
Abstract
Gastrointestinal stromal tumours (GISTs) are the most common gastrointestinal tract mesenchymal tumours. Tyrosine kinase inhibitors (TKIs) have transformed the management of advanced GIST. Imatinib was the first TKI to gain approval as management for patients with advanced GIST, establishing a new standard of care. Since then, as a result of several trials including the GRID and INVICTUS studies, we now have five lines of approved targeted therapy, including imatinib, sunitinib, regorafenib, ripretinib and avapritinib for the treatment of unresectable, advanced GISTs. In this review, the Australasian Gastrointestinal Trials Group (AGITG) provide an overview of the key trials that have changed clinical practice, discuss the molecular drivers of GISTs, the importance of molecular testing and directing therapy according to molecular targets, as well as future strategies in the management of advanced GISTs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The development of tyrosine kinase inhibitors (TKIs) has led to significant improvements in the management of inoperable, advanced gastrointestinal stromal tumours. |
Despite an initial response to TKIs, disease progression often occurs due to resistance mutations. |
Next-generation TKIs targeting secondary KIT mutations and other therapeutic targets are being investigated to overcome these resistance mutations. |
1 Introduction
Gastrointestinal stromal tumours (GISTs) originate from interstitial cells of Cajal (ICC) and constitute the most common gastrointestinal (GI) tract mesenchymal tumours. As such, GISTs are rare malignancies, accounting for 1–2% of all GI neoplasms [1–3]. GISTs can occur anywhere along the GI tract and are most commonly found in the stomach (55%), followed by small bowel (32%), large bowel (6%) and oesophagus (1%). Six percent of GIST cases occur outside of these locations [3].
The median age at diagnosis is 67 years. Rarely, GISTs can occur in the paediatric population and in young adults, where a genetic predisposition to the disease is often found [4–6].
2 Molecular Drivers of Gastrointestinal Stromal Tumours (GISTs)
Molecular alterations in GIST play an important role in the clinical management of patients with advanced disease. The response to tyrosine kinase inhibitor (TKI) therapy is often determined by the primary kinase genotype.
2.1 KIT Mutations
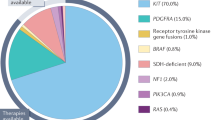
KIT is a proto-oncogene and a type III receptor tyrosine kinase that belongs to a family that includes platelet-derived growth factor receptor alpha (PDGFRA), platelet-derived growth factor receptor beta (PDGFRB), colony-stimulating factor 1 receptor (CSF1R), and FI cytokine receptor (FLT3) [7]. An activating KIT alteration is the driver event for approximately 80% of all GISTs (Fig. 1) [8–10]. KIT mutations in GISTs can occur in different exons of the gene. Exon 11, which encodes the juxtamembrane domain, is most commonly mutated in GISTs. Mutations (in-frame deletions, insertions and substitutions) in exon 11 lead to a disruption of the autoinhibitory domain of the receptor, which results in ongoing kinase activation [7]. GISTs harbouring KIT exon 11 mutations occur most commonly in the stomach. Histologically, KIT exon 11-mutated GISTs also tend to have spindled instead of epithelioid cell morphology and have a higher rate of recurrence following surgery [11]. The type of mutation in KIT exon 11 also has implications on the prognosis. GISTs harbouring exon 11 point mutations tend to have a better prognosis than deletions. A retrospective study from the Spanish Group for Sarcoma Research (GEIS) registry showed that GISTs harbouring KIT exon 11 deletion involving codons 557 and/or 558 tend to be more aggressive and have a poorer prognosis in untreated patients [12].

Driver genes in GIST. Created with BioRender.com. GIST gastrointestinal stromal tumours, SDH succinate dehydrogenase
A mutation in exon 9 that encodes the extracellular domain of KIT occurs in approximately 10% of all GISTs [7, 13, 14]. The majority of KIT exon 9-mutated GISTs occur in the intestine and rarely in the stomach. Approximately 30% of intestinal GISTs harbour a KIT exon 9 mutation [7, 11]. Mutations in exon 9 (typically due to nucleotide duplications) mimic the conformational change that the extracellular domain of the KIT receptor undergoes when bound to a ligand, resulting in constitutive kinase activation [11].
Mutations in KIT exon 13 and 17 are rare, occurring in 1–2% of all untreated GISTs. Exon 13 encodes the ATP-binding region of KIT, and the most common mutation is 1945A>G substitutions, which replace lysine with glutamic acid at codon 642 of the KIT protein [15]. GISTs harbouring exon 13 mutations arise from the stomach but can also occur anywhere along the GI tract. Exon 17 encodes the activation loop and the most common mutation is 2487T>A substitutions, leading to the replacement of asparagine with lysine at codon 822 of the KIT protein [15]. Exon 17-mutated GISTs are most commonly found in the small bowel [16].
2.2 Platelet-Derived Growth Factor Receptor Alpha (PDGFRA) Mutations
PDGFRA mutation is the second most common driver mutation in GISTs, seen in approximately 10% of all GISTs [17]. KIT and PDGFRA mutations are also mutually exclusive in GISTs [11].
The majority of GISTs with a PDGFRA mutation will harbour a mutation in exon 18, which encodes the activation loop. PDGFRA exon 18 D842V mutations make up approximately 70% of GISTs with a PDGFRA mutation. GISTs harbouring PDGFRA D842V mutations are also generally resistant towards conventional TKIs [18]. Although rare, PDGFRA mutations can also occur in exon 12, which encodes the activation loop, and exon 14, which encodes the ATP-binding domain [9, 17]. PDGFRA-mutated GISTs typically arise from the stomach and have an epithelioid or mixed epithelioid and spindle cell histology [19, 20].
2.3 Other Driver Mutations
GISTs that lack KIT and PDGFR mutation make up 10–15% of all GISTs and are sometimes referred to as receptor tyrosine kinase wild-type (RTK-WT) GISTs [11]. This is an heterogenous and genetically diverse group. RTK-WT GISTs may be due to succinate dehydrogenase (SDH) deficiency or alterations in the RAS-MAPK pathway.
One of the first descriptions of wild-type GISTs was provided by Carney in 1977 when he described a series of young patients (predominantly female) with gastric GISTs and concurrent paragangliomas or chondromas. This triad of gastric GISTs (initially referred to as gastric leiomyosarcoma), extra-adrenal paraganglioma and pulmonary chondroma was later referred to as the Carney triad [21, 22]. Subsequently, in 2002, Carney and Stratakis identified a separate familial condition (Carney–Stratakis syndrome) whereby individuals appear to be affected by GISTs and paraganglioma in an autosomal dominant pattern [23]. The Carney triad is often due to the result of hypermethylation of the SDHC gene [24], whereas Carney–Stratakis syndrome is a result of a pathogenic germline alteration in genes that encode the SDH complex (SDHA, SDHB, SDHC and SDHD subunits) [25, 26]. SDH-deficient GISTs comprise the majority of paediatric GISTs. Pathogenic germline alterations in genes that encode the SDH complex result in SDH dysfunction, and SDH deficiency leads to the intracellular accumulation of succinate. Succinate competitively inhibits hypoxia-inducible factor (HIF), which results in the stabilisation of HIF1a. In turn, the stabilisation of HIF1a leads to tumorigenesis and increases the risk of the development of GISTs [4, 27]. SDH-deficient GISTs arise predominantly from the stomach [4].
Activation of the RAS-RAF-MEK-ERK pathway due to gain-of-function RAS/BRAF mutations or loss-of-function neurofibromatosis type 1 mutations increases the risk of GISTs. Neurofibromin, which is encoded by NF1, is a tumour suppressor gene that downregulates the RAS-RAF-MEK-ERK signalling pathway. Most NF1-mutated GISTs arise in the small intestine [28], and GISTs with BRAF mutations also arise predominantly in the small intestine [29, 30].
In this review, we focus on the current evidence for the management of unresectable, advanced GIST. The timeline and published results for selected key studies of TKIs that have been approved by the US FDA for the treatment of unresectable or metastatic GISTs are summarised in Table 1 and Fig. 2. There are several excellent resources and guideline documents covering diagnosis, treatment and follow-up of GIST at all stages, including updated clinical practice guidelines from the European Society for Medical Oncology/European Reference Network for Rare Adult Solid Cancers/European Reference Network for Genetic Tumour Risk Syndromes (ESMO/EURACAN/GENTURIS) groups and the National Comprehensive Cancer Network (NCCN) [31, 32].

Timeline of US FDA approval for tyrosine kinase inhibitors for the treatment of advanced or metastatic GISTs. GISTs gastrointestinal stromal tumours
3 First-Line Management of Advanced or Metastatic Imatinib-Sensitive GIST
Imatinib was the first effective treatment for advanced GIST and has remained the standard treatment for over 20 years [31]. Prior to imatinib, chemotherapy and radiotherapy were often used, however clinical benefit was low [33]. Imatinib mesylate is a small-molecule TKI that was initially developed for the treatment of chronic myeloid leukaemia (CML). Efficacy in CML was mediated through inhibition of the fusion of the oncoprotein BCR-ABL [34]. It was then observed that imatinib also inhibits the transmembrane receptors KIT and PDGFR. Imatinib binds to the ATP-binding site, and thus competitively inhibits ATP binding and inhibits KIT signalling. In March 2000, a patient from Helsinki University Central Hospital with metastatic GIST who had not responded to numerous lines of cytotoxic chemotherapy was put on a trial of imatinib 400 mg daily. Within 1 month of treatment, the patient had significant radiological and histopathological response (proven on biopsy). While this was a single-patient study, this led to the development of several trials [35]. In 2001, a phase I trial provided evidence that imatinib at a dose of 400 mg twice daily was well tolerated [36]. The following year, the results of a phase II multicentre, randomised study of imatinib (400 mg vs. 600 mg) showed that imatinib resulted in a sustained objective response in approximately half of the patients with advanced unresectable or metastatic GISTs, with no significant differences in response rate between the two doses [37]. This pivotal study led to the FDA approval of imatinib for patients with advanced unresectable or metastatic GIST [38].
Two subsequent phase III trials confirmed this promising result. Overall, 946 patients were randomised in a 1:1 ratio in the European Organisation for Research and Treatment of Cancer (EORTC) 62005 trial to receive imatinib 400 mg daily or 400 mg twice daily. Response rates were similar in both arms. Complete response (CR) was 5% and 6% in the 400 mg daily arm and 400 mg twice-daily arm, respectively; however, toxicity was increased with the higher dose, and progression-free survival (PFS) was marginally higher in the twice-daily arm. At 760 days, 56% of patients in the once-daily arm developed progressive disease versus 50% of patients in the twice-daily arm, with a hazard ratio (HR) of 0.82 (p = 0.026) [39]. After a median follow-up of 10.9 years, the PFS observed was similar, i.e. 1.7 years in the 400 mg daily arm and 2.0 years in the 400 mg twice-daily arm. The median overall survival (OS) was 3.9 years in both arms [40].
The Southwest Oncology Group (SWOG) S0033/Cancer and Leukemia Group B (CALGB) 15105 was a similarly designed trial as the EORTC 62005. In this multicentre, randomised, phase III study, 694 patients with inoperable, advanced or metastatic GISTs were randomised to receive imatinib 400 mg daily or 400 mg twice daily [41]. The trial was designed to compare the PFS and OS rates for conventional dose imatinib versus a higher dose. The response rates were similar across both arms, with an overall response rate (ORR) of 45% in both cohorts; PFS was also similar in both arms. The median PFS was 18 months (95% confidence interval [CI] 16–21 months) and 20 months (95% CI 17–25 months) in the conventional-dose and higher-dose cohorts, respectively [41]. A further analysis showed that KIT exon 11-mutated GISTs had an improved treatment outcome when compared with GISTs with a KIT exon 9 mutant or wild-type GIST [42]; 71.7% of patients with a KIT exon 11-mutated GIST had a CR or partial response (PR) versus 44.4% of patients with KIT exon 9-mutated GISTs (p = 0.007) and 44.6% of wild-type GISTs (p = 0.0002). The median OS was 60.0 months versus 38.4 and 49.0 months, respectively. Improved response rates were seen in patients with exon 9-mutated GISTs treated with higher doses of imatinib (800 mg) versus a standard dose (400 mg), with a CR and PR rate of 67% and 17%, respectively (p = 0.02) [43].
Based on the results from the EORTC 62005 and SWOG S0033 trials, the standard dose of imatinib in the first-line setting for unresectable advanced GIST is 400 mg daily; however, in patients with GISTs that harbour KIT exon 9 mutations, a higher dose (800 mg daily) may result in a longer PFS [32]. A meta-analysis by the Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST) confirmed that patients with KIT exon 9-mutated disease derive a PFS benefit if they received a higher dose of imatinib [39, 41, 44]. As described earlier, exon 9 encodes the extracellular domain of KIT, and the kinase domain in exon 9-mutant KIT is similar to wild-type KIT, which may have an effect on inhibitor sensitivity [7]. Thus, the ESMO guidelines recommend starting patients with advanced GIST with an exon 9 mutation on imatinib 400 mg twice daily [45].
Further analysis performed on the EORTC 62005 trial also suggested that patients who progressed on a 400 mg daily dose may benefit from a dose escalation of their imatinib [46]. At the time of analysis, 247 of the 473 patients who were randomised to the 400 mg daily arm had disease progression, of whom 133 (55%) crossed over to receive imatinib 800 mg daily. Among those who crossed over to receive a higher dose of imatinib, 3 patients had a PR and 36 had stable disease (SD). Following crossover, the PFS was 81 days. Anaemia and fatigue were the most commonly reported adverse events. While this analysis has several limitations, there are some benefits in response rate and PFS in those who had dose escalation of their imatinib following disease progression.
It is important to stress that treatment with imatinib should continue until disease progression or if patients experience significant toxicities. Treatment interruption will often lead to disease progression. A prospective, randomised study that compared the PFS of patients who had treatment interruption with those who had ongoing treatment with imatinib for advanced GISTs demonstrated that treatment discontinuation or interruption may lead to rapid progression of disease [47]. The ESMO/EUROCAN/GENTURIS groups also recommend treatment with imatinib to be continued indefinitely, unless the patient progressed on treatment, because treatment interruption often results in rapid tumour progression [31].
4 Management of Imatinib-Resistant GIST
Resistance to treatment with a TKI such as imatinib can be divided into two categories: primary and secondary resistance [7].
4.1 Primary Resistance
PDGFRA D842V in exon 18 is the most common PDGFRA mutation in GIST and is resistant to imatinib and all other earlier approved KIT-targeted TKIs based on prior in vitro studies [17, 43, 48]. PDGFRA D842V mutation results in the active conformation of the kinase domain and this results in resistance to TKIs such as imatinib, which preferentially binds to the inactive conformation [18, 49]. Several studies have also provided evidence that a PDGFRA D842V mutation confers resistance to imatinib [39, 51]. Although PDGFRA D842V-mutated GIST tends to be more indolent, its resistance towards imatinib and other TKIs meant that there was no approved effective treatment for this molecular subtype of GIST until the recent development of avapritinib.
RTK-WT GISTs are also resistant to imatinib and may respond better to other targeted therapy. Examples include vascular endothelial growth factor receptor (VEGFR) inhibitors for SDH-mutant GIST, and BRAF-MEK inhibitors for GISTs with a BRAF mutation [31, 52].
4.2 Management of PDGFRA D842V-Mutated GIST
Avapritinib is a potent, highly selective oral inhibitor that targets the active conformation of KIT and PDGFRA. In 2020, results from the NAVIGATOR trial (a multicentre, phase I, dose-escalation/dose-expansion study) were published, confirming that avapritinib has clinical activity towards PDGFRA D842V-mutated GIST. In the PDGFRA D842V-mutated population, the ORR was 88%, with 9% of patients achieving a CR and 79% of patients achieving a PR. The recommended phase II dose was 300 mg daily [49]. Avapritinib is generally well tolerated, with a manageable adverse effect profile. The most common grade 3 or higher adverse event is anaemia, however avapritinib is associated with a higher frequency (40%) of cognitive effects, of which 5% are grade 3 or higher, with two patients having intracranial haemorrhage that improved upon treatment discontinuation [49].
The positive results from the NAVIGATOR study resulted in the FDA approval of avapritinib in January 2020 for adults with inoperable or metastatic GIST with PDGFRA exon 18 mutations, including the D842V mutation [53]. However, there are many limitations in that study. First, the sample size was small due to the rarity of PDGFRA D842V-mutant GISTs, and, second, this was a single-arm trial with no control group. We await randomised trials comparing avapritinib with other standard therapies to confirm these results.
4.3 Secondary Resistance
Despite an initial response to imatinib, patients often develop disease progression or secondary resistance towards imatinib. Acquired mutations in KIT account for most of the secondary resistance. Chen and colleagues first described a novel missense alteration in KIT kinase domain 1 which correlates with imatinib resistance in GIST. This was the first time an acquired KIT mutation in imatinib-resistant GISTs was described [54]. In 2006, Heinrich and colleagues published an analysis of the genomic mechanisms of imatinib resistance in a cohort of patients who were part of a randomised phase II study. In that study, 43 of the 92 patients with disease-related treatment failures consented to have their tumour samples studied. Sixty-seven percent of these patients had at least one secondary kinase mutation and these secondary mutations were not identified in the pretreatment specimens [43].
There has also been significant development in the way we assess radiological response with GISTs. Computed tomography (CT) has been used for diagnosis and therapy monitoring. The use of Response Evaluation Criteria in Solid Tumours (RECIST), which only takes into account the tumour size, has its limitations when it comes to monitoring response to TKIs because treatment with TKIs in GIST can result in only a small volume of tumour reduction on imaging, even when there is response to treatment [55]. Therefore, modified response criteria that takes into account tumour density and metabolic and functional parameters such as the Choi criteria has been developed [56]. F-fluorodeoxyglucose/positron emission tomography (FDG/PET) is also a useful imaging modality to assess tumour response but it should not be used in patients whose baseline scans are FDG-avid [56].
In an effort to delay resistance to imatinib, the Australasian Gastrointestinal Trials Group (AGITG) also conducted a phase II randomised study (ALT-GIST) to review whether alternating the regimen of imatinib and regorafenib delays resistance to imatinib and improves outcomes. Seventy-six patients were randomised to receive 21–25 days of oral imatinib 400 mg daily followed by a 3- to 7-day gap for washout, then followed by 21 days of oral regorafenib 160 mg daily and a 7-day gap for washout, or the control arm, i.e. continuous imatinib 400 mg daily. This was a negative study as there was no statistical or clinically meaningful benefit [57].
4.4 Management of Advanced, Imatinib-Resistant GISTs: Second-Line Setting
Sunitinib malate, an oral multitargeted receptor TKI, is a second-line option for patients who have disease progression while receiving imatinib or are intolerant to imatinib. However, for patients who have progressive disease but are tolerating standard dose imatinib and do not harbor resistant mutation, a dose increment of imatinib is recommended before switching therapy [45].
Sunitinib blocks receptor tyrosine kinase signalling by KIT, PDGFRs, all three isoforms of the VEGFRs (VEGFR 1–3), FMS-related tyrosine kinase-3 receptor (FLT3), and RET. As a result, sunitinib has demonstrated antiangiogenic and antitumour activities [58–61]. A phase I/II study of 97 patients demonstrated clinical benefit when sunitinib was used in imatinib-resistance GIST, although the objective rates of anti-tumour response were low [62]. Following this study, a randomised, phase III trial was designed to prospectively confirm the findings [63]. In this double-blind, placebo-controlled, clinical trial, patients with unresectable advanced GIST with confirmed treatment failure to previous imatinib therapy were randomised in a 2:1 ratio to receive sunitinib 50 mg or placebo daily for 4 consecutive weeks followed by a 2-week treatment break. Upon disease progression, participants were unblinded, and those found to be receiving placebo were allowed to crossover to open-label sunitinib treatment. This study met its primary endpoint. The median time to tumour progression was 27.3 week in the sunitinib arm versus 6.4 weeks in the placebo arm, with a clinically meaningful and statistically significant HR of 0.33 (95% CI 0.23–0.47; p < 0.0001). Patients in the sunitinib arm also had a much longer PFS of 24.1 weeks compared with 6 weeks in the placebo arm (HR 0.33, 95% CI 0.24–0.47; p < 0.0001). This trial led to the FDA approval of sunitinib for the treatment of GIST after disease progression or intolerance to imatinib [64].
Based on the phase III study by Demetri and colleagues, the approved treatment schedule for sunitinib is 50 mg daily for 4 consecutive weeks followed by a 2-week treatment break, comprising a 6-week cycle [63]. However, in 2009, a phase II study demonstrated that continuous daily oral sunitinib at a lower daily dose of 37.5 mg is not only effective but also well tolerated [65]. While no randomised trial has ever been carried out to compare both dosing strategies, continuous daily sunitinib dosing appears to be an acceptable alternative dosing strategy [45].
While biopsy of GIST upon imatinib progression is often not performed, it may be valuable to evaluate secondary mutations in imatinib-resistant disease. Secondary KIT mutations often arise in KIT exons 13/14 or exons 17/18. Sunitinib has demonstrated reduced activity against KIT exon 11 mutations coupled with secondary mutations in exon 17 or exon 18, whereas regorafenib has shown increased activity for secondary exon 17/18 mutations [66]. However, patients with exon 13 or 14 secondary KIT mutations treated with sunitinib had a longer PFS and OS [67].
4.5 Management of Advanced, Imatinib-Resistant GISTs: Third-Line Setting
For patients who develop progressive disease on imatinib and sunitinib, regorafenib is standard third-line therapy. Regorafenib is an oral multikinase inhibitor that inhibits the activity of angiogenic (VEGFR1–3 and TEK), stromal (PDGFR and fibroblast growth factor receptor [FGFR]) and oncogenic (KIT, RET, RAF1, BRAF, and BRAFV600E) receptor tyrosine kinases [68]. In preclinical models, regorafenib demonstrated anti-tumour activity, including growth inhibition of GIST cell lines [68]. In a multicentre, phase II study, regorafenib showed activity against imatinib- and sunitinib-resistant GIST. In this study, patients with advanced GIST who developed disease progression on imatinib and sunitinib received oral regorafenib 160 mg daily for 21 days of a planned 28-day cycle [69]. Clinical benefit (defined as CR, PR, and SD ≥16 weeks) was documented in approximately 75% of patients and the median PFS was 10 months.
The follow-on GRID study was an international, randomised, placebo-controlled, phase III trial investigating the efficacy and safety of regorafenib in patients with advanced GIST who did not respond to imatinib and sunitinib [70]. Overall, 199 patients with advanced GIST who progressed on prior imatinib and sunitinib treatment were randomised (2:1) to receive either regorafenib and best supportive care (BSC), or placebo and BSC. If disease progression occurred while on the trial, participants were unblinded and crossover was offered to those who were in the placebo arm. Participants who were in the regorafenib arm could continue on regorafenib even upon disease progression. The primary endpoint was PFS per modified Response Evaluation Criteria In Solid Tumors (RECIST) 1·1. Prospectively defined RECIST modifications were developed specifically for GISTs in this trial. These modified RECIST criteria were subsequently used by other GIST trials. The study met its primary endpoint, with a PFS of 4.8 months in the regorafenib arm and 0.9 months in the placebo arm (HR 0.27, 95% CI 0.19–0.39; p < 0.0001). The safety profile of regorafenib was similar to previous clinical trials. These results led to the FDA approval of regorafenib for the management of unresectable metastatic GIST after failure of imatinib and sunitinib [71].
Following this, the VOYAGER study compared avapritinib with regorafenib in patients with KIT/PDGRFA-mutant GIST who had disease progression after two or three lines of treatment. This randomised, open-label, phase III study enrolled 476 patients. The study did not meet its primary endpoint, with PFS in the avapritinib and regorafenib arms of 4.2 months and 5.6 months, respectively (HR 1.25, 95% CI 0.99–1.57) [72]. Regorafenib remains the standard third-line treatment for the management of unresectable, metastatic GISTs.
4.6 Management of Advanced, Imatinib-Resistant GISTs: Fourth-Line Setting and Beyond
In 2020, ripretinib was approved by the FDA for patients with advanced GIST who have received prior treatment with three or more kinase inhibitors [73]. Ripretinib is a switch-control TKI that locks the kinase in an inactive state by binding to both the switch pocket and the activation loop. This prevents downstream signalling and hence prevents cell proliferation [74].
The first-in-human, phase I study of ripretinib demonstrated promising activity in patients with refractory advanced GIST. Ripretinib is also well tolerated and the recommended phase II dose was 150 mg daily [75]. At a dose of 150 mg daily, the most frequent treatment-related toxicities were alopecia, fatigue, myalgia, nausea and palmar-plantar erythrodysaesthesia [75].
Following that, the INVICTUS trial, a double-blind, randomised, placebo-controlled, phase III trial was designed to evaluate the efficacy and safety of ripretinib as fourth or greater lines of therapy in patients with unresectable or metastatic GISTs [76]. The results from this trial were statistically and clinically significant. Overall, 129 patients with unresectable metastatic GIST with disease progression on at least imatinib, sunitinib and regorafenib were randomised in a 2:1 ratio to receive ripretinib 150 mg daily (n = 85) or placebo (n = 44). At the time of progressive disease, participants were unblinded therefore patients in the placebo arm could crossover to receive ripretinib. The study met its PFS primary endpoint of PFS, with a PFS of 6.3 months (95% CI 4.6–6.9) for ripretinib versus 1.0 month (95% CI 0.9–1.7) for placebo, and an HR of 0.15 (95% CI 0.09–0.25; p < 0.0001). The PFS at 6 months was estimated to be 51% (95% CI 39.4–61.4) for ripretinib and 3.2% (95% CI 0.2–13.8) for placebo. Eight patients who received ripretinib had PRs as assessed by blinded independent central review (BICR). The median OS was 15.1 months (95% CI 12.3–15.1) in the ripretinib arm versus 6.6 months (95% CI 4.1–11.6) in the placebo arm (HR 0.36, 95% CI 0.21–0.62), inclusive of the double-blind and open-label periods [76]. For the patients who crossed over to the ripretinib group from the placebo group, the median OS was 11.6 months versus 1.8 months in patients who did not crossover. The adverse effects of ripretinib were tolerable. Grade 3 or 4 treatment-related adverse events included elevated lipase, hypertension, fatigue, and hypophosphataemia. Patients in the INVICTUS study who had progressive disease on ripretinib 150 mg daily were also provided with the option of ripretinib dose escalation to 150 mg twice daily [77]. Forty-three patients underwent ripretinib intrapatient dose escalation (IPDE) to 150 mg twice daily. The median PFS was 4.6 months before (95% CI 2.7–6.4) and 3.7 months after (95% CI 3.1–5.3) ripretinib IPDE with an acceptable safety profile, indicating that dose escalation upon progression may provide additional clinical benefit [77].
The INVICTUS study also demonstrated that patients in the placebo arm had a rapid clinical deterioration in the absence of any TKI; hence, we recommend the short washout time from previous treatment. We also recommend not stopping the patient’s current TKI even in the presence of progressive disease while waiting for the next-line TKI to become available.
Ripretinib was also recently compared with sunitinib in the second-line setting in a randomised, multicentre, phase III trial [78]. Overall, 453 patients were randomised to either ripretinib 150 mg daily or sunitinib 50 mg daily for 4 weeks followed by 2 weeks without sunitinib. The INTRIGUE study in the second-line setting did not meet its primary endpoint of PFS [79]. We await the final publication of the results.
The promising results from the INVINCTUS study demonstrated that ripretinib is an appropriate treatment option for patients with advanced GISTs whose disease has progressed on imatinib, sunitinib and regorafenib.
5 Other Therapeutic Targets
Several therapeutic strategies focusing on the combined inhibition of KIT/PDGFRA and other targets have previously been studied. Inhibition of the mTOR pathway together with imatinib was studied in a phase I–II study. In the phase II part of the study, 70 patients received a combination of imatinib 600 mg daily and everolimus 2.5 mg daily [80]. Everolimus inhibits AKT/mTOR signalling downstream of KIT and PDGFRA. Patients were divided into two cohorts: those who received only one prior line of treatment (Cohort 1) and those who have received two or more lines of treatment (Cohort 2). The results were modest, with a median PFS of 1.9 months (95% CI 1.8–3.7) and 3.5 months (95% CI 1.9–5.2) in Cohorts 1 and 2, respectively. While the study was in progress, sunitinib was approved for treatment in the second-line setting for imatinib-resistant GIST, hence, this combination was not furthered explored.
A phase Ib, multicentre study also investigated the inhibition of the PI3K/AKT pathway together with imatinib [81]. Sixty patients with GISTs refractory to imatinib and sunitinib were enrolled in this study. Unfortunately, buparlisib (an oral PI3K inhibitor) in combination with imatinib failed to demonstrate additional clinical benefit, hence further development of buparlisib with imatinib was not recommended [81]. However, the combination of a PI3K inhibitor with imatinib has demonstrated anti-tumour activity in several preclinical studies involving patient-derived GIST xenograft models [82–84], highlighting the role of PI3K inhibitors in the management of advanced GISTs. Further studies exploring the inhibition of this pathway should be undertaken.
Despite preclinical rationale to suggest the use of combination treatment, several of these combination trials have had limited success in the clinical setting. One theory is that the use of imatinib combination therapy in imatinib-resistant disease may have resulted in the lack of clinical results. Future clinical trials may explore combination trials with a pan-KIT inhibitor such as ripretinib [85].
Another strategy used in overcoming KIT secondary mutations is by targeting important biological mechanisms involved in KIT oncoprotein stabilisation. Heat shock protein 90 (HSP90) is required for the function and stability of several conditionally activated and/or expressed signalling proteins. Several oncogenes such as KIT, PDGFRA and BRAF are client proteins of HSP90 [86, 87], and several HSP90 inhibitors have demonstrated preclinical activity in the management of GIST [8].
Pimitespib is a novel, oral selective HSP90α and HSP90β inhibitor that has anti-tumour effects. The first-in-human phase I study of pimetespib demonstrated that pimetespib is well tolerated, with adverse events including liver enzyme elevation, diarrhoea and some ocular toxicity [89]. In this study of 60 patients with solid organ malignancies, three confirmed PRs were seen, including a GIST case with no detectable KIT mutations. HSP90 inhibition was also confirmed by the induction of HSP70 expression. The recommended dose for phase II was determined to be 160 mg daily, with a dosing schedule of 5 days on, 2 days off per week [89].
A subsequent single-arm, phase II study was conducted in Japan, with 40 patients with unresectable or metastatic GISTs refractory to imatinib, sunitinib and regorafenib receiving 160 mg daily of pimitespib [90]. The centrally assessed median PFS was 4.4 months (95% CI 2.8–6.0). At ≥6 weeks, 34 (85%) patients still had SD. The median OS was 11.5 months (95% CI 7.0–not reached).
CHAPTER-GIST-301 is a randomised, double-blind, phase III trial comparing pimitespib with placebo in patients with GIST refractory to imatinib, sunitinib and regorafenib. The results from this study were recently reported [87]. Eighty-six patients were randomised in a 2:1 ratio to receive pimitespib or placebo, with the study yielding positive results. The median PFS was 2.8 months for the pimitespib arm compared with 1.4 months in the placebo arm (HR 0.51; p = 006). The median OS was not reached in both arms, although there was also an improvement in OS, with a median OS of 13.8 months (95% CI 9.2–not reached) in the pimitespib arm versus 9.6 months in the placebo arm (95% CI 5.5–not reached) [HR 0.63; p = 0.081). Among those who received placebo, 60.7% of patients crossed over to pimitespib, with a secondary PFS of 2.7 months (95% CI 0.7–4.1). Treatment-related adverse events included diarrhoea, with 13.8% of patients receiving pimitespib having grade 3 or higher diarrhoea [87]. The use of a different therapeutic approach should be further explored in patients with advanced, unresectable GISTs.
Lastly, next-generation TKIs targeting secondary KIT mutations are being investigated and have had some success in early-phase trials. As discussed above, despite an initial response to TKIs, disease progression often occurs due to resistance mutations. Currently approved TKIs such as imatinib, sunitinib and regorafenib are type II kinase inhibitors, which bind to the inactive conformation of the KIT kinase. PLX9486, a selective type I KIT inhibitor, harbours activities against primary KIT mutations and secondary mutations (exons 17 and 18). An early-phase Ib/II trial investigated the combination of PLX9486, and sunitinib demonstrated that patients with refractory GIST may derive clinical benefit from combination treatment [91]. In part 1 of this trial, 21 patients received PLX9486 monotherapy at a dose of 250, 350, 500 or 1000 mg daily. In part 2 of the trial, 18 patients received PLX9486 at a dose of 500 or 1000 mg combined with sunitinib at 25 or 37.5 mg daily [91]. Patients who received monotherapy with PLX9486 at a dose of 500 mg or less (n = 7) had no objective responses and the median PFS was 1.74 months (95% CI 1.55–1.84). The median PFS of those receiving PLX9486 monotherapy 1000 mg daily (n = 12) was 5.75 (95% CI 0.99–11.0). The combination of sunitinib 25 or 37.5 mg with PLX9486 500 or 1000 mg resulted in a longer median PFS of 12.1 months (95% CI 1.35–not available) [91]. The results from this study suggest that PLX9486 can be safely combined with sunitinib. Further randomised clinical trials of PLX9486 and sunitinib will determine if a combination of type 1 and type II TKIs leads to improved clinical outcomes.
Several trials are ongoing to investigate other therapeutic options for the management of advanced GISTs, as summarised in Table 2 [78, 92, 93, 94, 95, 96–99, 104–108].
6 Conclusion
In the last two decades, clinical and translational research have contributed to the growing understanding of the molecular biology of GISTs and has resulted in significant achievements made in the treatment of GISTs. GISTs are a heterogenous group of tumours with distinct clinical and molecular characteristics. The use of tumour genotype has helped determine the optimal treatment of GISTs.
The last two decades have seen the introduction of imatinib followed by the second-, third- and fourth-line TKIs sunitinib, regorafenib and ripretinib for the management of advanced GISTs, and avapritinib for GISTs with an exon 18 PDGFRA D842V mutation. However, imatinib-sensitive GISTs often develop secondary resistance caused by acquired mutations, and the median time to progression is 24 months [40, 41]. For patients who have progressed on currently available treatments, options are limited, making this setting an area of unmet need. Future research should focus on overcoming resistance towards these TKIs or exploring other therapeutic approaches such as targeting biological mechanisms involved in KIT oncoprotein stabilisation.
Lastly, patients with GISTs with mutations in BRAF, NF1 and FGFR, as well as SDH-deficient GISTs, do not respond to imatinib. The management of this subset of patients should be different. However, only a small population of patients with GISTs have an SDH deficiency or a mutation in BRAF, NF1 and FGFR. These patients are often eligible for the same clinical trials as these are more common molecular subtypes. This approach, as well as the rarity of these cancers, make it challenging to define their treatment strategies. A phase II clinical trial investigating the mitogen-activated protein kinase (MEK1/2) inhibitor selumetinib (AZD6244 hydrogen sulfate) in patients with NF1-mutated GISTs was withdrawn due to slow accrual 111. Similarly, a phase II trial investigating the DNA methyltransferase inhibitor guadecitabine (SGI-110) in children and adults with wild-type GIST, pheochromocytoma and paraganglioma associated with SDH deficiency and hereditary leiomyomatosis and renal cell carcinoma (HLRCC)-associated kidney cancer was also withdrawn due to slow accrual 112. There is a pressing need for international collaborations to help determine the best treatment strategy for this subset of patients.
Change history
19 July 2022
Missing Open Access funding information has been added in the Funding Note.
References
Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152(5):1259–69.
Parab TM, DeRogatis MJ, Boaz AM, Grasso SA, Issack PS, Duarte DA, et al. Gastrointestinal stromal tumors: a comprehensive review. J Gastrointest Oncol. 2019;10(1):144–54. https://doi.org/10.21037/jgo.2018.08.20.
Søreide K, Sandvik OM, Søreide JA, Giljaca V, Jureckova A, Bulusu VR. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016;40:39–46. https://doi.org/10.1016/j.canep.2015.10.031.
Mei L, Smith SC, Faber AC, Trent J, Grossman SR, Stratakis CA, et al. Gastrointestinal stromal tumors: the GIST of precision medicine. Trends Cancer. 2018;4(1):74–91. https://doi.org/10.1016/j.trecan.2017.11.006.
Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2(7):922–8. https://doi.org/10.1001/jamaoncol.2016.0256.
Benesch M, Wardelmann E, Ferrari A, Brennan B, Verschuur A. Gastrointestinal stromal tumors (GIST) in children and adolescents: a comprehensive review of the current literature. Pediatr Blood Cancer. 2009;53(7):1171–9. https://doi.org/10.1002/pbc.22123.
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–78. https://doi.org/10.1038/nrc3143.
Lasota J, Miettinen M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin Diagn Pathol. 2006;23(2):91–102. https://doi.org/10.1053/j.semdp.2006.08.006.
Kelly CM, Gutierrez Sainz L, Chi P. The management of metastatic GIST: current standard and investigational therapeutics. J Hematol Oncol. 2021;14(1):2. https://doi.org/10.1186/s13045-020-01026-6.
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–80. https://doi.org/10.1126/science.279.5350.577.
Bannon AE, Klug LR, Corless CL, Heinrich MC. Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert Rev Mol Diagn. 2017;17(5):445–57. https://doi.org/10.1080/14737159.2017.1308826.
Martín J, Poveda A, Llombart-Bosch A, Ramos R, López-Guerrero JA, García del Muro J, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005;23(25):6190–8. https://doi.org/10.1200/JCO.2005.19.554.
Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, Demetri G, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol. 2000;156(3):791–5. https://doi.org/10.1016/S0002-9440(10)64946-2.
Oppelt PJ, Hirbe AC, Van Tine BA. Gastrointestinal stromal tumors (GISTs): point mutations matter in management, a review. J Gastrointest Oncol. 2017;8(3):466–73. https://doi.org/10.21037/jgo.2016.09.15.
Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology. 2008;53(3):245–66. https://doi.org/10.1111/j.1365-2559.2008.02977.x.
Lasota J, Corless CL, Heinrich MC, Debiec-Rychter M, Sciot R, Wardelmann E, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008;21(4):476–84. https://doi.org/10.1038/modpathol.2008.2.
Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–64. https://doi.org/10.1200/JCO.2005.14.068.
Indio V, Astolfi A, Tarantino G, Urbini M, Patterson J, Nannini M, et al. Integrated molecular characterization of gastrointestinal stromal tumors (GIST) harboring the rare D842V mutation in PDGFRA gene. Int J Mol Sci. 2018;19(3):732. https://doi.org/10.3390/ijms19030732.
Lasota J, Stachura J, Miettinen M. GISTs with PDGFRA exon 14 mutations represent subset of clinically favorable gastric tumors with epithelioid morphology. Lab Investig. 2006;86(1):94–100. https://doi.org/10.1038/labinvest.3700360.
Chetty R, Serra S. Molecular and morphological correlation in gastrointestinal stromal tumours (GISTs): an update and primer. J Clin Pathol. 2016;69(9):754–60. https://doi.org/10.1136/jclinpath-2016-203807.
Carney JA, Sheps SG, Go VLW, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296(26):1517–8. https://doi.org/10.1056/NEJM197706302962609.
Carney JA. Carney triad. In: Stratakis CA, editor. Endocrine tumor syndromes and their genetics, Frontiers of hormone research, vol. 41. Basel: Karger; 2013. p. 92–110. https://doi.org/10.1159/000345672.
Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108(2):132–9. https://doi.org/10.1002/ajmg.10235.
Zhang L, Smyrk TC, Young WFJ, Stratakis CA, Carney JA. Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol. 2010;34(1):53–64. https://doi.org/10.1097/PAS.0b013e3181c20f4f.
Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79–88. https://doi.org/10.1038/sj.ejhg.5201904.
Gaal J, Stratakis CA, Carney JA, Ball ER, Korpershoek E, Lodish MB, et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Mod Pathol. 2011;24(1):147–51. https://doi.org/10.1038/modpathol.2010.185.
Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11(2):101–11. https://doi.org/10.1038/nrendo.2014.188.
Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30(1):90–6. https://doi.org/10.1097/01.pas.0000176433.81079.bd.
Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010;133(1):141–8. https://doi.org/10.1309/AJCPPCKGA2QGBJ1R.
Agaram NP, Wong GC, Guo T, Maki RG, Singer S, DeMatteo RP, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47(10):853–9.
Casali PG, Blay JY, Abecassis N, Bajpai J, Bauer S, Biagini R, et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2022;33(1):20–33. https://doi.org/10.1016/j.annonc.2021.09.005.
NCCN clinical practice guidelines in oncology- gastrointestinal stromal tumors (GISTs) version 1.2022–21 January 2022. https://www.nccn.org/professionals/physician_gls/pdf/gist.pdf. Accessed 20 Jan 2022.
Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002;33(5):466–77. https://doi.org/10.1053/hupa.2002.124122.
O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. https://doi.org/10.1056/NEJMoa022457.
Joensuu H, Roberts PJ, Sarlomo-Rikala M, Andersson LC, Tervahartiala P, Tuveson D, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–6. https://doi.org/10.1056/NEJM200104053441404.
van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato di Paola E, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358(9291):1421–3. https://doi.org/10.1016/s0140-6736(01)06535-7.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–80. https://doi.org/10.1056/NEJMoa020461.
Dagher R, Cohen M, Williams G, Rothmann M, Gobburu J, Robbie G, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034–8.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–34. https://doi.org/10.1016/S0140-6736(04)17098-0.
Casali PG, Zalcberg J, Le Cesne A, Reichardt P, Blay JY, Lindner LH, et al. Ten-year progression-free and overall survival in patients with unresectable or metastatic gi stromal tumors: long-term analysis of the european organisation for research and treatment of cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Tr. J Clin Oncol. 2017;35(15):1713–20. https://doi.org/10.1200/JCO.2016.71.0228.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–32. https://doi.org/10.1200/JCO.2007.13.4452.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CDM, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–7. https://doi.org/10.1200/JCO.2008.17.4284.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–74. https://doi.org/10.1200/JCO.2006.06.2265.
Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28(7):1247-1253. doi: https://doi.org/10.1200/JCO.2009.24.2099
Casali PG, Abecassis N, Bauer S, Biagini R, Bielack S, Bonvalot S, et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv68–78. https://doi.org/10.1093/annonc/mdy095.
Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer. 2005;41(12):1751–7. https://doi.org/10.1016/j.ejca.2005.04.034.
Le Cesne A, Ray-Coquard I, Bui BN, Adenis A, Rios M, Bertucci F, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol. 2010;11(10):942–9. https://doi.org/10.1016/S1470-2045(10)70222-9.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, Von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9. https://doi.org/10.1200/JCO.2003.04.190.
Heinrich MC, Jones RL, von Mehren M, Schöffski P, Serrano C, Kang YK, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21(7):935–46. https://doi.org/10.1016/S1470-2045(20)30269-2.
Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-kit/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40(5):689–95. https://doi.org/10.1016/j.ejca.2003.11.025.
Cassier PA, Fumagalli E, Rutkowski P, Schöffski P, Van Glabbeke M, Debiec-Rychter M, et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin Cancer Res. 2012;18(16):4458–64. https://doi.org/10.1158/1078-0432.CCR-11-3025.
Falchook GS, Trent JC, Heinrich MC, Beadling C, Patterson J, Bastida CC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4(2):310–5. https://doi.org/10.18632/oncotarget.864.
US FDA. FDA approves avapritinib for gastrointestinal stromal tumor with a rare mutation. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-avapritinib-gastrointestinal-stromal-tumor-rare-mutation. Accessed 17 Aug 2021.
Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64(17):5913–9. https://doi.org/10.1158/0008-5472.CAN-04-0085.
Dimitrakopoulou-Strauss A, Ronellenfitsch U, Cheng C, Pan L, Sachpekidis C, Hohenberger P, et al. Imaging therapy response of gastrointestinal stromal tumors (GIST) with FDG PET, CT and MRI: a systematic review. Clin Transl Imaging. 2017;5(3):183–97. https://doi.org/10.1007/s40336-017-0229-8.
Choi H, Charnsangavej C, de Castro FS, Tamm EP, Benjamin RS, Johnson MM, et al. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol. 2004;183(6):1619–28. https://doi.org/10.2214/ajr.183.6.01831619.
Yip D, Zalcberg JR, Blay JY, Eriksson M, Espinoza D, Price TJ, et al. ALT-GIST: Randomized phase II trial of imatinib alternating with regorafenib versus imatinib alone for the first-line treatment of metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol. 2019;37(15 Suppl):11023. https://doi.org/10.1200/JCO.2019.37.15_suppl.11023.
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KWH, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–605. https://doi.org/10.1182/blood-2002-07-2307.
Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2(5):471–8.
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–37.
Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7(3):225–33. https://doi.org/10.1007/s10456-004-3149-y.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Fletcher JA, Fletcher CD, et al. Sunitinib (SU) response in imatinib-resistant (IM-R) GIST correlates with KIT and PDGFRA mutation status. J Clin Oncol. 2006;24(18 Suppl):9502. https://doi.org/10.1200/jco.2006.24.18_suppl.9502.
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. https://doi.org/10.1016/S0140-6736(06)69446-4.
Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JVS, et al. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13(5):1367–73. https://doi.org/10.1158/1078-0432.CCR-06-2328.
George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, Deprimo SE, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–68. https://doi.org/10.1016/j.ejca.2009.02.011.
Napolitano A, Vincenzi B. Secondary KIT mutations: the GIST of drug resistance and sensitivity. Br J Cancer. 2019;120(6):577–8. https://doi.org/10.1038/s41416-019-0388-7.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–9. https://doi.org/10.1200/JCO.2007.15.7461.
Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–55. https://doi.org/10.1002/ijc.25864.
George S, Wang Q, Heinrich MC, Corless CL, Zhu M, Butrynski JE, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–7. https://doi.org/10.1200/JCO.2011.39.9394.
Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. https://doi.org/10.1016/S0140-6736(12)61857-1.
Ingram I. FDA Approves Regorafenib (Stivarga) for GIST. https://www.cancernetwork.com/view/fda-approves-regorafenib-stivarga-gist. Accessed 16 Aug 2021.
Kang YK, George S, Jones RL, Rutkowski P, Shen L, Mir O, et al. Avapritinib versus regorafenib in locally advanced unresectable or metastatic GI stromal tumor: a randomized, open-label phase III study. J Clin Oncol. 2021;39(28):3128–39. https://doi.org/10.1200/JCO.21.00217.
FDA approves ripretinib for advanced gastrointestinal stromal tumor | FDA. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ripretinib-advanced-gastrointestinal-stromal-tumor. Accessed 14 Aug 2021.
QINLOCK® (ripretinib) clinical overview. Spec Ther. 2021.
Janku F, Abdul Razak AR, Chi P, Heinrich MC, von Mehren M, Jones RL, et al. Switch control inhibition of KIT and PDGFRA in patients with advanced gastrointestinal stromal tumor: a phase I study of ripretinib. J Clin Oncol. 2020;38(28):3294–303. https://doi.org/10.1200/JCO.20.00522.
Blay JY, Serrano C, Heinrich MC, Zalcberg J, Bauer S, Gelderblom H, et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21(7):923–34. https://doi.org/10.1016/S1470-2045(20)30168-6.
Zalcberg JR, Heinrich MC, George S, Bauer S, Schöffski P, Serrano C, et al. Clinical benefit of ripretinib dose escalation after disease progression in advanced gastrointestinal stromal tumor: an analysis of the INVICTUS study. Oncologist. 2021;26(11):e2053–60. https://doi.org/10.1002/onco.13917.
A Study of DCC-2618 vs Sunitinib in Advanced GIST Patients After Treatment With Imatinib. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03673501. Accessed 21 Aug 2021.
Deciphera Pharmaceuticals announces top-line results from the INTRIGUE phase 3 clinical study. 2021. https://investors.deciphera.com/news-releases. Accessed 18 Jan 2022.
Schöffski P, Reichardt P, Blay JY, Dumez H, Morgan JA, Ray-Coquard I, et al. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990–8. https://doi.org/10.1093/annonc/mdq076.
Gelderblom H, Jones RL, George S, Valverde Morales C, Benson C, Blay JY, et al. Imatinib in combination with phosphoinositol kinase inhibitor buparlisib in patients with gastrointestinal stromal tumour who failed prior therapy with imatinib and sunitinib: a Phase 1b, multicentre study. Br J Cancer. 2020;122(8):1158–65. https://doi.org/10.1038/s41416-020-0769-y.
Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, et al. A potent combination of the novel PI3K Inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res. 2013;19(3):620–30. https://doi.org/10.1158/1078-0432.CCR-12-2853.
Van Looy T, Wozniak A, Floris G, Sciot R, Li H, Wellens J, et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: rationale and efficacy. Clin Cancer Res. 2014;20(23):6071–82. https://doi.org/10.1158/1078-0432.CCR-14-1823.
Zook P, Pathak HB, Belinsky MG, Gersz L, Devarajan K, Zhou Y, et al. Combination of imatinib mesylate and AKT inhibitor provides synergistic effects in preclinical study of gastrointestinal stromal Tumor. Clin Cancer Res. 2017;23(1):171–80. https://doi.org/10.1158/1078-0432.CCR-16-0529.
Lostes-Bardaji MJ, García-Illescas D, Valverde C, Serrano C. Ripretinib in gastrointestinal stromal tumor: the long-awaited step forward. Ther Adv Med Oncol. 2021;13:1758835920986498. https://doi.org/10.1177/1758835920986498.
Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013;14(9):e358–69. https://doi.org/10.1016/S1470-2045(13)70169-4.
Honma Y, Kurokawa Y, Sawaki A, Naito Y, Iwagami S, Baba H, et al. Randomized, double-blind, placebo (PL)-controlled, phase III trial of pimitespib (TAS-116), an oral inhibitor of heat shock protein 90 (HSP90), in patients (pts) with advanced gastrointestinal stromal tumor (GIST) refractory to imatinib (IM), sunitinib (SU) and regorafenib (REG). J Clin Oncol. 2021;39(15 Suppl):11524. https://doi.org/10.1200/JCO.2021.39.15_suppl.11524.
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–61. https://doi.org/10.1158/0008-5472.CAN-06-0165.
Shimomura A, Yamamoto N, Kondo S, Fujiwara Y, Suzuki S, Yanagitani N, et al. First-in-human phase I study of an oral HSP90 inhibitor, TAS-116, in patients with advanced solid tumors. Mol Cancer Ther. 2019;18(3):531–40. https://doi.org/10.1158/1535-7163.MCT-18-0831.
Doi T, Kurokawa Y, Sawaki A, Komatsu Y, Ozaka M, Takahashi T, et al. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: a phase II, single-arm trial. Eur J Cancer. 2019;121:29–39. https://doi.org/10.1016/j.ejca.2019.08.009.
Wagner AJ, Severson PL, Shields AF, Patnaik A, Chugh R, Tinoco G, et al. Association of combination of conformation-specific kit inhibitors with clinical benefit in patients with refractory gastrointestinal stromal tumors: a phase 1b/2a nonrandomized clinical trial. JAMA Oncol. 2021;7(9):1343–50. https://doi.org/10.1001/jamaoncol.2021.2086.
A Drug-Drug Interaction Study to Evaluate the Effect of Ripretinib on the Pharmacokinetics of a CYP2C8 Probe Substrate in Patients with Advanced GIST ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04530981?recrs=abdf&cond=GIST&draw=5&rank=3. Accessed 19 Oct 2021.
A Study of HQP1351 in Patients With GIST or Other Solid Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03594422. Accessed 19 Oct 2021.
Single Agent Regorafenib in First-line for Metastatic/Unresectable KIT/PDGFR Wild Type GIST. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02638766?recrs=abdf&cond=GIST&draw=6&rank=13. Accessed 19 Oct 2021.
Multicentre Placebo-controlled Double-blinded Phase II Study of Lenvatinib Efficacy in Patients With Locally Advanced or Metastatic GIST (Gastrointestinal Stromal Tumor) After Imatinib/Sunitinib Failure. ClinicalTrials.govc. https://clinicaltrials.gov/ct2/show/NCT04193553?recrs=abdf&cond=GIST&draw=6&rank=14. Accessed 19 Oct 2021.
A Study of Avelumab In Combination With Axitinib in Patients With Unresectable/Metastatic Gastrointestinal Stromal Tumor After Failure of Standard Therapy. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04258956?recrs=abdf&cond=GIST&draw=9&rank=42. Accessed 19 Oct 2021.
Selinexor as a Single Agent and in Combination With Imatinib in Patients With Metastatic and/or Unresectable Gastrointestinal Stromal Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04138381?recrs=abdf&cond=GIST&draw=10&rank=54. Accessed 19 Oct 2021.
Phase Ib Study of TNO155 in Combination With Spartalizumab or Ribociclib in Selected Malignancies. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04000529?recrs=abdf&cond=GIST&draw=11&rank=64. Accessed 19 Oct 2021.
A Phase I/II Study of Regorafenib Plus Avelumab in Solid Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03475953?recrs=abdf&cond=GIST&draw=11&rank=68. Accessed 19 Oct 2021.
Ipilimumab and Imatinib Mesylate in Advanced Cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01738139. Accessed 19 Oct 2021.
Nivolumab and Ipilimumab in Treating Patients With Rare Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02834013?recrs=abdf&cond=GIST&draw=12&rank=80.
CGT9486 (formerly known as PLX9486) as a Single Agent and in Combination With PLX3397 (Pexidartinib) or Sunitinib in Participants With Advanced Solid Tumors. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02401815. Accessed 19 Oct 2021.
PDR001 Plus Imatinib for Metastatic or Unresectable GIST. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03609424?recrs=abdf&cond=GIST&draw=6&rank=20. Accessed 19 Oct 2021.
Paclitaxel in Patients With GIST With Low P-glycoprotein Expression After Failure of at Least Imatinib and Sunitinib, and Regorafenib. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03944304?recrs=abdf&cond=GIST&draw=7&rank=22. Accessed 19 Oct 2021.
Temozolomide (TMZ) In Advanced Succinate Dehydrogenase (SDH)-Mutant/Deficient Gastrointestinal Stromal Tumor (GIST). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03556384?recrs=abdf&cond=GIST&draw=7&rank=24. Accessed 19 Oct 2021.
Testing the Anti-cancer Drug, Rogaratinib (BAY 1163877), for Treatment of Advanced Sarcoma With Alteration in Fibroblast Growth Factor Receptor (FGFR 1-4), and in Patients With SDH-deficient Gastrointestinal Stromal Tumor (GIST). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04595747?recrs=abdf&cond=GIST&draw=8&rank=31. Accessed 19 Oct 2021.
Nivolumab With or Without Ipilimumab in Treating Patients With Gastrointestinal Stromal Tumor That Is Metastatic or Cannot Be Removed by Surgery. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02880020?recrs=abdf&cond=GIST&draw=8&rank=33. Accessed 19 Oct 2021.
Treatment of Patients With Metastatic or Unresectable Gastrointestinal Stromal Tumors in First Line With Nilotinib. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT00756509?recrs=abdf&cond=GIST&draw=8&rank=37. Accessed 19 Oct 2021.
A Study of CS3007 in Subjects With Gastrointestinal Stromal Tumor. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04254939?recrs=abdf&cond=GIST&draw=8&rank=39. Accessed 19 Oct 2021.
Efficacy and Safety of Famitinib Versus Sunitinib in the Treatment of Advanced Gastrointestinal Stromal Tumour Patients After Failure of Imatinib. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04409223?recrs=abdf&cond=GIST&draw=9&rank=41. Accessed 19 Oct 2021.
Mitogen Activated Protein Kinase Kinase (MEK1/2) Inhibitor selumetinib (AZD6244 Hydrogen Sulfate) in people with neurofibromatosis type 1 (NF1) mutated gastrointestinal stromal tumors (GIST). ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03109301. Accessed 19 Oct 2021.
A phase II trial of the DNA methyl transferase inhibitor, guadecitabine (SGI-110), in children and adults with wild type GIST, pheochromocytoma and paraganglioma associated with succinate dehydrogenase deficiency and HLRCC-associated kidney cancer. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03165721. Accessed 19 Oct 2021.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflict of interest
Tiffany Foo, David Goldstein, Eva Segelov, Jeremy Shapiro, Nick Pavlakis, Jayesh Desai, Desmond Yip, John Zalcberg, Adnan Nagrial, Lorraine Chantrill, Matt Burge, Christos S. Karapetis, Niall Tebbutt, and Amitesh C. Roy declare they have no conflicts of interest that might be relevant to the contents of this manuscript. Timothy Price reports advisory roles for MSD, Servier and BioGene.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Authors' contributions
All authors contributed to the conception and writing of the paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Foo, T., Goldstein, D., Segelov, E. et al. The Management of Unresectable, Advanced Gastrointestinal Stromal Tumours. Targ Oncol 17, 95–110 (2022). https://doi.org/10.1007/s11523-022-00869-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-022-00869-y