
Abstract
Purpose of Review
Regulatory T cell (Treg) biology continues to evolve at a rapid pace. The role of Tregs in solid organ transplantation offers a unique window into Treg ontogeny and function as well as limitless possibilities for clinical application. Here we review recent significant discoveries and key translational work.
Recent Findings
Advances in transplantation deepen understanding of Treg differentiation, expansion, transcription, co-stimulation, and signaling. T cell receptor (TCR) sequencing and single-cell analytics allow unprecedented insight into Treg repertoire diversity and phenotypic heterogeneity. Efforts to replace conventional immunosuppression with Treg adoptive immunotherapy are underway and coalescing around strategies to increase efficiency through development of donor-reactive Tregs.
Summary
Adoptive immunotherapy with Tregs is a leading tolerogenic strategy. Early clinical trials suggest that Treg infusion is safe and reports on efficacy will soon follow.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Solid organ transplantation provides optimal therapy for patients with end-stage organ failure. The scarcity of available donor organs constitutes a pressing need within the field. Efforts to promote living donation, efficiently capture all eligible deceased donors, utilize marginal donor organs, and rehabilitate injured organs all offer hope. Equally important are efforts to maximize the longevity of each individual organ transplanted working toward the goal of “one organ for life.” In 2017 12.1% of kidney and 9.9% of liver transplants were performed on prior recipients of the same organ [1]. Graft survival is markedly reduced by chronic immune-mediated graft injury and toxic effects of current best available immunosuppressive drugs. There is urgent clinical need to dampen destructive alloimmunity, enhance regulatory immunity, and replace traditional pharmaceuticals with nontoxic cell-based immunotherapy.
Originally termed “suppressor” T cells, regulatory T cells have been studied for at least 50 years, frequently in the context of transplantation. “Modern” CD4+ Tregs are defined by surface expression of the IL-2 receptor CD25, activity of the Forkhead box P3 (FOXP3) transcription factor, characteristic hypomethylation of genes regulated by FOXP3, and the ability to suppress immune responses in vitro and in vivo. The so-called “natural” Tregs (nTregs) originate in the thymus, while “induced” or “peripheral” Tregs (iTregs or pTregs) are generated through the reprogramming of conventional T cells. Myriad additional subtypes of Tregs are described including Th1-, Th2-, and Th17-like and follicular Tregs (Tfr) [2, 3]. All are major histocompatibility complex (MHC) restricted, and Tregs are known to suppress pro-inflammatory immune responses using both T cell receptor (TCR)-dependent and TCR-independent mechanisms including secretion of anti-inflammatory soluble factors, inhibitory co-stimulation, IL-2 sequestration, antigen-presenting cell (APC) modulation, and direct cytotoxicity [4, 5].
In solid organ transplantation, Tregs are widely viewed as a solution to the challenge of inhibiting destructive donor-reactive immunity while sparing protective host defenses. Epidemiologic studies of patients with Foxp3 mutations clearly establish the importance of Tregs in preventing autoimmune disease [6]. Elucidating the role of Tregs in solid organ transplantation has been more complicated because transplantation is, of itself, a deviation from “natural history.” Limited studies do suggest that patients with Foxp3 mutations have worse transplantation outcomes [7,8,9] and tolerant kidney transplant recipients have increased indirect pathway regulatory anti-donor T cell responses [10]. There is overwhelming evidence in rodent and primate models that Treg activity can be modulated to prolong allograft survival. Adoptive immunotherapy with Tregs offers the additional potential appeal of replacing nephrotoxic and diabetogenic calcineurin inhibitors with a nontoxic cellular alternative. Here we review major discoveries in the past 3 years that enhance our understanding of Treg function in solid organ transplant and explore ongoing efforts to develop Treg adoptive immunotherapy.
New Molecular Targets and Novel Mechanistic Insights
Despite overwhelming data supporting a role for Tregs in murine allogeneic tolerance, parallel findings supporting causality in human transplant recipients are less common. A recent longitudinal analysis of Treg frequency in living-donor kidney transplant recipients demonstrates that activated alloreactive CD4+CD25highFOXP3+GARP+ Tregs increase in number approximately 3 months following transplantation [11]. Consistent with the belief that enhanced Treg numbers are favorable, a variety of molecular targets have been manipulated to augment the expansion and survival of Tregs. In a single MHC mismatched skin transplant model, combined administration of donor-specific Tregs and IL-2 synergistically prolonged graft survival and increased numbers of Kd-specific Tregs [12]. Induced expression of the mTOR binding partner DEPTOR in CD4+ regulatory T cells stabilized FOXP3 expression, increased survival and suppressive potency of Tregs, and prolonged survival of fully MHC mismatched murine cardiac allografts [13]. In a murine model of Treg-dependent cardiac allograft survival, overexpression of the complement receptor C5aR2 augmented iTreg induction and prolonged allograft survival [14]. Lastly, in mice and human living-donor kidney transplant recipients, adoptive immunotherapy with human regulatory macrophages enhanced induction of IL-10 producing FOXP3+TIGIT+ iTregs [15].
In related studies, additional cell surface, signaling, and transcriptional targets have been utilized to subtly shift the Treg/Teff balance in favor of allo-acceptance. A prime example is the recent demonstration that the CD45 isoform CD45RC is not expressed on CD4+FOXP3+ Tregs and transient administration of anti-CD45RC in a rat cardiac allotransplantation model induced transplant tolerance [16]. Of note, the ability to mount T cell-dependent B cell responses to keyhole limpet hemocyanin were preserved even during anti-CD45C administration. At the signaling level, deletion of both the γ and δ variants of PI3 kinase prolonged murine heart allograft survival, but PI3Kδ deletion also reduced Treg survival, suggesting that selective PI3Kγ targeting will be favored in transplant [17]. At the transcriptional level, it is well-known that FOXP3 is essential for expression of lineage-specific target genes in CD4 Tregs, but the roles of other transcription factors are under active investigation. Treg cell-specific conditional knockouts of c-Rel and p65 were used to investigate the role of NF-ĸB in Treg function [18]. Double conditional knockouts displayed a severe autoimmune Scurfy-like [19] phenotype, and subsequent RNA-seq experiments confirmed that NF-ĸB helps maintain the identity and function of mature Tregs. Both the NF-ĸB and IRF transcriptional pathways are potential targets in transplantation, and recently deletion of transcription factor IRF4 in CD4+ T cells caused upregulation of the Treg-associated markers Helios and PD-1, resulting in disordered immunity and transplant acceptance [20].
Co-stimulatory blockade continues to evolve as a strategy in transplantation. With > 10 years of clinical data now available, belatacept is now familiar in renal transplantation, and αCD40/CD40L therapy, originally plagued by problems with thromboembolism, is resurfacing [21]. Selectively disrupting checkpoints while preserving Treg function is essential, and to that end Wood and colleagues compared CTLA4-Ig with selective antibody blockade of CD28 in a humanized murine skin transplant model [22•]. Anti-CD28 demonstrated superiority, likely in part by leaving CD80/86 available to engage CTLA4 present on Tregs.
Perhaps most unexpected are recent reports on donor-derived Tregs. Pettigrew et al. demonstrate persistence of donor-derived nTregs in human lung transplant recipients [23•]. Pursuing this observation in a murine cardiac transplant model, they demonstrate that depletion of donor CD4 nTregs before organ recovery accelerated allograft rejection and show that donor-derived nTregs were more efficacious than recipient-derived nTregs in restoring allograft survival. In similar experiments, Sachs and colleagues report that long-term tolerant swine kidney grafts confer infectious tolerance when re-transplanted implying the presence of a strong intra-graft regulatory element [24]. In recognition of the importance of Treg locale, culture conditions favoring CXCR3, α4β7 integrin, and CCR9 were used to tailor the homing capacity of Tregs to tissue sites of interest [25].
Because Tregs are able to suppress through both TCR-dependent and TCR-independent mechanisms, the transplant community has sought to utilize both polyclonal and donor antigen reactive Tregs in a therapeutic capacity. Consensus opinion now seems to accept that efficacy will be greatest when donor-reactive Tregs are utilized but larger questions remain concerning the true size of the human alloresponse and the diversity of the Treg TCR repertoire compared with that of conventional T cells. Advances in next-generation sequencing and big data analysis have enabled recent breakthroughs with relevance across disciplines. Shen and colleagues revisited the age-old question of alloreactive frequency using modern approaches and found 0.5–6% of the circulating TCR repertoire reactive to just two different allogeneic stimulators, reproducing the antiquated conventional estimate of 1–10% with remarkable accuracy [26•]. The TCR repertoire of Tregs is as diverse, if not more so, than the repertoire on naïve CD4+ T cells [27], and thus, we can infer broad clonal diversity within populations of donor-reactive Tregs. Lastly, Benoist and colleagues used single-cell RNA-seq to profile thousands of mouse and human Tregs and found that while extensive phenotypic diversity exists, the main features of Treg heterogeneity are similar in mice and humans [28], providing validation to the relevance of murine studies.
Active Strategies for Translation
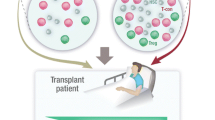
Efforts to convert our evolving understanding of Tregs in transplantation to safe human therapies primarily involve Treg adoptive immunotherapy (Fig. 1). This includes bulk transfer of polyclonal Tregs, transfer of “tailored” donor-reactive populations, combined kidney bone marrow transplantation, and adoptive transfer of T cells engineered to express TCRs, antibodies, or protein antigens that direct Treg function in an antigen-specific manner.

Schematic illustrating four distinct approaches to Treg immunotherapy. (1) Infusion of polyclonal Tregs, (2) infusion of Tregs with known anti-donor reactivity, (3) combined kidney and bone marrow transplantation with mixed chimerism, and (4) infusion of T cells bearing transgenic receptors (T cell receptors, chimeric antigen receptors, B cell antigen receptors) engineered with anti-donor reactivity. (Figure created using Biorender.com)
Adoptive Immunotherapy with Polyclonal and Donor-Reactive Tregs
Recipient peripheral blood is the primary source of Tregs for ex vivo expansion and subsequent adoptive immunotherapy; however, reports utilizing umbilical cord blood-derived Tregs are emerging [29, 30], and West and colleagues report the intriguing prospect of using human thymus routinely removed during pediatric cardiothoracic surgery as a source of nTregs [31]. Numerous manuscripts addressing the technical aspects of clinical Treg manufacture including cryopreservation [32] and automation [33] have appeared. Major outstanding issues are optimization of culture conditions for ex vivo Treg expansion, strategies for directing donor reactivity, and compatibility of various Treg products with conventional immunosuppression required in clinical trials.
Typical Treg expansion strategies involve magnetic bead or flow cytometric enrichment of a CD4+CD25+ cell population (sometimes with additional selection based upon CD127 or CD45RA), which is then expanded several thousand-fold in culture bags or bioreactors containing serum-enhanced media, IL-2, and TCR and co-stimulatory signals most often provided via αCD3/αCD28 antibodies. Media is further modified with rapamycin, cytokines, the vitamin A metabolite all-trans retinoic acid, amino acids, and short chain fatty acids to enhance purity and tailor Treg functionality. In comparing CD45RA positive and negative CD4+CD127-/loCD25+ cells cultured in the presence of tacrolimus, Wood and colleagues found that although CD45RA− Tregs have greater suppressive capacity post-expansion, they do not retain a stable TSDR demethylated phenotype raising concerns that these cells might become pathogenic in transplant recipients [34]. Marti and colleagues compared ex vivo Treg expansion with rapamycin and everolimus and found, despite differing kinetics, equivalence in the final Treg product supporting consistent in and ex vivo use of the clinically favored drug everolimus [35]. Early investigation into the mTOR inhibitory activity of azithromycin shows no clear advantage over rapamycin [36], and stimulation of naïve CD4+ T cells in media containing low tryptophan and kynurenines has been shown to foster development of iTregs [37]. Markmann’s group reports successful Treg generation from peripheral blood of uremic pre-transplant candidates using ex vivo MLR and belatacept co-stimulatory blockade [38].
Efforts to promote anti-donor reactivity add considerable complexity to Treg expansion protocols. While unmodified peripheral blood has been successfully used as a source of donor antigen-presenting cells [38,39,40], donor B cells activated with CD40L-expressing feeder cells are typically used to capitalize on the relative abundance (vs. dendritic cells) and potent stimulatory capacity of B cells. To bypass concerns that CD40L+ feeder cells contaminating Treg preparations would cause indiscriminant activation of alloreactive effector T cells, Leventhal and colleagues tested B cell activation and expansion using soluble 4-trimer CD40 ligand and successfully converted naïve CD4 T cells to demethylated Tregs with a constricted donor-reactive TCR repertoire [41].
Adoptive immunotherapy (AI) and conventional immunosuppression will be co-administered, at least until non-inferiority of Treg AI is proven, and thus there is great interest in understanding how conventional immunosuppression affects endogenous Treg numbers and synergizes with adoptive Treg therapy. Amirzargar et al. compared Treg number and phenotype in 24 renal transplant recipients treated with either tacrolimus/mycophenolate/prednisone or tacrolimus/sirolimus/prednisone therapy and proved that the latter was favorable in augmenting Treg numbers and reducing RORγt expression associated with Treg conversion to a pro-inflammatory Th17 phenotype [42]. Lombardi and colleagues show that rapamycin-treated ex vivo-expanded human Tregs maintain a stable Treg phenotype in the presence of tacrolimus, mycophenolate, and methylprednisolone; however, tacrolimus altered chemokine receptor expression and reduced IL-10 production [43]. All three agents reduced viability, function, and proliferative capacity relative to rapamycin in a humanized mouse model.
Published Clinical Trials
Of the four published clinical trials utilizing Treg adoptive immunotherapy, two involve liver transplant which is widely accepted as a less immunogenic transplant than kidney. Okumura and colleagues co-cultured recipient lymphocytes and irradiated donor lymphocytes in the presence of αCD80/86 monoclonal antibodies which generated Tregs with donor reactivity in mixed lymphocyte reactions [44]. Ten splenectomized living-donor liver transplant recipients received this Treg product 13 days after transplantation and, notably, after administration of 40 mg/kg of cyclophosphamide on postoperative day 5. Immunosuppression was weaned between months 6 and 18 post-transplant, and seven of ten patients successfully discontinued immunosuppression. The trial was halted because three patients with primary biliary cirrhosis or primary sclerosing cholangitis as the cause of their liver failure developed treatable acute cellular rejection. Lombardi and colleagues add to this with their recently published open-label, dose escalation, phase I clinical trial of autologous polyclonal Treg therapy [45]. Polyclonal Tregs were grown from recipient peripheral blood in the presence of αCD3/CD28, IL-2, and rapamycin, and 0.5–1 million Tregs/kg or 3–4.5 million Tregs/kg were administered to nine cadaveric liver transplant recipients at least 3 months after transplantation. No attempts were made to wean immunosuppression. One patient experienced an infusion-related cytokine storm. Infusion transiently increased the pool of circulating Tregs, and the study was not powered to address therapeutic efficacy.
Two differing approaches to Treg adoptive immunotherapy in renal transplantation have been published to date. Vincenti and colleagues tested the safety and feasibility of autologous polyclonal Treg therapy in patients with subclinical inflammation on 6-month surveillance biopsies [46]. Three renal transplant recipients with 6-month biopsies demonstrating 5–25% inflammation (Banff i0 or i1) and no evidence of rejection (Banff i < 2, t < 2) received 320 × 106 autologous CD4+CD127loCD25+ polyclonal Tregs isolated from peripheral blood via FACS sorting and expanded in the presence of αCD3/αCD28, IL-2, and deuterated glucose. Infusion was well-tolerated, and infused Tregs were detectable in the peripheral blood for 3 months post-infusion. Future studies will be powered to detect changes in graft inflammation. Leventhal and colleagues report a related phase I trial in which nine alemtuzumab-induced living-donor renal transplant recipients received 0.5, 1, or 5 × 109 autologous polyclonal Tregs at day 60 post-transplant prepared using magnetic bead technology and expanded in the presence of αCD3/αCD28, IL-2, TGFβ, and sirolimus [47]. Treg infusion was safe. Conventional immunosuppression with sirolimus and mycophenolate was maintained. In 2 years of follow-up, one patient developed subclinical rejection, and two patients developed de novo DSA.
Overall, the published clinical trials to date suggest that both polyclonal and donor-reactive Treg products can be safely manufactured and administered. Feared complications including over-immunosuppression, infection, malignancy, and conversion of infused Tregs to a destructive alloreactive phenotype have not been observed. The efficacy of Treg adoptive immunotherapy and optimal approach in each clinical scenario are open questions, and a large number of additional clinical trials are currently in progress (Table 1).
Combined Kidney Bone Marrow Transplantation
Bone marrow transplantation is perhaps the most extreme form of adoptive immunotherapy, and combined kidney bone marrow transplantation (CKBMT) with transient mixed chimerism has been shown to induce long-term tolerance in human recipients [48,49,50]. The long-term success of this strategy relies upon deletion of donor-reactive clones [51]. Investigation into the complex mechanisms allowing for clonal deletion allows opportunity to study Tregs with proven clinical efficacy. In the nonhuman primate CKBMT model, CD4+FOXP3+ T cells proliferating in response to donor antigens in the CFSE mixed lymphocyte reaction were shown to be iTregs converted from conventional T cells [52]. In human CKBMT recipients, both new thymic emigration and lymphopenia-driven proliferation were shown to account for the marked early enrichment of CD4+CD25highCD127lowFoxp3+ cells in peripheral blood [53]. Sykes and colleagues have introduced the novel technique of using activated B cells to expand donor-reactive Tregs pre-transplant [54]. Expansion facilitated deep sequencing and allowed for clonal tracking which ultimately demonstrated that preexisting donor-reactive Tregs were expanded at 6 months post-transplant in tolerant human CKBMT recipients and failed to expand in a non-tolerant recipient. Kawai’s group used a series of allograft biopsies from nonhuman primate CKBMT recipients to establish an mRNA signature of tolerance that included a large number of Treg-associated transcripts including FOXP3, IL10, TGFβ, and GATA3 [55]. Further, they have demonstrated that their combined CKBMT approach does not induce tolerance to islet allografts in nonhuman primates [56]. Lastly, they have recently demonstrated that the addition of αCD40 monoclonal antibody to their mixed chimerism approach abrogates tolerance induction and they speculate that the mechanism involves a defect in antigen presentation to regulatory cells [57].
TCRs, CARs, and BARs
“Manufacturing” recipient-derived donor-reactive Tregs poses significant challenges, particularly in the setting of cadaveric transplantation where the time interval between donor selection and transplantation can be short. Redirecting the specificity of Tregs via gene transfer of donor-reactive TCRs, antibody-based fusion proteins specific for allo-MHC (CAR), or the antigenic targets of B cells (BAR) are promising alternative strategies under development. Attempting to capitalize on the prevalence of HLA-A2 in many donor populations, two groups report creation of HLA-A2-specific Treg CARs that display potent suppressive capacity in vitro and the ability to suppress GVHD and skin transplant rejection in humanized mouse models [58, 59]. Boardman et al. have begun to explore mechanisms of Treg CAR function and utilize regulatory CARs with mutated intracellular signaling domains to show convincingly that signaling through the CAR is essential for suppressive function [60]. Meyer and colleagues experiment with transient transfection and offer an elegant platform in which a single CAR can accommodate multiple specificities [61]. By coupling CD28 and CD3ς signaling domains with an anti-FITC scFv, FITC-conjugated antibodies of various specificities can be added to modulate this single “platform.” Using FITC-conjugated anti-donor HLA class I monoclonal antibodies, they facilitate the homing of Tregs to pancreatic islets placed under the kidney capsule. Surprisingly, these “mAbCAR” Tregs remain localized near the islets long after expression of the transgene is lost suggesting that transient CAR expression “parades” polyclonal Tregs through the effector site with retention and/or proliferation of donor-reactive clones. Concerns surrounding CAR therapy involve insertional oncogenesis, graft versus host disease, and off-target expression of effector function, but to date these have not been problematic in animal models. BAR therapy, intended to be useful in recruiting regulatory cells to germinal centers to prevent anti-donor antibody formation, is also under active development [62].
Summary and Future Challenges
Alloimmune responses involve a balance between effector and regulatory T cell activity. Recent work highlighted here enhances understanding of Treg origin, development, and effector function. Enhancing Treg activity in solid organ transplantation offers the hope of reducing or eliminating current noxious immunosuppressive drugs. Although donor-reactive Tregs display broad clonal and phenotypic diversity, strategies to harness donor-reactive Tregs for adoptive immunotherapy are plausible, and early clinical trials suggest that the approach is safe. Highly anticipated results from a number of ongoing trials are likely to enable a new era of biologic immunotherapy. Critical challenges include (1) the need for strategies to create donor-reactive Tregs that can be administered at the time of transplantation within the logistic constraints imposed by both living-donor and cadaveric-donor transplantation, (2) identification of ideal Treg phenotypes and optimization of ex vivo expansion conditions that preserve these phenotypes, and (3) the need to understand compatibility with existing immunosuppressive regimens to ensure that adoptive immunotherapy trials remain both safe and rational.
Abbreviations
- APC:
-
Antigen-presenting cell
- BAR:
-
B cell antigen receptor
- CAR:
-
Chimeric antigen receptor
- CKBMT:
-
Combined kidney bone marrow transplantation
- CFSE:
-
Carboxyfluorescein succinimidyl ester
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DSA:
-
Donor-specific antigen
- FACS:
-
Fluorescence-activated cell sorting
- FITC:
-
Fluorescein isothiocyanate
- FOXP3:
-
Forkhead box P3 transcription factor
- GARP:
-
Glycoprotein A repetitions predominant
- IRF:
-
Interferon regulatory transcription factor
- MHC:
-
Major histocompatibility complex
- mTOR:
-
Mammalian target of rapamycin
- NFκB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PD-1:
-
Programmed cell death protein 1
- Treg :
-
Regulatory T cell
- Tfr :
-
Follicular regulatory T cell
- iTreg :
-
Induced regulatory T cell
- nTreg :
-
Natural or thymic-derived regulatory T cell
- pTreg :
-
Peripheral regulatory T cell
- RORγT:
-
Nuclear receptor retinoic acid receptor-related orphan receptor gamma
- scFv:
-
Single-chain variable fragment
- TCR:
-
T cell receptor
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- TSDR:
-
Treg-specific demethylated region
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
2017 Annual Data Report. Scientific Registry of Transplant Recipients. http://srtr.transplant.hrsa.gov/annual_reports/Default.aspx. Accessed 12/12/2019.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64.
Halim L, Romano M, McGregor R, Correa I, Pavlidis P, Grageda N, et al. An atlas of human regulatory T helper-like cells reveals features of Th2-like Tregs that support a tumorigenic environment. Cell Rep. 2017;20(3):757–70.
Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol. 2012;3:51.
Zhao H, Liao X, Kang Y. Tregs: where we are and what comes next? Front Immunol. 2017;8:1578.
Abdel-Motal UM, al-Shaibi A, Elawad M, Lo B. Zero tolerance! A perspective on monogenic disorders with defective regulatory T cells and IBD-like disease. Immunol Rev. 2019;287(1):236–40.
Qiu XY, Jiao Z, Zhang M, Chen JP, Shi XJ, Zhong MK. Genetic association of FOXP3 gene polymorphisms with allograft rejection in renal transplant patients. Nephrology (Carlton). 2012;17(4):423–30.
Engela AU, Boer K, Roodnat JI, Peeters AM, Eilers PH, Kal-van Gestel J, et al. Genetic variants of FOXP3 influence graft survival in kidney transplant patients. Hum Immunol. 2013;74(6):751–7.
Misra MK, Mishra A, Pandey SK, Kapoor R, Sharma RK, Agrawal S. Association of functional genetic variants of transcription factor Forkhead box P3 and nuclear factor-kappaB with end-stage renal disease and renal allograft outcome. Gene. 2016;581(1):57–65.
Haynes LD, Jankowska-Gan E, Sheka A, Keller MR, Hernandez-Fuentes MP, Lechler RI, et al. Donor-specific indirect pathway analysis reveals a B-cell-independent signature which reflects outcomes in kidney transplant recipients. Am J Transplant. 2012;12(3):640–8.
Mederacke YS, Vondran FW, Kollrich S, Schulde E, Schmitt R, Manns MP, et al. Transient increase of activated regulatory T cells early after kidney transplantation. Sci Rep. 2019;9(1):1021.
Ratnasothy K, Jacob J, Tung S, Boardman D, Lechler RI, Sanchez-Fueyo A, et al. IL-2 therapy preferentially expands adoptively transferred donor-specific Tregs improving skin allograft survival. Am J Transplant. 2019;19(7):2092–100.
Wedel J, Bruneau S, Liu K, Kong SW, Sage PT, Sabatini DM, et al. DEPTOR modulates activation responses in CD4(+) T cells and enhances immunoregulation following transplantation. Am J Transplant. 2019;19(1):77–88.
Verghese DA, et al. T cell expression of C5a receptor 2 augments murine regulatory T cell (TREG) generation and TREG-dependent cardiac allograft survival. J Immunol. 2018;200(6):2186–98.
Riquelme P, Haarer J, Kammler A, Walter L, Tomiuk S, Ahrens N, et al. TIGIT(+) iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun. 2018;9(1):2858.
Picarda E, Bézie S, Boucault L, Autrusseau E, Kilens S, Meistermann D, et al. Transient antibody targeting of CD45RC induces transplant tolerance and potent antigen-specific regulatory T cells. JCI Insight. 2017;2(3):e90088.
Uehara M, et al. Regulation of T cell alloimmunity by PI3Kgamma and PI3Kdelta. Nat Commun. 2017;8(1):951.
Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity. 2017;47(3):450–65 e5.
Ramsdell F, Ziegler SF. FOXP3 and scurfy: how it all began. Nat Rev Immunol. 2014;14(5):343–9.
Wu J, Zhang H, Shi X, Xiao X, Fan Y, Minze LJ, et al. Ablation of transcription factor IRF4 promotes transplant acceptance by driving allogenic CD4(+) T cell dysfunction. Immunity. 2017;47(6):1114–28 e6.
Kim SC, Wakwe W, Higginbotham LB, Mathews DV, Breeden CP, Stephenson AC, et al. Fc-silent anti-CD154 domain antibody effectively prevents nonhuman primate renal allograft rejection. Am J Transplant. 2017;17(5):1182–92.
• Zaitsu M, et al. Selective blockade of CD28 on human T cells facilitates regulation of alloimmune responses. JCI Insight. 2017;2(19) This work suggests a means to block CD28 - CD80/86 interactions while preserving Tregfunction.
• Harper IG, et al. Prolongation of allograft survival by passenger donor regulatory T cells. Am J Transplant. 2019;19(5):1371–9 This study explores the previously overlooked role of donor-derived Tregsin prolonging cardiac allograft survival.
Villani V, Yamada K, Scalea JR, Gillon BC, Arn JS, Sekijima M, et al. Adoptive transfer of renal allograft tolerance in a large animal model. Am J Transplant. 2016;16(1):317–24.
Hoeppli RE, MacDonald K, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplant. 2019;19(1):62–76.
• DeWolf S, et al. Quantifying size and diversity of the human T cell alloresponse. JCI Insight. 2018;3(15) This study revisits the classic question of alloreactive frequency using cutting edge technique.
Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25(2):249–59.
Zemmour D, Zilionis R, Kiner E, Klein AM, Mathis D, Benoist C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat Immunol. 2018;19(3):291–301.
Brunstein CG, Miller JS, McKenna D, Hippen KL, DeFor T, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. 2016;127(8):1044–51.
Do JS, Zhong F, Huang AY, van't Hof W, Finney M, Laughlin MJ. Foxp3 expression in induced T regulatory cells derived from human umbilical cord blood vs. adult peripheral blood. Bone Marrow Transplant. 2018;53(12):1568–77.
Dijke IE, Hoeppli RE, Ellis T, Pearcey J, Huang Q, McMurchy A, et al. Discarded human thymus is a novel source of stable and long-lived therapeutic regulatory T cells. Am J Transplant. 2016;16(1):58–71.
Golab K, et al. Cell banking for regulatory T cell-based therapy: strategies to overcome the impact of cryopreservation on the Treg viability and phenotype. Oncotarget. 2018;9(11):9728–40.
Marin Morales JM, et al. Automated clinical grade expansion of regulatory T cells in a fully closed system. Front Immunol. 2019;10:38.
Arroyo Hornero R, et al. CD45RA distinguishes CD4+CD25+CD127−/low TSDR Demethylated regulatory T cell subpopulations with differential stability and susceptibility to tacrolimus-mediated inhibition of suppression. Transplantation. 2017;101(2):302–9.
Gedaly R, et al. mTOR inhibitor everolimus in regulatory T cell expansion for clinical application in transplantation. Transplantation. 2019;103(4):705–15.
Bergstrom M, et al. Comparing the effects of the mTOR inhibitors azithromycin and rapamycin on in vitro expanded regulatory T cells. Cell Transplant. 2019:963689719872488.
Hippen KL, O'Connor RS, Lemire AM, Saha A, Hanse EA, Tennis NC, et al. In vitro induction of human regulatory T cells using conditions of low tryptophan plus kynurenines. Am J Transplant. 2017;17(12):3098–113.
Guinan EC, Cole GA, Wylie WH, Kelner RH, Janec KJ, Yuan H, et al. Ex vivo costimulatory blockade to generate regulatory T cells from patients awaiting kidney transplantation. Am J Transplant. 2016;16(7):2187–95.
Peters JH, Hilbrands LB, Koenen HJ, Joosten I. Ex vivo generation of human alloantigen-specific regulatory T cells from CD4(pos)CD25(high) T cells for immunotherapy. PLoS One. 2008;3(5):e2233.
Koenen HJ, Fasse E, Joosten I. CD27/CFSE-based ex vivo selection of highly suppressive alloantigen-specific human regulatory T cells. J Immunol. 2005;174(12):7573–83.
Mathew JM, Voss JH, McEwen S, Konieczna I, Chakraborty A, Huang X, et al. Generation and characterization of alloantigen-specific regulatory T cells for clinical transplant tolerance. Sci Rep. 2018;8(1):1136.
Jamali S, et al. Sirolimus vs mycophenolate moftile in tacrolimus based therapy following induction with antithymocyte globulin promotes regulatory T cell expansion and inhibits RORgammat and T-bet expression in kidney transplantation. Hum Immunol. 2019;80(9):739–47.
Scotta C, et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica. 2016;101(1):91–100.
Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64(2):632–43.
Sanchez-Fueyo A, et al. Applicability, Safety And Biological Activity Of Regulatory T Cell Therapy In Liver Transplantation. Am J Transplant. 2019.
Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. 2017;17(11):2945–54.
Mathew JM, H-Voss J, LeFever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. 2018;8(1):7428.
Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. 2008;358(4):353–61.
Kawai T, Sachs DH, Sykes M, Cosimi AB, Immune Tolerance Network. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med. 2013;368(19):1850–2.
Kawai T, Sachs DH, Sprangers B, Spitzer TR, Saidman SL, Zorn E, et al. Long-term results in recipients of combined HLA-mismatched kidney and bone marrow transplantation without maintenance immunosuppression. Am J Transplant. 2014;14(7):1599–611.
Morris H, et al. Tracking donor-reactive T cells: evidence for clonal deletion in tolerant kidney transplant patients. Sci Transl Med. 2015;7(272):272ra10.
Hotta K, et al. Induced regulatory T cells in allograft tolerance via transient mixed chimerism. JCI Insight. 2016;1(10).
Sprangers B, DeWolf S, Savage TM, Morokata T, Obradovic A, LoCascio S, et al. Origin of enriched regulatory T cells in patients receiving combined kidney-bone marrow transplantation to induce transplantation tolerance. Am J Transplant. 2017;17(8):2020–32.
Savage TM, et al. Early expansion of donor-specific Tregs in tolerant kidney transplant recipients. JCI Insight. 2018;3(22).
Matsunami M, Rosales IA, Adam BA, Oura T, Mengel M, Smith RN, et al. Long-term kinetics of intragraft gene signatures in renal allograft tolerance induced by transient mixed chimerism. Transplantation. 2019;103(11):e334–44.
Oura T, Ko DS, Boskovic S, O'Neil JJ, Chipashvili V, Koulmanda M, et al. Kidney versus islet allograft survival after induction of mixed chimerism with combined donor bone marrow transplantation. Cell Transplant. 2016;25(7):1331–41.
Oura T, Hotta K, Rosales I, Dehnadi A, Kawai K, Lee H, et al. Addition of anti-CD40 monoclonal antibody to nonmyeloablative conditioning with belatacept abrogated allograft tolerance despite induction of mixed chimerism. Transplantation. 2019;103(1):168–76.
MacDonald KG, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126(4):1413–24.
Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. 2017;17(4):917–30.
Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. 2017;17(4):931–43.
Pierini A, et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight. 2017:2(20).
Sicard A, Levings MK, Scott DW. Engineering therapeutic T cells to suppress alloimmune responses using TCRs, CARs, or BARs. Am J Transplant. 2018;18(6):1305–11.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Immunology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El-Ayachi, I., Washburn, W.K. & Schenk, A.D. Recent Progress in Treg Biology and Transplant Therapeutics. Curr Transpl Rep 7, 131–139 (2020). https://doi.org/10.1007/s40472-020-00278-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40472-020-00278-y