
Abstract
The treatment of metastatic gastrointestinal stromal tumors (GISTs) changed dramatically with the introduction of imatinib into the therapeutic lexicon in 2001. Over the past 15 years, tyrosine kinase inhibitors in the adjuvant and metastatic settings have remained the standard of care for this disease, though alternate classes of agents and new therapeutic targets are being actively explored in clinical trials. Although data are limited, the use of surgical and non-surgical locoregional techniques for the treatment of GIST metastases has increased and given reports of promising and durable responses. Herein we provide an overview of the contemporary therapeutic landscape of metastatic GIST.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumors (GISTs) are digestive tract sarcomas originating from the interstitial cells of Cajal [1]. Most GISTs develop in the stomach and small intestine and frequently metastasize to the liver and peritoneum. Lymph node and lung metastases are rare [2].
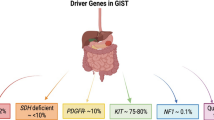
Activating mutations of the KIT oncogene are the pathogenic hallmark of GISTs and are present in 75–85% of tumors [1–3]. Approximately 65% of KIT mutations involve the juxtamembrane domain of the KIT receptor encoded by exon 11, and 7–10% involve exon 9, which encodes the extracellular domain [3]. Ten percent of GISTs harbor mutations in the platelet-derived growth factor receptor alpha (PDGFRA) gene, which occur mutually exclusive of KIT mutations, and another 10–15% are KIT/PDGFRA wild-type [3]. Up to 50% of these previously termed wild-type GISTs demonstrate defects in succinate dehydrogenase (SDH), which is associated with Carney–Stratakis syndrome, one of the few heritable GIST syndromes associated with pediatric GISTs [4]. Mutations in PIK3CA and NF1 (neurofibromatosis type 1) have also been identified very infrequently [3].
The connection between KIT mutations and GISTs coincided with the clinical development of what was then called STI571, an oral small molecular inhibitor of Abl, KIT, and PDGFRs, which showed compelling activity in chronic myelogenous leukemia [5, 6]. In 2001, Joensuu et al. [7] described the dramatic response of a patient with rapidly progressive, chemorefractory GIST to STI571. Subsequent clinical trials confirmed the antineoplastic activity of STI571, now called imatinib, in metastatic GIST , leading to its approval by the FDA in 2001 (Fig. 1).

Suggested treatment algorithm for metastatic gastrointestinal stromal tumor
In the 15 years that have since passed, the therapeutic repertory for metastatic GIST has expanded significantly and includes additional targeted agents as well as new locoregional techniques. This review will discuss the current therapeutic landscape for metastatic GISTs.
Systemic Therapy for Metastatic Gist
Imatinib
In phase I, II, and III studies, imatinib induced objective responses in 45–55% of patients, 3–5% of whom had a complete response [8–11]. The median time to response was ~3–4 months, though some responses were observed up to 2 years later [11]. Median progression-free survival (PFS) and overall survival (OS) were 18–24 months and 50–60 months, respectively [10–12]. Clinical outcomes were similar across different doses ranging from 400 mg QD to 400 mg BID.
Tumor genotype influences sensitivity to imatinib. KIT exon 11 mutations are associated with significantly better response and survival outcomes compared to exon 9 mutated and wild-type GISTs [13–16]. Because high dose imatinib (400 mg BID) has been shown to increase the ORR and PFS in patients with exon 9 mutations [13, 16], patients with this genotype should be closely monitored for progression and dose-escalated as tolerated [17]. PDGFRA mutations are associated with variable sensitivity to imatinib with the D842V mutation resistant and other mutations generally sensitive to imatinib [18].
Secondary progression on imatinib is usually the result of acquired secondary KIT mutations in exons 13, 14, 17, and 18, which hinder drug-receptor interactions [3, 19]. KIT amplification, loss of KIT protein expression, and PDGFRA D842V mutations also confer resistance [3, 19]. Imatinib dose escalation may “rescue” some patients, around 30% of whom regain disease control [10, 20]. Even patients who progress on multiple agents may benefit from a retrial of imatinib given that some tumors retain imatinib-sensitive clones [17, 21].
Several unique aspects concerning the clinical use of imatinib deserve mention. First, among patients experiencing toxicity, it is recommended to dose reduce rather than discontinue therapy [17]. Rapid progression occurs in >80% of patients within a year of stopping imatinib and is associated with a poor prognosis [22]. Second, there is no established role for on-therapy monitoring of plasma drug levels. Although one study linked imatinib trough levels <1100 ng/mL with poorer outcomes, there was significant interpatient variability and therapeutic compliance was not monitored [17, 23]. Third, the sensitivity of conventional response criteria in solid tumors (RECIST) to detect radiological change on imatinib is limited. While most imatinib-sensitive GISTs demonstrate shrinkage, some tumors may enlarge while becoming more hypodense on CT [24]. By conventional RECIST, such tumors would be labelled as progressing when they are in fact responding. Likewise, conventional RECIST may fail to detect “resistant clonal nodules”, an early sign of progression in which a new enhancing focus appears within pre-existing tumor mass that is otherwise stable in size [25]. Recognition that a significant decrease in metabolic activity on PET scan predicts subsequent response on CT or MRI led to the development of the “Choi criteria” in which a 10% decrease in tumor size or 15% decrease in tumor density on CT correlates with a favorable response on PET [26]. The Choi criteria outperform conventional RECIST with respect to sensitivity in identifying PET responders (97% vs 52%), and in predicting disease control and survival outcomes on imatinib [24, 26].
Sunitinib
Sunitinib targets KIT, PDGFR, vascular endothelial growth factor receptor (VEGFR), Fms-like tyrosine kinase 3 (FLT3), and RET [27]. Based on preclinical and phase I/II studies showing promising activity in imatinib-refractory GIST [28, 29], a phase III trial was launched. The primary endpoint, time to progression (TTP) was significantly longer with sunitinib versus placebo (27.3 vs 6.4 weeks, HR 0.33, p < 0.0001) [27]. Objective responses were uncommon but favored sunitinib (7% vs 0%, p = 0.006). Median OS was significantly improved in this study that allowed cross-over from placebo to drug (HR 0.49, p = 0.007). The most common toxicities were fatigue, gastrointestinal, skin rash or discoloration, anorexia, and stomatitis, which were mostly grade 1–2. Comparable outcomes have been reported with sunitinib given on a continuous (37.5 mg QD) or intermittent schedule (50 mg QD for 4 weeks every 6 weeks) [27, 30].
Patients with KIT/PDGFRA wild-type or KIT exon 9 mutated GISTs fared better on sunitinib than those with exon 11 mutations [31]. Secondary KIT exon 13 or 14 mutations were also associated with better outcomes than exon 17 or 18 mutations [31]. Sunitinib was FDA-approved for imatinib-resistant GIST in 2006.
Regorafenib
Regorafenib inhibits KIT, VEGFR, PDGFRA, fibroblast growth factor receptor 1 (FGFR1), TIE-2, RET, RAF, and MAP kinases [32]. In a phase II trial in patients with GIST resistant to both imatinib and sunitinib, median PFS was 10 months and 75% of patients had disease control lasting ≥16 weeks [32]. The main side effects were mostly grade 1–2 hand foot skin reaction, hypertension, fatigue, and diarrhea. PFS appeared superior in patients with KIT exon 11 mutations compared to exon 9 mutations.
The phase III GRID trial randomized patients with imatinib and sunitinib-resistant GIST to regorafenib (160 mg QD for 3 weeks of a 4-week cycle) or placebo [33]. The primary endpoint, median PFS, significantly favored regorafenib (4.8 vs 0.9 months, HR 0.27, p < 0.0001). The ORR and stable disease rate with regorafenib were 4.5% and 71%, respectively. There was no difference in median OS (HR 0.77, p = 0.199). Outcomes were similar in patients with KIT exon 9 and 11 mutations. Toxicities mirrored those reported in the phase II trial and responded to dose reductions. In 2013, regorafenib was FDA-approved for the treatment of metastatic GIST refractory to imatinib and sunitinib.
Nilotinib
Nilotinib is a more potent inhibitor of BCR-Abl than imatinib, and also inhibits KIT, PDGFR, and discoidin domain receptor (DDR) tyrosine kinases [34]. Nilotinib was evaluated in GIST resistant to imatinib and sunitinib in phase II and III studies [35–37]. Median PFS and OS were 12–16 weeks and 34–47 weeks, respectively. The ORR was ≤10%, but 37–66% of patients had stable disease. In the phase III trial randomizing patients to nilotinib or best supportive care ± imatinib or sunitinib, PFS was significantly longer with nilotinib by local assessment (119 vs 70 days, HR 0.58, p = 0.0007). So-called “clinical benefit rates” (CBR; complete response + partial response + stable disease) were similar between arms (53% vs 45%, p = 0.28), and median OS was longer with nilotinib (332 vs 280 days, HR 0.79, p = 0.29). A post hoc analysis restricted to the 80% patients with clearly documented progression on imatinib and sunitinib only yielded a significant prolongation in OS by >4 months (p = 0.02) with nilotinib [37].
A phase III trial randomizing treatment naïve patients to imatinib versus nilotinib was terminated early due to a higher occurrence of progression and death events in the nilotinib arm [34]. Mutational subgroup analysis revealed differential responses to nilotinib: 2-year PFS was superior with imatinib in patients with KIT exon 9 mutations, but was similar between arms in the exon 11 mutation subgroup (67.5% and 70%). This finding, along with the non-overlapping toxicity profile, has led some to propose nilotinib as an alternative in patients with KIT exon 11 mutations who are intolerant to imatinib [38]. Nevertheless, nilotinib is not FDA-approved for GIST, and there are no plans for further clinical development in this disease.
Sorafenib
Sorafenib inhibits VEGFR, PDGFR, KIT, RAF, and MAP kinases. The activity of sorafenib in multi-TKI (imatinib, sunitinib, nilotinib) resistant GIST was evaluated in phase II studies and in a retrospective series [39–42]. Partial response rates ranged from 10 to 24% and 16–60% of patients experienced stable disease. Median PFS and OS were 4.9–7.2 months and 9.7–15.2 months, respectively.
The most common toxicities were HFSR, rash, abdominal pain, hypertension, and diarrhea, which were mostly grade 1/2 [39, 41, 42]. In one study, patients who required a dose reduction for HFSR tended to have a longer PFS than those who did not (8.0 vs 4.7 months, p = 0.07) [41].
Masitinib
Masitinib is a potent and selective KIT inhibitor that also targets PDGFRs, FGFR3, and intracellular Lyn kinase [43]. A phase II study of first-line masitinib reported a 5-year OS of 61.5% and median PFS was 41.3 months [44]. In patients with imatinib-resistant GIST, masitinib was associated with a better toxicity profile and significantly longer OS than sunitinib [45]. Randomized phase III studies of mastinib in first and second-line are ongoing (NCT00812240, NCT01694277).
Vatalinib
Vatalinib inhibits KIT, PDGFR, and VEGFR. In phase II studies of patients with imatinib ± sunitinib-resistant GIST, the disease control rate was 40–67% and median TTP was 4.5–8.5 months [46, 47]. The most common grade 1/2 side effects were hypertension, nausea, dizziness, proteinuria, diarrhea, pyrexia, and abdominal pain. Grade 3/4 hypertension, abdominal pain, asthenia, transaminases, hypercalcemia, and headache occurred in 2–7% of patients.
Dasatinib
Dasatinib inhibits BCR-Abl, KIT, PDGFR, and Src family kinases. In a phase II study of first-line dasatinib, median PFS was 13.6 months and 4-year survival was 74%. Nearly 75% of patients had metabolic responses on PET scan [48].
Dasatinib was also assessed in imatinib and sunitinib-resistant GIST in a phase II study [49]. Nearly a third of patients had a partial response by Choi criteria, and median PFS and OS were 2 and 19 months, respectively. In patients with wild-type GIST, median PFS was 8.2 months. Grade 3/4 myelosuppression, pain, constitutional, gastrointestinal, and respiratory toxicities were reported in 30% of patients.
Linsitinib
Linsitinib inhibits insulin-like growth factor receptor-1, which is highly expressed in wild-type GISTs demonstrating a loss of SDH, and also weakly inhibits insulin receptor signaling [50]. In a phase II study restricted to patients with wild-type GIST, 35% of patients had a partial or stable metabolic responses on PET. Nine-month PFS and OS were 52% and 80%, respectively [50]. The most common toxicities were nausea, vomiting, fatigue, and liver enzymes.
Dovitinib
Dovitinib targets VEGFR 1-3, FGFR 1-3, KIT, PDGFR-beta, and fetal liver tyrosine kinase receptor 3 [51]. In a phase II study conducted in imatinib and sunitinib-refractory GIST, 13% had metabolic partial responses and 50% had stable disease on PET scan [51]. Median PFS was 3.6 months and median OS was 9.7 months. Diarrhea, nausea, asthenia, anemia, and increased alanine aminotransferase occurred in 50–63% of patients and were mostly grade 1/2 in severity. Grade 3/4 asthenia, myelosuppression and dyslipidemia occurred in 10–20% of patients.
Ponatinib
Ponatinib is a pan-BCR-Abl inhibitor that is also active against KIT mutant isoforms associated with TKI resistance [52]. Initial results from a phase II study in patients with TKI refractory GIST reported higher CBR, ORR, PFS, and OS in patients with a KIT exon 11 mutation than those without a mutation [53]. Rash, fatigue, myalgia, dry skin, headache, abdominal pain, and constipation occurred in 34–54% of patients. One patient died of pneumonia possibly related to ponatinib.
Pazopanib
Pazopanib inhibits KIT, VEGFR, and PDGFR and has been evaluated in two phase II trials of TKI-resistant GIST [54, 55]. In a randomized study of pazopanib ± best supportive care, 4-month PFS was significantly better with pazopanib (47.7% vs 19.5%, HR 0.56, p = 0.02) but there was no difference in survival [55, 56]. There were no objective responses but 84% had stable disease compared to 71% on best supportive care only. Serious toxicities were more frequent with pazopanib (52.5% vs 14.6%) and included gastrointestinal disorders, decreased global health status, and pulmonary embolism. Absence of prior gastrectomy was independently associated with improved PFS [56]. A patient with SDH-deficient wild-type GIST had prolonged disease control [54]. Results from the single-arm phase II PAGIST trial of pazopanib in imatinib and sunitinib-resistant GIST (NCT 01524848) are pending.
Binimetinib
The ETV1 transcription factor mediates GIST oncogenesis, maintenance, and survival, and is part of a positive feedback loop with KIT [57]. Inhibition of MAPK signaling using the MEK inhibitor binimetinib destabilizes KIT and synergizes with imatinib. In a phase Ib study of binimetinib and imatinib, the CBR was 65% and ORR was 41% by Choi criteria [58]. Median PFS was 8 weeks. Two patients had stable disease lasting over a year; one was SDH deficient and the other had a KIT exon 11 mutation. Grade ≥ 3 fatigue, edema, rash, HFSR, gastrointestinal toxicities, electrolyte disturbances, and increased liver enzymes occurred in 17–28% of patients, and 89% had asymptomatic grade 3/4 elevations of CPK. A phase II trial is ongoing (NCT01991379).
PI3K/Akt/mTOR Inhibitors
mTOR activation is a mechanism of secondary resistance to imatinib and occurs in 70% of PDGFRA-mutated and “wild-type” GISTs [59, 60]. Based on preclinical data showing synergy [61], everolimus was combined with imatinib in several phase II studies [62, 63]. Median PFS and OS were 1.9 and 14.9 months, respectively, in second line, and 3.5 and 10.7 months, respectively, in third-line [61]. Thirty to 40% of patients had stable disease [62, 63].
Preclinical data suggest suppression of PI3K or Akt may yield greater therapeutic benefit [64]. In a phase II trial, the Akt inhibitor perifosine plus imatinib yielded a CBR of 36% in patients with imatinib-resistant disease, especially those with wild-type tumors. Median PFS and OS were 2.2 and 18.3 months, respectively [65]. Several clinical trials of PI3K inhibitors in GIST are underway (NCT01468688, NCT01735968) [64].
Heat Shock Protein 90 Inhibitors
Heat shock protein 90 (HSP90) is a chaperone molecule that protects protein integrity, including mediators of oncogenesis, growth, and maintenance [66]. Preclinical studies have shown that the antineoplastic effects of HSP90 inhibition in GIST cell lines are mediated by KIT degradation [67].
The HSP90 inhibitor retaspimycin has been evaluated in several clinical trials. In a phase I study, ORR was 3% by RECIST and 38% on PET [68]. Median PFS and TTP were 10.6 and 12 weeks, respectively. One patient with imatinib and sunitinib-resistant disease remained on retaspimycin for 5 years. The most common toxicities were fatigue, nausea, headache, diarrhea, and vomiting. Grade 1–2 arrhythmias and QTc prolongation occurred in 2–20% of patients. A phase III study of retaspimycin versus placebo in TKI-resistant GIST was terminated early due to four drug-related deaths [69].
Other HSP90 inhibitors such as BIIB021, AT13387, and ganetespib have shown good tolerability and disease-stabilizing activity in GISTs [70–72].
Immunotherapy
The GIST microenvironment contains regulatory, cytotoxic, proinflammatory, and anti-inflammatory cells, which appear to create a net immunosuppressive effect [73, 74]. GIST infiltrating lymphocytes demonstrate upregulation of the programmed-death receptor-1 (PD-1) and ligand (PD-L1), which dampen immune responses [75]. Imatinib increases the ratio of cytotoxic to regulatory T cells and enhances cross-talk between dendritic and natural killer cells [76, 77], providing a rationale for evaluating immunotherapy combined with TKIs in metastatic GIST.
In a phase Ib trial of dasatinib and the anti-CTLA4 inhibitor ipilimumab, four out of eight GIST patients had durable disease control [78]. PD-1/PD-L1 axis inhibition demonstrated an additive antiproliferative effect on murine GISTs when combined with imatinib [75]. In a cohort of eight patients, imatinib and pegylated interferon-alpha 2b yielded an ORR of 100% and OS was 100% after a median follow-up of 3.6 years [79].
A summary of these agents and their associated outcomes is provided in Table 1.
Locoregional Therapy for Metastatic Disease
Although TKIs are the therapeutic mainstay for metastatic GIST, locoregional approaches can play a complementary role by extending the progression-free interval in patients responding to TKIs, and controlling focal areas of progression due to expansion of resistant clones.
Surgery
Cytoreductive surgery may prevent or delay the onset of resistance and progression in patients on systemic therapy by eliminating viable cells and treatment-resistant clones. Patients with responding disease who undergo complete cytoreduction have the best outcomes; 2-year OS and PFS are 90–100% and 60–70%, respectively [80–82]. Median survival times exceeding 8 years have been reported, even in patients with liver and peritoneal metastases [83].
Selected patients with limited progression, gastric GISTs, smaller tumor size and number, and completely cytoreducible disease may also benefit from surgery [84]. Those with generalized disease do not benefit from cytoreduction [82, 83]. Imatinib should be resumed post-operatively to reduce the risk of recurrence or progression.
Radiotherapy (RT)
Though long considered radioresistant, GIST metastases can be effectively palliated by RT [85–87]. In a prospective study of 25 patients with TKI-refractory GIST, 80% had stability of target lesions lasting a median of 16 months and median OS was 19 months [87]. Median TTP of target lesions was significantly longer in patients who did not continue TKIs during RT than in those who did (23 vs 11 months, p = 0.014). One patient developed grade 4 biliary tract necrosis while taking sorafenib during RT.
Hepatic Artery Embolization (HAE)
HAE involves the selective catheterization and delivery of microspheres through a tumor-feeding artery, inducing ischemia and apoptosis. Transarterial chemoembolization (TACE) is HAE performed with chemotherapy. Response rates using modified RECIST guidelines, which only measure arterially enhancing disease instead of the entire tumor diameter, are 45–54% [88, 89]. Median liver PFS and OS times range from 3.4 to 8.2 months and 9.7–23.8 months, respectively. The absence of extrahepatic metastases, multiple versus single embolizations, fewer liver metastases, and disease control following embolizations are independently associated with improved survival on multivariate analyses [88, 90, 91].
The main side effect of HAE/TACE is post-embolization syndrome consisting of abdominal pain, fatigue, fever, nausea and vomiting. Moderate to severe complications including liver failure, abscess, respiratory failure, pulmonary embolism, cholecystitis and hemorrhage occur in 5–14% of patients [88, 90, 91]. When used, the authors recommend bland HAE rather than TACE given the lack of sensitivity to standard chemotherapy agents, even at very high concentrations.
Radiofrequency Ablation (RFA)
RFA induces coagulative necrosis of tumors using thermal energy.
In small retrospective series, complete ablation was achieved in >90% of treated tumors, and 3- and 5-year local tumor progression rates were 0% and 12.5%, respectively [92–94]. As with cytoreductive surgery, patients who undergo RFA for residual disease after maximal response to imatinib experienced the best outcomes [92].
Radioembolization
Radioembolization with yttrium-90 selectively delivers high dose radiation to target lesions while sparing the normal hepatic parenchyma. In a small retrospective series of nine evaluable patients with progressive GIST on ≥2 prior TKIs, 33% had a complete response, 56% had a partial response and 11% had stable disease [95]. Median hepatic PFS was 16 months and OS was nearly 30 months. Toxicities primarily consisted of increased liver enzymes which were mostly grade 1, although one patient developed a chronic gastric ulcer and required surgery.
Outcomes associated with various locoregional therapies are summarized in Table 2.
Future Directions and Conclusions
Patients with metastatic GIST have more treatment options than ever before. Since the introduction of imatinib in 2001, a panoply of newer generation TKIs and agents targeting intracellular signaling cascades, stress response mediators, epigenetic modifiers, and the immune system have entered the picture. In addition, the judicious use of locoregional therapies can extend the benefit of TKIs that are otherwise working for the bulk of disease in a given patient. Growing knowledge of the molecular pathophysiology of GISTs guides the rational design of combination strategies and later-line therapies based on known mechanisms of resistance.
Areas of future research include the ongoing assessment of therapeutic targets beyond the current spectrum of receptor tyrosine kinases and the development of agents to improve the outcomes of molecular subpopulations. The identification of biomarkers beyond KIT and PDGFRA mutations to facilitate patient selection and improve the cost-effectiveness of treatment is another priority. Prospective studies are also needed to delineate the optimal timing and use of locoregional strategies, and to ascertain their impact on outcomes in different disease contexts.
In summary, the outcomes of patients with metastatic GIST are optimized through a contemporary framework that combines systemic and locoregional therapies, established standards of care, and investigational approaches.
References
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):557–80.
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813–25.
Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(Suppl 1):S1–16.
Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108(1):314–8.
Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–6.
Druker BJ, Lydon NB. Lessons learned from the development of an abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Investig. 2000;105:3–7.
Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–6.
van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358(9291):1421–3.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80.
Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergoup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32.
Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364(9440):1127–34.
Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard-versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–5.
Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–7.
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9.
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–103.
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1640 patients. J Clin Oncol. 2010;28(7):1247–53.
Maki RG, Blay J-Y, Demetri GD, et al. Key issues in the clinical management of gastrointestinal stromal tumors: an expert discussion. Oncologist. 2015;20(7):823–30.
Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–64.
Debiec-Rychter M, Cools J, Cumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib resistant mutants. Gastroenterology. 2005;128(20):270–9.
Zalcberg JR, Verweij J, Casali PG, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41(12):1751–7.
Kang YK, Ryu MH, Yoo C, et al. Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2013;14(12):1175–82.
Patrikidou A, Chabaud S, Ray-Coquard I, et al. Influence of imatinib interruption and rechallenge on the residual disease in patients with advanced GIST: results of the BFR14 prospective French Sarcoma Group randomized, phase III trial. Ann Oncol. 2013;24(4):1087–93.
Demetri GD, Wang Y, Wehrle E, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;29(19):3141–7.
Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25(13):1760–4.
Desai J, Shankar S, Heinrich MC, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res. 2007;13(18 Pt 1):5398–405.
Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25(13):1753–9.
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38.
Prenen H, Cools J, Mentens N, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res. 2006;12(8):2622–7.
Demetri GD, Heinrich MC, Fletcher JA, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15(18):5902–9.
George S, Blay JY, Casali PG, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–68.
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitnib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–9.
George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–7.
Demetri G, Reichardt P, Kang Y, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicenter randomized, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302.
Blay JY, Shen L, Kang YK, et al. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): a randomised phase 3 trial. Lancet Oncol. 2015;16(5):550–60.
Sawaki A, Nishida T, Doi T, et al. Phase 2 study of nilotinib as third-line therapy for patients with gastrointestinal stromal tumor. Cancer. 2011;117(20):4633–41.
Montemurro M, Schoffski P, Reichardt P, et al. Nilotinib in the treatment of advanced gastrointestinal stromal tumours resistant to both imatinib and sunitinib. Eur J Cancer. 2009;45:2293–7.
Reichardt P, Blay JY, Gelderblom H, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol. 2012;23:1680–7.
Kanda T, Ishikawa T, Takahashi T, Nishida T. Nilotinib for treatment of gastrointestinal stromal tumors: out of the equation? Expert Opin Pharmacother. 2013;14(13):1859–67.
Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012;30(6):2377–83.
Kindler HL, Campbell NP, Wroblewski K, et al. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitini (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): final results of a University of Chicago Phase II Consortium trial. J Clin Oncol. 2011;29(suppl; abstr):10009.
Montemurro M, Gelderblom H, Bitz U, et al. Sorafenib as third- or fourth-line treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: a retrospective analysis. Eur J Cancer. 2013;49(5):1027–31.
Kefeli U, Benekli M, Sevinc A, et al. Efficacy of sorafenib in patients with gastrointestinal stromal tumros in the third or fourth-line treatment: a retrospective multicenter experience. Oncol Lett. 2013;6(2):605–11.
Dubreuil P, Letard S, Ciufolini M, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258.
Le Cesne A, Blay JY, Nguyen BB, et al. Masitinib in imatinib-naïve advanced gastrointestinal stromal tumor (GIST): five-year follow-up of the French Sarcoma Group phase II trial. J Clin Oncol. 2012;30(suppl; abstr):10089.
Adenis A, Blay JY, Bui-Nguyen B, et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: a randomized controlled open-label trial. Ann Oncol. 2014;25(9):1762–9.
Joensuu H, De Braud F, Coco P, et al. Phase II, open-label study of PTK787/ZK222584 for the treatment of metastatic gastrointestinal stromal tumors resistant to imatinib mesylate. Ann Oncol. 2008;19(1):173–7.
Joensuu H, De Braud F, Grignagni G, et al. Vatalinib for metastatic gastrointestinal stromal tumour (GIST) resistant to imatinib: final results of a phase II study. Br J Cancer. 2011;104(11):1686–90.
Montemurro M, Cioffi A, Domont J, et al. Long term outcome of dasatinib first-line treatment in gastrointestinal stromal tumors: a multicenter two stage phase II trial SAKK 56/07. Ann Oncol. 2014;25(suppl 4):iv494–iv510. doi:10.1093/annonc/mdu354.
Trent JC, Wathen K, von Mehren M, et al. A phase II study of dasatinib for pateients with imatinib-resistant gastrointesitnal stromal tumor (GIST). J Clin Oncol. 2011;29(suppl; abstr):10006.
von Mehren M, George S, Heinrich MC, et al. Results of SARC 022, a phase II multicenter study of linsitinib in pediatric and adult wild-type (WT) gastrointestinal stromal tumors (GIST). J Clin Oncol. 2014;32(suppl; abstr):10507.
Kang YK, Yoo C, Ryoo BY, et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br J Cancer. 2013;109:2309–15.
Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res. 2014;20(22):5745–55.
Heinrich MC, von Mehren M, Demetri GD, et al. A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: initial report. J Clin Oncol. 2014;32(suppl; abstr):10506.
Ganjoo KN, Villalobos VM, Kamaya A, et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann Oncol. 2014;25(1):236–40.
Blay J, Mollimard M, Cropet C, et al. Final results of the multicenter randomized phase II PAZOGIST trial evaluating the efficacy of pazopanib (P) plus best supportive care (BSC) vs BSC alone in resistant unresectable metastatic and/or locally advanced gastrointestinal stromal tumors (GIST). J Clin Oncol. 2015;33(suppl; abstr):10506.
Blay J, Domont J, Cropet C, et al. LBA45—a randomized multicenter phase II study of pazopanib plus best supportive care (BSC) vs BSC alone in metastatic gastrointestinal stromal tumors (GIST) resistant to imatinib and sunitinib. Ann of Oncol. 2014;25(5):1–41. doi:10.1093/annonc/medu438.
Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;456(7317):849–53.
Chi P, Qin L-X, D’Angelo SP, et al. A phase Ib/II study of MEK162 (binimetinib [BINI] in combination with imatinib in patients with advanced gastrointestinal stromal tumor (GIST). J Clin Oncol. 2015;33(suppl; abstr):10507.
Wang CM, Huang K, Zhou Y, et al. Molecular mechanisms of secondary imatinib resistance in patients with gastrointestinal stromal tumors. J Cancer Res Clin Oncol. 2010;136(7):1065–71.
Sapi Z, Fule T, Hajdu M, et al. The activated targets of mTOR signaling pathway are characteristics for PDGFRA mutant and wild-type rather than KIT mutant GISTs. Diagn Mol Pathol. 2011;20(1):22–33.
Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signalint mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26(54):7560–8.
Hohenberger P, Bauer S, Gruenwald V, et al. Multicenter, single-arm, two-stage phase II trial of everolimus (RAD001) with imatinb in imatinib-resistant patients (pts) with advanced GIST. J Clin Oncol. 2010;28(suppl; abstr):10048.
Schoffski P, REichardt P, Blay JY, et al. A phase I–II study of everolimus (RAD001) in combination with imatinib in patietns with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990–8.
Songdej N, von Mehren M. GIST treatment options after tyrosine kinase inhibitors. Curr Treat Options Oncol. 2014;15(3):493–506.
Conley AP, Araujo D, Ludwig J, et al. A randomized phase II study of perifosine (P) plus imatinib for patients with imatinib-resistnat gastrointestinal stromal tumor (GIST). J Clin Oncol. 2009;27(suppl; abstr):10563.
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–72.
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–61.
Wagner AJ, Chugh R, Rosen LS, et al. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft tissue sarcomas. Clin Cancer Res. 2013;19(21):6020–9.
Demetri GD, Le Cesne A, Von Mehren M, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. In: 2010 Gastrointestinal Cancers Symposium, abstract 64.
Demetri GD, Heinrich MC, Chmielowski B, et al. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. J Clin Oncol. 2011;29(suppl; abstr):10011.
Dickson MA, Okuno SH, Keohan ML, et al. Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol. 2013;24(1):252–7.
Mahadevan D, Shapiro G, Kurtin SE, et al. Activity of AT13387, a novel, non-ansamycin inhibitor of heat shock protein 90, against gastrointestinal stromal tumors (GIST). J Clin Oncol. 2013;31(suppl 4; abstr):105.
Tan Y, Garcia-Buitrago MT, Trent JC, Rosenberg AE. The immune system and gastrointestinal stromal tumor: a wealth of opportunities. Curr Opin Oncol. 2015;27(4):338–42.
van Dongen M, Savage ND, Jordanova ES, et al. Anti-inflammatory M2 type macrophages characterize metastasized and tyrosine kinase inhibitor-treated gastrointestinal stromal tumors. Int J Cancer. 2010;127(4):899–909.
Seifert AM, Kim TS, Greer JB, et al. PD-1/PD-L1 blockade enhances the efficacy of imatinib in gastrointestinal stromal tumor (GIST). J Am College Surgeons. 2014;219(3):S129.
Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100.
Delahaye NF, Rusakiewicz S, Martins I, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. 2011;17(6):700–7.
Shoushtari AN, D’Angelo SP, Keohan ML, et al. Combined KIT and CTLA-4 blockade in patients with refractory GIST and other sarcomas. J Clin Oncol. 2014;32(suppl; abstr):10521.
Chen LL, Chen X, Choi H, et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol Immunother. 2012;61(7):1113–24.
Gronchi A, Fiore M, Miselli F, et al. Surgery of residual disease following molecular-targeted therapy with imatinib mesylate in advanced/metastatic GIST. Ann Surg. 2007;245(3):341–6.
Raut CP, Posner M, Desai J, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol. 2006;24(15):2325–31.
DeMatteo RP, Maki RG, Singer S, Gonen M, Brennan MF, Antonescu CR. Results of tyrosine kinase inhibitor therapy followed by surgical resection for metastatic gastrointestinal stromal tumor. Ann Surg. 2007;245(3):347–52.
Bauer S, Rutkowski P, Hohenberger P, et al. Long-term follow-up of patients with GIST undergoing metastasectomy in the era of imatinib—analysis of prognostic factors (EORTC-STBSG collaborative study). Eur J Surg Oncol. 2014;40(4):412–9.
Hasegawa J, Kanda T, Hirota S, et al. Surgical interventions for focal progression of advanced gastrointestinal stromal tumors during imatinib therapy. Int J Clin Oncol. 2007;12(3):212–7.
Corbin KS, Kindler HL, Liauw SL. Considering the role of radiation therapy for gastrointestinal stromal tumor. Onco Targets Ther. 2014;7:713–8.
Cuaron JJ, Goodman KA, Lee N, Wu AJ. External beam radiation therapy for locally advanced and metastatic gastrointestinal stromal tumors. Radiat Oncol. 2013;8:274.
Joensuu H, Eriksson M, Collan J, Balk MH, Leyvraz S, Montemurro M. Radiotherapy for GIST progressing during or after tyrosine kinase inhibitor therapy: a prospective study. Radiother Oncol. 2015;116(2):233–8.
Kobayashi K, Szklaruk J, Trent JC, et al. Hepatic arterial embolization and chemoembolization for imatinib-resistant gastrointestinal stromal tumors. Am J Clin Oncol. 2009;32(6):574–81.
Takaki H, Litchman T, Covey A, et al. Hepatic artery embolization for liver metastasis of gastrointestinal stromal tumor following imatinib and sunitinib therapy. J Gastrointest Cancer. 2014;45(4):494–9.
Cao G, Li J, Shen L, Zhu X. Transcatheter arterial chemoembolization for gastrointestinal stromal tumors with liver metastases. World J Gastroenterol. 2012;18(42):6134–40.
Kobayashi K, Gupta S, Trent JC, et al. Hepatic artery chemoembolization for 110 gastrointestinal stromal tumors: response, survival and prognostic factors. Cancer. 2006;107(12):2833–41.
Jones RL, McCall J, Adam A, et al. Radiofrequency ablation is a feasible therapeutic option in the multi modality management of sarcoma. Eur J Surg Oncol. 2010;36(5):477–82.
Hakime A, Le Cesne A, Deschamps F, et al. A role for adjuvant RFA in managing hepatic metastases from gastrointestinal stromal tumors (GIST) after treatment with targeted systemic therapy using kinase inhibitors. Cardiovasc Intervent Radiol. 2014;37(1):132–9.
Yamanaka T, Takaki H, Nakatsuka A, et al. Radiofrequency ablation for liver metastasis from gastrointestinal stromal tumor. J Vasc Interv Radiol. 2013;24(3):341–6.
Rathmann N, Diehl SJ, Dinter D, et al. Radioembolization in patients with progressive gastrointestinal stromal tumor liver metastases undergoing treatment with tyrosine kinase inhibitors. J Vasc Interv Radiol. 2015;26(2):231–8.
Acknowledgments
No funding or sponsorship was received for the publication of this review. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published.
Disclosures
Robert Maki has done consulting work and received honoraria from Bayer. Celina Ang had received an honorarium for participation in an advisory board for Bayer.
Compliance with Ethics Guidelines
This article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by either of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ang, C., Maki, R.G. Contemporary Management of Metastatic Gastrointestinal Stromal Tumors: Systemic and Locoregional Approaches. Oncol Ther 4, 1–16 (2016). https://doi.org/10.1007/s40487-015-0014-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-015-0014-7