradation. Nat Med. 2008;14:1333–42.
Pietersen AM, van Lohuizen M. Stem cell regulation by polycomb repressors: postponing commitment. Curr Opin Cell Biol. 2008;20:201–7.
Marinelli A, Bossi D, Pelicci PG, Minucci S. Redundant function of retinoic acid receptor isoforms in leukemogenesis unravels a prominent function of genome topology and architecture in the selection of mutagenic events in cancer. Leukemia. 2009;23:417–9.
Osumi T, Tsujimoto SI, Tamura M, et al. Recurrent RARB translocations in acute promyelocytic leukemia lacking RARA translocation. Cancer Res. 2018;78:4452–8.
Such E, Cervera J, Valencia A, et al. A novel NUP98/RARG gene fusion in acute myeloid leukemia resembling acute promyelocytic leukemia. Blood. 2011;117:242–5.
Marinelli A, Bossi D, Pelicci PG, Minucci S. A redundant oncogenic potential of the retinoic receptor (RAR) a, b and c isoforms in acute promyelocytic leukemia. Leukemia. 2007;21:647–50.
Chen SJ, Zhou GB, Zhang XW, Mao JH, de Thé H, Chen Z. From an old remedy to a magic bullet: molecular mechanisms underlying the therapeutic effects of arsenic in fighting leukemia. Blood. 2011;117:6425–37.
Chen Z, Chen SJ. Poisoning the devil. Cell. 2017;168:557–60.
Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H. How acute promyelocytic leukemia revived arsenic. Nat Rev Cancer. 2002;2:705–13.
Thomas X, Troncy J. Arsenic: a beneficial therapeutic poison—a historical overview. Adler Museum Bull. 2009;35:3–13.
Jolliffe DM. A history of the use of arsenicals in man. J R Soc Med. 1993;86:287–9.
Sears DA. History of the treatment of chronic myelocytic leukemia. Am J Med Sci. 1988;296:85–6.
Sun HD, Ma L, Hu XC, Zhang TD. Ai-lin 1 treated 32 cases of acute promyelocytic leukemia. Chin J Integrat Chin West Med. 1992;12:170–1.
Zhang P, Wang SY, Hu LH. Arsenic trioxide treated 72 cases of acute promyelocytic leukemia. Chin J Hematol. 1996;17:58–62.
Chen GQ, Shi XG, Tang W, et al. Use of arsenic trioxide (AS2O3) in the treatment of acute promyelocytic leukemia (APL), I: AS2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–53.
Shen ZX, Chen GQ, Ni JH, et al. Use of arsenic trioxide (AS2O3) in the treatment of acute promyelocytic leukemia (APL), II: clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–60.
Niu C, Yan H, Yu T, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic patients. Blood. 1999;94:3315–24.
Zhang P. On arsenic trioxide in the clinical treatment of acute promyelocytic leukemia. Leuk Res Rep. 2017;7:29–32.
Soignet SL, Frankel SR, Douer D, et al. United States multicenter study of arsenic trioxide in relapsed acute promyelocytic leukemia. J Clin Oncol. 2001;19:3852–60.
Shigeno K, Naito K, Sahara N, et al. Arsenic trioxide therapy in relapsed or refractory Japanese patients with acute promyelocytic leukemia: updated outcomes of the phase II study and postremission therapies. Int J Hematol. 2005;82:224–9.
Shen Y, Shen ZX, Yan H, et al. Studies on the clinical efficacy and pharmacokinetics of low-dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: a comparison with conventional dosage. Leukemia. 2001;15:735–41.
Kwong YL, Au WY, Chim CS, Pang A, Suen C, Liang R. Arsenic trioxide- and idarubicin-induced remissions in relapsed acute promyelocytic leukaemia: clinicopathological and molecular features of a pilot study. Am J Hematol. 2001;66:274–9.
Leoni F, Gianfaldoni G, Annunziata M, et al. Arsenic trioxide therapy for relapsed acute promyelocytic leukemia: a bridge to transplantation. Haematologica. 2002;87:485–9.
Ohnishi K, Yoshida H, Shigeno K, et al. Arsenic trioxide therapy for relapsed or refractory Japanese patients with acute promyelocytic leukemia: need for careful electrocardiogram monitoring. Leukemia. 2002;16:617–22.
Lazo G, Kantarjian H, Estey E, Thomas D, O’Brien S, Cortes J. Use of arsenic trioxide (AS2O3) in the treatment of patients with acute promyelocytic leukemia: the MD Anderson experience. Cancer. 2003;97:2218–24.
Raffoux E, Rousselot P, Poupon J, et al. Combined treatment with arsenic trioxide and all-trans-retinoic acid in patients with relapsed acute promyelocytic leukemia. J Clin Oncol. 2003;21:2326–34.
Carmosino I, Latagliata R, Avvisati G, et al. Arsenic trioxide in the treatment of advanced acute promyelocytic leukemia. Haematologica. 2004;89:615–7.
Thomas X, Pigneux A, Raffoux E, Huguet F, Caillot D, Fenaux P. Superiority of an arsenic trioxide-based regimen over a historic control combining all-trans retinoic acid plus intensive chemotherapy in the treatment of relapsed acute promyelocytic leukemia. Haematologica. 2006;91:996–7.
Aribi A, Kantarjian HM, Estey EH, et al. Combination therapy with arsenic trioxide, all-trans retinoic acid, and gemtuzumab ozogamicin in recurrent acute promyelocytic leukemia. Cancer. 2007;109:1355–9.
Alimoghaddam K, Ghavamzadeh A, Jahani M, et al. Treatment of relapsed acute promyelocytic leukemia by arsenic trioxide in Iran. Arch Iran Med. 2011;14:167–9.
Lengfelder E, Hofmann WK, Nowak D. Impact of arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2012;26:433–42.
Chen Z, Zhao WL, Shen ZX, et al. Arsenic trioxide and acute promyelocytic leukemia: clinical and biological. Curr Top Microbiol Immunol. 2007;313:129–44.
Joe Y, Jeong JH, Yang S, et al. ATR, PML, and CHK2 play a role in arsenic trioxide-induced apoptosis. J Biol Chem. 2006;281:28764–71.
Camacho LH, Soignet SL, Chanel S, et al. Leukocytosis and the retinoic acid syndrome in patients with acute promyelocytic leukemia treated with arsenic trioxide. J Clin Oncol. 2010;18:2620–5.
De Thé G, Rivière M, Bernhard W. Examen au microscope électronique de la tumeur VX2 du lapin domestique dérivée du papillome de Shope. Bull Cancer. 1960;47:570–84.
Jeanne M, Lallemand-Breitenbach V, Ferhi O, et al. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010;18:88–98.
Zhang XW, Yan XJ, Zhou ZR, et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328:240–3.
Lallemand-Breitenbach V, Jeanne M, Benhenda S, et al. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat Cell Biol. 2008;10:547–55.
Tatham MH, Geoffroy MC, Shen L, et al. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol. 2008;10:538–46.
Ablain J, Rice K, Soilihi H, et al. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat Med. 2014;20:167–74.
Ghavamzadeh A, Alimoghaddam K, Rostami S, et al. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J Clin Oncol. 2011;29:2753–7.
Mathews V, George B, Chendamarai E, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J Clin Oncol. 2010;28:3866–71.
Lehmann-Che J, Bailly C, de Thé H. Therapy resistance in APL. N Engl J Med. 2014;371:1171–2.
DosSantos GA, Kats L, Pandolfi PP. Synergy against PML-RARa: targeting transcription, proteolysis, differentiation, and self-renewal in acute promyelocytic leukemia. J Exp Med. 2013;210:2793–802.
Au WY, Tam S, Fong BM, Kwong YL. Elemental arsenic entered the cerebrospinal fluid during oral arsenic trioxide treatment of meningeal relapse of acute promyelocytic leukemia. Blood. 2006;107:3012–3.
Goto E, Tomita A, Hayakawa F, et al. Missense mutations in PML-RARA are critical for the lack of responsiveness to arsenic trioxide treatment. Blood. 2011;118:1600–8.
Roboz GJ, Ritchie EK, Carlin RF, et al. Prevalence, management, and clinical consequences of QT interval prolongation during treatment with arsenic trioxide. J Clin Oncol. 2014;32:3723–8.
Kiguchi T, Yoshino Y, Yuan B, et al. Speciation of arsenic trioxide penetrates into cerebrospinal fluid in patients with acute promyelocytic leukemia. Leuk Res. 2010;34:403–5.
Hu J, Liu YF, Wu CF, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2009;106:3342–7.
Zhu H, Hu J, Chen L, et al. The 12-year follow-up of survival, chronic adverse effects, and retention of arsenic in patients with acute promyelocytic leukemia. Blood. 2016;128:1525–8.
Estey E, Garcia-Manero G, Ferrajoli A, et al. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood. 2006;107:3469–73.
Ravandi F, Estey E, Jones D, et al. Effective treatment of acute promyelocytic leukemia with all-trans retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol. 2009;27:504–10.
Powell BL, Moser B, Stock W, et al. Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood. 2010;116:3751–7.
Gore SD, Gojo I, Sekeres MA, et al. Single cycle of arsenic trioxide-based consolidation chemotherapy spares anthracycline exposure in the primary management of acute promyelocytic leukemia. J Clin Oncol. 2010;28:1047–53.
Adès L, Thomas X, Guerci Bresler A, et al. Arsenic trioxide is required in the treatment of newly diagnosed acute promyelocytic leukemia. Analysis of a randomized trial (APL 2006) by the French-Belgian-Swiss APL group. Haematologica. 2018;1:1. https://doi.org/10.3324/haematol.2018.198614.
Lallemand-Breitenbach V, Guillemin MC, Janin A, et al. Retinoic acid and arsenic synergize to eradicate leukemic cells in a mouse model of acute promyelocytic leukemia. J Exp Med. 1999;189:1043–52.
Jing Y, Wang L, Xia L, et al. Combined effect of all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia cells in vitro and in vivo. Blood. 2001;97:264–9.
Shen ZX, Shi ZZ, Fang J, et al. All-trans retinoic acid/AS2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA. 2004;101:5328–35.
Abaza Y, Kantarjian H, Garcia-Manero G, et al. Long-term outcome of acute promyelocytic leukemia treated with all-trans retinoic acid, arsenic trioxide, and gemtuzumab. Blood. 2017;129:1275–83.
Platzbecker U, Avvisati G, Cicconi L, et al. Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non-high-risk acute promyelocytic leukemia: final results of the randomized Italian-German APL0406 trial. J Clin Oncol. 2017;35:605–12.
Iland HJ, Bradstock K, Supple SG, et al. All-trans retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood. 2012;120:1570–80.
Ikeda M, Nagashima T, Bhattacharjee AK, et al. Quantitative analysis of hyperosmotic and hypothermic blood–brain barrier opening. Acta Neurochir Suppl. 2003;86:559–63.
Wang H, Cao FL, Li JM, et al. Arsenic trioxide and mannitol for the treatment of acute promyelocytic leukemia relapse in the central nervous system. Blood. 2014;124:1998–2000.
Cicconi L, Breccia M, Franceschini L, et al. Prolonged treatment with arsenic trioxide (ATO) and all-trans-retinoic acid (ATRA) for relapsed acute promyelocytic leukemia previously treated with ATRA and chemotherapy. Ann Hematol. 2018;97:1797–802.
Rego EM, He LZ, Warrell RP Jr, Wang ZG, Pandolfi PP. Retinoic acid (RA) and As2O3 treatment in transgenic models of acute promyelocytic leukemia (APL) unravel the distinct nature of the leukemogenic process induced by the PML-RARalpha and PLZF-RARalpha oncoproteins. Proc Natl Acad Sci USA. 2000;97:10173–8.
De Thé H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010;10:775–83.
Insinga A, Monestiroli S, Ronzoni S, et al. Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. EMBO J. 2004;23:1144–54.
Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661.
Arteaga MF, Mikesch JH, Qiu J, et al. The histone demethylase PHF8 governs retinoic acid response in acute promyelocytic leukemia. Cancer Cell. 2013;23:376–89.
Cole CB, Verdoni AM, Ketkar S, et al. PML-RARA requires DNA methyltransferase 3A to initiate acute promyelocytic leukemia. J Clin Investig. 2016;126:85–98.
Li K, Wang F, Cao WB, et al. TRIB3 promotes APL progression through stabilization of the oncoprotein PML-RARalpha and inhibition of p53-mediated senescence. Cancer Cell. 2017;31:697–710.
Mehdipour P, Santoro F, Botrugno OA, et al. HDAC3 activity is required for initiation of leukemogenesis in acute promyelocytic leukemia. Leukemia. 2017;31:995–7.
Prunier C, Zhang MZ, Kumar S, et al. Disruption of the PHRF1 tumor suppressor network by PML-RARalpha drives acute promyelocytic leukemia pathogenesis. Cell Rep. 2015;10:883–90.
Zhu J, Lallemand-Breitenbach V, de Thé H. Pathways of retinoic acid- or arsenic trioxide-induced PML/RARalpha catabolism, role of oncogene degradation in disease remission. Oncogene. 2001;20:7257–65.
Schafernak KT, Corey SJ. Histiocytic clearance of neoplastic cells in acute promyelocytic leukemia. Blood. 2014;124:3020.
Korf K, Wodrich H, Haschke A, et al. The PML domain of PML-RARalpha blocks senescence to promote leukemia. Proc Natl Acad Sci USA. 2014;111:12133–8.
Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11:503–11.
Sterndorf T, Phan VT, Maunakea ML, et al. Forced retinoic acid receptor a homodimer prime mice for APL-like leukemia. Cancer Cell. 2006;9:81–94.
Minucci S, Maccarana M, Cioce M, et al. Oligomerization of RAR and AML1 transcription factors as a novel mechanism of oncogenic activation. Mol Cell. 2000;5:811–20.
Occhionorelli M, Santoro F, Pallavicini I, et al. The self-association coiled-coil domain of PML is sufficient for the oncogenic conversion of the retinoic acid receptor (RAR) alpha. Leukemia. 2011;25:814–20.
Wei S, Kozono S, Kats L, et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med. 2015;21:457–66.
NCCN. NCCN guidelines on acute myeloid leukemia, version 1. 2015. National Comprehensive Cancer Network, Fort Washington, PA, USA. http://www.nccn.org/professionals/physician_gls/pdf/aml.pdf. Accessed Aug 2018.
Lehmann S, Ravn A, Carlsson L, et al. Continuing high early death rate in acute promyelocytic leukemia: a population-based report from the Swedish Adult Acute Leukemia Registry. Leukemia. 2011;25:1128–34.
Park JH, Qiao B, Panageas KS, et al. Early death rate in acute promyelocytic leukemia remains high despite all-trans retinoic acid. Blood. 2011;118:1248–54.
Paulson K, Serebrin A, Lambert P, et al. Acute promyelocytic leukemia is characterized by stable incidence and improved survival that is restricted to patients managed in leukemia referral centres: a pan-Canadian epidemiological study. Br J Haematol. 2014;166:660–6.
Rahmé R, Thomas X, Recher C, et al. Early death in acute promyelocytic leukemia (APL) in French centers: a multicenter study in 399 patients. Leukemia. 2014;28:2422–4.
Micol JB, Raffoux E, Boissel N, et al. Management and treatment results in patients with acute promyelocytic leukaemia (APL) not enrolled in clinical trials. Eur J Cancer. 2014;50:1159–68.
Jillela AP, Kota VK. The global problem of early deaths in acute promyelocytic leukemia: a strategy to decrease induction mortality in the most curable leukemia. Blood Rev. 2018;32:89–95.
Mantha S, Goldman DA, Devlin SM, et al. Determinants of fatal bleeding during induction therapy for acute promyelocytic leukemia in the ATRA era. Blood. 2017;129:1763–7.
Altman JK, Rademaker A, Cull E, et al. Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res. 2013;37:1004–9.
Breccia M, Latagliata R, Cannella L, et al. Early hemorrhagic death before starting therapy in acute promyelocytic leukemia: association with high WBC count, late diagnosis and delayed treatment initiation. Haematologica. 2010;95:853–4.
Zhang Y, Hou W, Wang P, et al. Development of a risk grading system to identify patients with acute promyelocytic leukemia at high risk of early death. Cancer Manag Res. 2018;10:3619–27.
Kayser S, Schlenk RF, Platzbecker U. Management of patients with acute promyelocytic leukemia. Leukemia. 2018;32:1277–94.
Mi JQ, Chen SJ, Zhou GB, et al. Synergistic targeted therapy for acute promyelocytic leukaemia: a model of translational research in human cancer. J Intern Med. 2015;278:627–42.
Imaizumi M, Tawa A, Hanada R, et al. Prospective study of a therapeutic regimen with all-trans retinoic acid and anthracyclines in combination of cytarabine in children with acute promyelocytic leukaemia: the Japanese Childhood Acute Myeloid Leukaemia Cooperative Study. Br J Haematol. 2011;152:89–98.
Cheng Y, Zhang L, Wu J, et al. Long-term prognosis of childhood acute promyelocytic leukaemia with arsenic trioxide administration in induction and consolidation chemotherapy phases: a single-center experience. Eur J Haematol. 2013;91:483–9.
Creutzig U, Dworzak MN, Bochennek K, et al. First experience of the AML-Berlin-Frankfurt-Munster group in pediatric patients with standard-risk acute promyelocytic leukemia treated with arsenic trioxide and all-trans retinoic acid. Pediatr Blood Cancer. 2017;64:e26451.
Strocchio L, Gurnari C, Santoro N, et al. Arsenic trioxide and all-trans retinoic acid treatment for childhood acute promyelocytic leukaemia. Br J Haematol. 2018. https://doi.org/10.1111/bjh.15507 [Epub ahead of print].
Lo-Coco F, Cimino G, Breccia M, et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104:19995–9.
Stein EM, Walter RB, Erba HP, et al. A phase 1 trial of vadastuximab talirine as monotherapy in patients with CD33-positive acute myeloid leukemia. Blood. 2018;131:387–96.
Jurcic JG, DeBlasio T, Dumont L, Yao TJ, Scheinberg DA. Molecular remission induction with retinoic acid and anti-CD33 monoclonal antibody HuM195 in acute promyelocytic leukemia. Clin Cancer Res. 2000;6:372–80.
Sohal J, Phan VT, Chan PV, et al. A model of APL with FLT3 mutation is responsive to retinoic acid and a receptor tyrosine kinase inhibitor, SU11657. Blood. 2003;101:3188–97.
Tobita T, Takeshita A, Kitamura K, et al. Treatment with new synthetic retinoid, Am 80, of acute promyelocytic leukemia relapsed from complete remission induced by all-trans retinoic acid. Blood. 1997;90:967–73.
Sanford D, Lo-Coco F, Sanz M, et al. Tamibarotene in patients with acute promyelocytic leukaemia relapsing after treatment with all-trans retinoic acid and arsenic trioxide. Br J Haematol. 2015;171:471–7.
Takeshita A, Asou N, Atsuta Y, et al. Tamibarotene maintenance improved relapse-free survival of acute promyelocytic leukemia: a final result of prospective, randomized, JALSG-APL204 study. Leukemia. 2018. https://doi.org/10.1038/s41375-018-0233-7 [Epub ahead of print].
Kumana CR, Au WY, Lee NS, et al. Systemic availability of arsenic from oral arsenic-trioxide used to treat patients with hematological malignancies. Eur J Clin Pharmacol. 2002;58:521–6.
Siu CW, Au WY, Yung C, et al. Effects of oral arsenic trioxide therapy on QT intervals in patients with acute promyelocytic leukemia: implications for long-term cardiac safety. Blood. 2006;108:103–6.
Zhu HH, Liu YR, Jia JS, et al. Oral arsenic and all-trans retinoic acid for high-risk acute promyelocytic leukemia. Blood. 2018;131:2987–8.
Zhu HH, Wu DP, Jin J, et al. Oral tetra-arsenic tetra-sulfide formula versus intravenous arsenic trioxide as first-line treatment of acute promyelocytic leukemia: a multicenter randomized controlled trial. J Clin Oncol. 2013;31:4215–21.
Zhu HH, Wu DP, Du X, et al. Oral arsenic plus retinoic acid versus intravenous arsenic plus retinoic acid for non-high-risk acute promyelocytic leukaemia: a non-inferiority, randomized phase 3 trial. Lancet Oncol. 2018;19:871–9.
Yang MH, Wan WQ, Luo JS, et al. Multicenter randomized trial of arsenic trioxide and Realgar-Indigo naturalis formula in pediatric patients with acute promyelocytic leukemia: interim results of the SCCLG-APL clinical study. Am J Hematol. 2018;93:1467–73. https://doi.org/10.1002/ajh.25271.
Au WY, Kumana CR, Kou M, et al. Oral arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia. Blood. 2003;102:407–8.
Zhu HH, Huang XJ. Oral arsenic and retinoic acid for non-high-risk acute promyelocytic leukemia. N Engl J Med. 2014;371:2239–41.
Au WY, Kumana CR, Lee HK, et al. Oral arsenic trioxide-based maintenance regimens for first complete remission of acute promyelocytic leukemia: a 10-year follow-up study. Blood. 2011;118:6535–43.
Muchtar E, Vidal L, Ram R, et al. The role of maintenance therapy in acute promyelocytic leukemia in the first complete remission. Cochrane Database Syst Rev 2013;CD009594.
Iland HJ, Seymour JF. Role of arsenic trioxide in acute promyelocytic leukemia. Curr Treat Options Oncol. 2013;14:170–84.
Cicconi L, Fenaux P, Kantarjian H, Tallman M, Sanz MA, Lo-Coco F. Molecular remission as a therapeutic objective in acute promyelocytic leukemia. Leukemia. 2018;32:1671–8.
Correa de Araujo Koury L, Ganser A, Berliner N, et al. Treating acute promyelocytic leukaemia in Latin America: lessons from the International Consortium on Acute Leukaemia experience. Br J Haematol. 2017;177:979–83.
Efficace F, Mandelli F, Avvisati G, et al. Randomized phase II trial of retinoic acid and arsenic trioxide versus retinoic acid and chemotherapy in patients with acute promyelocytic leukemia: health-related quality-of-life outcomes. J Clin Oncol. 2014;32:3406–12.
Acknowledgements
Funding
No funding or sponsorship was received for this study or the publication of this article.
Authorship
The author meets the International Committee of Medical Journal Editors criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and has given his approval for this version to be published.
Disclosures
Xavier Thomas is a member of the journal’s Editorial Board.
Compliance with Ethics Guidelines
The article is based on previously conducted studies and does not contain any studies with human participants or animals performed by the author.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7479977.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Thomas, X. Acute Promyelocytic Leukemia: A History over 60 Years—From the Most Malignant to the most Curable Form of Acute Leukemia. Oncol Ther 7, 33–65 (2019). https://doi.org/10.1007/s40487-018-0091-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-018-0091-5