
Abstract
Purpose of Review
Over the past two decades, significant strides made in our understanding of the etiology of antibody-mediated rejection (AMR) in transplantation have put the complement system in the spotlight. Here, we review recent progress made in the field of pharmacologic complement inhibition in clinical transplantation and aim to understand the impact of this therapeutic approach on outcomes in transplant recipients.
Recent Findings
Encouraged by the success of agents targeting the complement cascade in disorders of unrestrained complement activation like paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS), investigators are testing the safety and efficacy of pharmacologic complement blockade in mitigating allograft injury in conditions ranging from AMR to recurrent post-transplant aHUS, C3 glomerulopathies and antiphospholipid anti-body syndrome (APS). A recent prospective study demonstrated the efficacy of terminal complement inhibition with eculizumab in the prevention of acute AMR in human leukocyte antigen (HLA)-incompatible living donor renal transplant recipients. C1 esterase inhibitor (C1-INH) was well tolerated in two recent studies in the treatment of AMR and was associated with improved renal allograft function.
Summary
Pharmacologic complement inhibition is emerging as valuable therapeutic tool, especially in the management of highly sensitized renal transplant recipients. Novel and promising agents that target various elements in the complement cascade are in development.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Improved understanding of the role of the immune system in allograft rejection, elucidation of the molecular mechanisms underlying graft failure and advances in immunosuppressive therapy have led to significant progress in the field of kidney transplantation in the last 50 years [1]. Kidney transplantation is now considered the treatment of choice for patients with end-stage renal disease (ESRD) since it offers greater long-term survival and improved quality of life at a considerably lower health care cost when compared to dialysis [2,3,4].
In 1969, Patel and Terasaki’s landmark study established that detection of preformed circulating donor reactive cytotoxic antibodies identified patients at risk for developing immediate graft failure due to hyper-acute rejection with a high degree of certainty [5••]. This led to the practice of avoidance of transplantation in patients with donor reactive antibodies detected by the complement dependent cytotoxicity test. Therefore, until the mid-1980s, acute cellular rejection, as opposed to rejection mediated by humoral factors, was considered the major barrier to successful transplantation [6]. The advent of calcineurin inhibitor (CNI)-based maintenance immunosuppression resulted in significant decline in acute rejection rates and a concurrent improvement in graft survival rates [7•]. Today, cellular rejection seldom causes graft loss [6]. However, investigation of contemporary data suggests that these gains have not led to sustained improvement in long-term graft survival [8••]. A similar arc of impressive decades-long progress tempered by intractable long-term allograft attrition rates has also been observed in liver, lung, heart, intestine, and pancreas transplantation [9]. Reasons for the lack of improvement in long-term graft survival remained unclear and most late graft losses were attributed to either chronic allograft nephropathy (CAN) or death with a functioning graft [10]. However, CAN is not a monolithic clinicopathologic entity, but rather a descriptive histologic term for interstitial fibrosis and tubular atrophy that is the culmination of a heterogeneous group of pathologic processes [11]. In 2005, the Banff classification system for renal allograft pathology did away with the non-specific term CAN in order to encourage the recognition of histologic features that signify specific causes of chronic graft dysfunction [12]. It is now apparent that, with adequate clinical and histologic information, most cases of kidney allograft failure can be attributed to specific etiologies and that the origin of interstitial fibrosis/tubular atrophy can be traced to prior insults such as cellular and/or antibody mediated rejection (AMR), BK virus nephropathy and CNI toxicity [13]. Recurrent glomerulonephritis is also recognized as an important cause of allograft loss in those with renal failure due to glomerulonephritis [14]. But it is antibody-mediated injury that appears to be the culprit in a majority of the patients with new-onset late kidney allograft dysfunction [10]. AMR is increasingly being diagnosed in the transplant population [15].
The development of more sensitive and specific methods of detection of donor reactive antibodies by means of flow cytometry cross-matching and solid-phase luminex platform-based assays supported the notion that donor specific antibodies (DSA) were more prevalent than previously appreciated [16, 17]. Discovery of prominent vascular deposition of the complement split product, C4d, in renal allografts undergoing rejection led to the recognition that antibody mediated attack of allograft vascular endothelial cells leads to activation of the classical complement pathway [18••]. Eventually, standardized diagnostic criteria for AMR were added to the Banff classification of renal allograft pathology in 2003 [15]. The presence of covalently bound inert complement split product, C4d in peritubular capillaries is considered a footprint of donor reactive antibody interaction with allograft vascular endothelial cells and is one of the diagnostic criteria for AMR [19].
The role of complement in antibody-mediated allograft injury was highlighted in a study in which the C1q binding DSA were useful in identifying patients at increased risk of kidney allograft loss. In this population-based study, patients who developed complement-binding DSA after transplantation had significantly lower 5-year graft survival when compared with patients with non–complement-binding DSA as well as patients without DSA [20]. Others have suggested that C1q binding activity of DSA is a function of antibody strength and does not distinguish qualitatively different DSA that predict increased risk of AMR [21]. In addition to its role in AMR, recent evidence suggests that the complement cascade modulates T cell allo-immunity and is involved in pathogenesis of DGF mediated by IR injury [22, 23]. There is accumulating evidence that AMR is the dominant cause of late renal allograft failure [24]. In this article, we provide an overview of the existing evidence supporting the use of pharmacologic complement inhibition as a therapeutic strategy in various settings in organ transplantation.
The Complement System
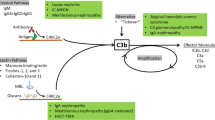
Although complement was identified as a heat-labile fraction in the serum that mediated antibody-dependent bacterial defense, it is now appreciated that the complement cascade consists of more than 30 soluble and cell-bound proteins involved in innate and adaptive immunity. Three pathways of activation of the complement system are recognized—the classical, alternative, and mannose-binding lectin pathways [25]. These pathways all converge to form C3 convertase that cleaves complement protein C3 to C3a and C3b [26]. The classical pathway is activated by cross-linking of antibodies by the C1q, r, s complex. This leads to the cleavage of C4 and C2 and the subsequent assembly of C4b2a. This complex acts as the classical pathway’s C3 convertase and cleaves C3 to C3a and C3b. The alternative pathway is continually active due to the spontaneous hydrolysis (“tick-over”) of C3 to generate C3a and C3b [26]. C3b binds covalently to hydroxyl groups on carbohydrates and proteins in the vicinity. This membrane bound C3b associates with complement factor-B. Subsequent activation of complement factor-B (CFB) by complement factor-D (CFD) leads to the formation of C3bBb which also acts as a C3 convertase [25]. An amplification loop is setup by cleavage of more C3 molecules by C3bBb. Thus, C3 convertase acts as a nodal point in the complement cascade [26]. Association of another C3b molecule with either C3 convertase (C4b2a or C3bBb) leads to the formation of C5 convertase, which cleaves C5 into C5a and C5b. The complement cascade culminates in the activation of the terminal complement pathway and the formation of the C5b–C9 membrane-attack complex (MAC) on cell surfaces that results in cell lysis. The complement cascade is tightly regulated by complement regulatory proteins such as complement factor-H (CFH), complement factor-I (CFI), membrane cofactor protein (CD46), and decay accelerating factor (CD55). These inhibitors prevent injury to normal cells and tissues. Complement dysregulation can result in a variety of pathological processes [22, 25, 26].
Complement Blockade in Disorders of Complement Dysregulation
Intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria (PNH) is a consequence of the absence of GPI-linked complement regulatory protein, CD59. Eculizumab is a recombinant humanized monoclonal antibody that targets terminal complement component C5 [27••]. It is a hybrid IgG2/IgG4 molecule that has been engineered to diminish Fc-mediated properties such as recruitment of inflammatory cells and complement activation [28]. In a randomized, double-blind phase 3 clinical trial in patients with PNH, eculizumab was shown to stabilize hemoglobin levels and eliminate the need for blood transfusion [27••].
In atypical HUS (aHUS), defects in regulation of the complement system result in systemic thrombotic microangiopathy involving the kidney, central nervous system, heart, and gastrointestinal tract. In a phase 2 trial of patients with aHUS, eculizumab treatment was associated with significant time-dependent improvement in renal function [29••]. Eculizumab was originally approved by the FDA for PNH, and now has been approved for the treatment of pediatric and adult patients with aHUS [28, 29••]. Hereditary angioedema (HAE) is an autosomal dominant disorder that is characterized by recurrent episodes of life-threatening angioedema due to C1 esterase inhibitor (C1-INH) deficiency. Two randomized trials that demonstrated the efficacy of nanofiltered C1 inhibitor concentrate from human serum in the treatment and prophylaxis of attacks of angioedema resulted in the approval of C1-INH for use in HAE [30•, 31].
In the past decade, better understanding of the role of the complement system in the pathogenesis of several glomerular diseases has led to refined diagnostic algorithms and approaches to their treatment. An important development has been the reclassification of membranoproliferative glomerulonephritis (MPGN) into immunoglobulin-mediated disease caused by the activation of the classical complement pathway and non-immunoglobulin mediated disease, C3 glomerulopathy, driven by dysregulation of the alternative complement pathway. In C3 glomerulopathy, akin to pathogenic events leading to aHUS, mutations in and/or autoantibodies to complement components that regulate the alternative pathway’s C3 convertase lead to deposition of C3 breakdown products, assembly of MAC and subsequent endothelial injury. Evidence supporting pharmacologic complement inhibition with eculizumab in the therapy of C3 glomerulopathy is scarce and mostly in the form of case reports. Prospective studies are needed to ascertain the efficacy of eculizumab in this disease [32].
Complement Blockade in Post-Transplant Recurrence of Disorders of Complement Dysregulation
aHUS recurs in 60% of patients following transplantation and in this population, graft failure occurs in excess of 90% cases. Given the high likelihood of renal allograft loss due to recurrence of aHUS, renal transplantation is fraught with difficulty. Genetic testing may be predictive of risk of post-transplant recurrence. Mutations in CFH and CFI are associated with a higher risk of recurrence than mutations in MCP [33]. Since complement components and regulatory factors such as C3, CFB, CFH, and CFI are derived from the liver, simultaneous liver and kidney transplantation has been attempted in ESRD patients with aHUS with the rationale that the liver allograft would supply the requisite normal complement components to prevent recurrence of aHUS in the transplanted kidney [34, 35]. However, dual organ transplantation in this setting carries significant risks due to heightened postoperative complement activation and greater complexity of the surgery [34].
Complement blockade may be a viable option for prevention of post-transplant recurrence in patients with aHUS as evidenced by results documented in several pediatric case reports with demonstrable genetic abnormalities in the CFH gene [34, 36,37,38]. Matar et al. reported outcomes in 12 consecutive patients with aHUS at a single-center in a 10-year retrospective study [39•]. Half of these patients had one or more identified genetic mutations associated with aHUS including four patients with CFH mutations. Three fourths of the patients had lost a prior renal allograft to recurrent aHUS. They reported successful prevention of recurrent aHUS in all 4 of these 12 patients who received prophylactic eculizumab. Of these 4 patients, eculizumab was discontinued after 6 months of therapy in 3 patients and one continued to receive eculizumab with an intent for life-long therapy. Three of the 12 patients in this cohort received eculizumab after they developed recurrent aHUS, and one of them experienced graft failure despite this therapy. The authors underscored this variability in treatment response and ascribed it to disparate genetic mutations leading to differences in the phenotype and severity of disease [39•]. However, eculizumab has emerged as a reliable therapeutic agent for both the treatment and prevention of recurrent aHUS involving native as well as transplanted kidneys [28].
In the case of C3 glomerulopathy, which, like aHUS, is also a disorder of alternative complement pathway dysregulation, the role of eculizumab has not been clearly established. In a small series of 6 patients with C3 glomerulopathy, including 3 patients with recurrent disease afflicting renal transplants, Bomback et al. reported outcomes of therapy with 1 year of eculizumab therapy. Clinical and histopathologic improvement was noted in some but not all patients but the authors noted that elevation of serum membrane attack complex (sMAC) prior to treatment may be predictive of response to therapy [40]. Others have reported success with eculizumab in the treatment of aggressive forms of C3 glomerulopathy-dense deposit disease (DDD) with glomerular crescent formation [41]. Close monitoring of renal transplant recipients with a history of C3 glomerulopathy is essential and at this time, there no consensus regarding preventive therapy with eculizumab or any other anti-complement therapy in renal transplant recipients in whom C3 glomerulopathy led to ESRD [28].
Complement Blockade in AMR
In the context of AMR in organ transplantation, vigorous and brisk injury to allograft endothelium occurs by complement activation via the classic pathway by donor reactive antibodies. Success of pharmacologic complement blockade in disorders of complement regulation has heralded interest in the use of similar strategies in prevention and treatment of AMR in kidney transplant recipients [31].
Complement Blockade in the Prevention of AMR in HLA Incompatible Kidney Transplantation
ESRD patients who are sensitized to HLA have a prolonged wait for a transplant and a reduced transplantation rate. In a single center study performed at a high-volume center, live donor renal transplantation after the depletion of donor-specific anti-HLA antibodies was shown to provide significant survival benefit when compared to waiting for a compatible organ. In this study, desensitization was performed using a regimen consisting of plasmapheresis and low dose intravenous immunoglobulin (100 mg/kg) [42••]. These findings were also corroborated in a large multicenter study across varying levels of pre-transplant donor-specific antibody strengths [43••]. In another study, a combination of high-dose intravenous immunoglobulin (1 g/kg) and rituximab was shown to be effective as a desensitization regimen for recipients of living donor or a deceased donor kidney transplant [44]. However, some groups have reported an incidence of early post-transplant AMR as high as 40% in recipients of HLA incompatible kidney transplants. A high percentage of episodes of AMR are difficult to treat and may cause immediate graft loss or delayed transplant glomerulopathy [45].
The efficacy of terminal complement inhibition with eculizumab in HLA incompatible living donor renal transplant recipients was evaluated in a study of 26 highly sensitized patients and 51 historical controls by Stegall et al. [46••]. Both groups received a plasma exchange-based desensitization prior to transplant. The incidence of AMR within 3 months after transplant was significantly lower in the eculizumab group (7.7 vs. 41.2%) [47••]. Examination of outcomes beyond the 1 year in the same cohort of patients reiterated the effectiveness of eculizumab in prevention of acute clinical AMR (6.7 vs. 43.8%) [47••]. All episodes of clinical AMR occurred in the first 3 months post-transplant. However, the eculizumab treated patients did not differ from controls with respect to chronic AMR and death-censored allograft survival. These data suggest that chronic AMR can occur without antecedent acute AMR. Importantly, transplant glomerulopathy and chronic AMR were more common in patients with persistent high levels of DSA. In this study, no planned additional plasma exchange was performed after the first dose of eculizumab and so patients who had strong antibody before the transplant, in many cases, maintained high levels of antibody throughout the study period.
Based on their findings, the authors of the study inferred several plausible mechanisms of chronic AMR. It is conceivable that events preceding the activation of terminal complement elements may be responsible for observed chronic injury. Eculizumab does not prevent the formation of the anaphylatoxins C3a and C4a. This would suggest that blockade of more proximal elements of the complement cascade may avert chronic AMR. Another possible explanation is that sub-lytic levels of the MAC due to low-level activation of C5 might cause endothelial cell activation and chronic injury. Alternatively, chronic AMR could result from complement independent mechanisms: the recruitment of NK cells through the Fcɣ receptor of endothelial bound DSA or the direct activation of endothelial cells by DSA via HLA mediated signaling [47••]. Notably, the safety and efficacy of eculizumab in the prevention of AMR in HLA-sensitized living-donor kidney transplant recipients was studied in a recently concluded randomized, open-label, multicenter, phase 2 clinical trial. However, a composite endpoint of biopsy-proven AMR, graft loss, patient death, or loss to follow-up at week 9 post-transplant did not reach statistical significance. While the primary composite endpoint rate in the eculizumab arm was consistent with rates expected from earlier studies, the rate in the control arm was lower than expected [48•].
C1 esterase inhibitor (C1-INH) inhibits both the classical and lectin pathways of complement activation. It is a serine protease inhibitor, and its ability to inhibit complement activation derives from its ability to prevent the assembly of C1s and C1r in the classic pathway. A phase I/II, placebo-controlled study examined the safety and efficacy of C1-INH in highly sensitized renal transplant recipients for prevention of AMR. Patients underwent desensitization with intravenous immunoglobulin (IVIg) + rituximab ± plasma exchange prior to transplantation; and a total of 20 patients were randomized 1:1 to either C1-INH or placebo arm. No patient in the C1-INH arm developed AMR during the study as compared to the one patient in the placebo arm who developed AMR. Delayed graft function developed less frequently in C1-INH treated patients than those that received the placebo (1 vs. 4). The authors concluded that this data suggests a potential benefit from C1-INH for prevention of AMR [31, 49•].
Complement Blockade in Treatment of AMR
Although a combination of plasmapheresis and low dose IVIg is considered standard of care (SOC) for the treatment of AMR, there is little consensus regarding the details of treatment regimens including the type of replacement fluid or number of sessions of plasmapheresis and the dose or formulation of IVIg [50]. Rituximab, a chimeric monoclonal antibody that targets CD20 and proteasome inhibitor, bortezomib have been used in conjunction with plasmapheresis and/or IVIg for the treatment of AMR [50]. Splenectomy combined with plasmapheresis and low dose IVIg has been reported to be effective as rescue therapy for severe, early AMR in recipients of HLA incompatible live donor renal transplants. In patients experiencing severe AMR, the burden of donor-specific antibodies (DSA) may overwhelm the ability of plasmapheresis to remove antibody and prevent irreversible injury to the allograft. Anti-CD20 therapy with rituximab would also be rendered ineffective in such circumstances since antibody producing plasmablasts and plasma cells do not express CD20. Although splenectomy, by de-bulking plasma cells and rapidly diminishing antibody production may be employed to salvage renal allografts in such dire circumstances, not all patients are good candidates for an invasive procedure such as this [51]. Therefore, there remains an unmet need for more effective medical therapy [52•]. Successful salvage of a renal allograft undergoing AMR refractory to SOC therapy with the use of eculizumab was first reported in 2009 [53•]. Since then, several isolated instances of use of eculizumab as rescue therapy with varying results have been reported [54,55,56,57,58,59]. The heterogeneity of the clinical scenarios of the reported cases and the assortment of therapies employed in each of these instances makes it impossible to establish the efficacy of eculizumab or the lack thereof in the treatment of AMR.
In a retrospective study of 267 consecutive HLA incompatible renal transplant recipients, Orandi et al. compared the efficacy of splenectomy alone (n = 14), eculizumab alone (n = 5), or splenectomy plus eculizumab (n = 5) in rescuing allografts experiencing severe, oliguric AMR. All patients received plasmapheresis and low-dose IVIg in addition to splenectomy and/or eculizumab as part of treatment for AMR. Four out of 14 splenectomy alone patients, 4 out of 5 eculizumab alone patients and none of the splenectomy plus eculizumab patients experienced graft failure. There appeared to be trend toward a greater proportion of patients with transplant glomerulopathy in the former two groups as compared to the splenectomy plus eculizumab group [60].
A prospective, randomized, open-label trial examining the safety and efficacy of eculizumab for the treatment of AMR in kidney transplant recipients was terminated due to lack of demonstrable efficacy of the study drug. The primary outcome that this study set out to measure was the percent change in estimated glomerular filtration rate at 3 months post-treatment [61•].
Two recent studies examined the role of C1-INH in the treatment of AMR [52•, 62•]. Viglietti et al. enrolled 6 patients with acute AMR and allograft dysfunction that was refractory to SOC therapy (high-dose IVIg and rituximab) in a prospective, single-arm pilot study between April 2013 and July 2014. Patients received C1-INH (Berinert, CSL Behring) in addition to high-dose IVIg for a duration of 6 months. The drug was well tolerated, and they demonstrated an improvement in estimated glomerular filtration rate (eGFR) at the 6 month follow-up mark [38.7 +/− 17.9 to 45.2+/− 21.3 mL/min/1.73 m2 (p = 0.0277)]. The authors highlight a significant decrease in prevalence of C1q binding anti-HLA DSA and the proportion of patients with positive C4d staining in peritubular capillaries following therapy with C1-INH. However, C1-INH therapy, in this cohort, did not result in amelioration of key histologic features of AMR such as glomerulitis, peritubular capillaritis, and allograft glomerulopathy [62•]. These findings are akin to those noted by Cornell et al. (cited earlier in this review) in their cohort of HLA incompatible kidney transplant recipients who received eculizumab, to prevent AMR [47••]. These patients developed persistent microcirculatory inflammation and had no improvement in long-term graft survival.
Montgomery et al. also evaluated the use of C1-INH in a recently concluded phase 2, multicenter double-blind randomized placebo-controlled study of 18 patients with AMR (C1-INH n = 9, placebo n = 9). Patients received 20,000 units of C1 INH (CINRYZE, Shire ViroPharma Incorporated) or placebo in divided doses every other day for 2 weeks as adjunct therapy to SOC with plasmapheresis, low-dose IVIg, and/or anti-CD20. The drug was well tolerated with no reported serious adverse events, graft losses or deaths. There was no demonstrable difference between groups with respect to the primary end points of 20-day graft survival or histologic findings. However, the C1-INH group was noted to have a trend toward sustained improvement in renal function [63]. In contrast to the findings of Viglietti, among 14 patients who underwent 6-month allograft biopsies, no transplant glomerulopathy (TG) was seen in any of the seven who received C1-INH [52•, 62•]. Three of the seven placebo patients had TG [52•]. TG portends a poor prognosis and patients with this histologic finding have a significantly reduced graft survival [63]. While this signal suggesting lower TG rates is highly encouraging, larger studies are required before it can be concluded that C1-INH can avert the evolution of TG in patients with AMR. Notable distinctions between these two pilot studies by Viglietti and Montgomery et al. include the use of a different C1 inhibitor product (Berinert vs. CINRYZE) and differing study design (single arm vs. randomized double-blind placebo-controlled trial) [52•, 62•]. The authors of this review have recently initiated a phase 2, single arm, open-label pilot study to further examine the safety and efficacy of Berinert in the treatment of refractory AMR [64].
Complement Blockade in Renal Transplant Recipients with Anti-Phospholipid Syndrome
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by arterial and venous thrombotic events [65]. Obstetric manifestations include recurrent loss of morphologically normal fetuses and premature birth due eclampsia and pre-eclampsia [66]. While it was first described in patients with systemic lupus erythematosus (SLE), it is now recognized that APS can be associated with other autoimmune disorders and can also present as an independent affliction [67]. Sustained presence of antiphospholipid antibodies (aPLs) in high titers plus a thrombotic event involving a single organ system is required to make a diagnosis of APS. Detection of anticardiolipin antibodies or anti-β2-glycoprotein I antibodies by enzyme-linked immunosorbent assay (ELISA) or a positive lupus-anticoagulant assay is required to ascertain the presence of aPL [66]. In patients with APS, in addition to the thrombotic complications, vasculopathy characterized by fibrosis of the intima and media develop in renal vascular beds leading to nephropathy. Indeed, progression to ESRD is common in patients with APS [67]. Survival of this population on dialysis is poor in part because thrombophilia predisposes to vascular access failure [68, 69•]. However, renal transplantation in patients with APS is challenging due to increased risk for thrombosis and allograft failure [69•]. In the setting of renal transplantation, historically, systemic anticoagulation has been the only effective treatment for prevention of APS-associated thrombosis and renal allograft loss, however, it prevents disease recurrence in less than half of the patients [69•]. A potentially lethal, accelerated version of this disorder, called catastrophic antiphospholipid syndrome (CAPS), occurs in less than 1 % of patients with APS. It is characterized by micro-vascular thrombosis in multiple vascular beds (at least 3 within a week’s period) leading to rapid development of multiorgan failure [70]. Infection, trauma, and recent surgery are recognized precipitants of CAPS [66]. Unfortunately, due to the fear of triggering CAPS, associated graft loss and calamitous outcomes following surgery, a history of CAPS has been considered a contraindication to renal transplantation [69•].
Fischetti et al. demonstrated platelet-leukocyte aggregates and thrombotic occlusions in the mesentery of rats receiving aPL immunoglobulin G (IgG) derived from patients with APS [71]. They noted that complement components C3 and C9 colocalized with aPL IgG in the mesenteric vasculature and concluded that aPLs precipitate coagulation in a complement dependent manner [71]. Based on murine studies, pharmacologic targeting of C3 and C5 to treat obstetric complications of APS has been suggested [66]. Lonze et al. described the first successful use of eculizumab in a patient with CAPS and ESRD undergoing live donor renal transplantation [72]. In a subsequent study by the same group, 3 additional patients with APS/CAPS were treated with systemic anticoagulation and eculizumab prior to live donor renal transplantation [69•]. The regimen involved administration of 1200 mg of eculizumab 1 day prior to transplantation followed by 900 mg on postoperative day (POD) 1, and weekly for the first month. Patients continued to receive 1200 mg every 2 weeks thereafter. As is often the case in patients with APS/CAPS, two of these patients were highly sensitized to HLA due to prior blood transfusions and/or failed transplantation and required plasmapheresis prior to and following transplantation to deplete DSA. Since plasmapheresis removes eculizumab from the serum, based on data from pharmacokinetic studies, additional doses of 600 mg were administered immediately following plasmapheresis treatments. All these patients had immediate graft function following transplantation and after follow-up periods ranging from 4 months to 4 years, all have functioning renal allografts with no occurrence of systemic thrombotic events [69•]. Although definitive conclusions favoring the preemptive and long-term therapy with eculizumab cannot be drawn given the diminutive sample size and the observational nature of this case series, the report is encouraging in an otherwise despairing scenario for this patient population.
Complement Blockade in Delayed Graft Function
Multiple disparate definitions have been used for DGF, but it is most frequently defined as suboptimal graft function necessitating dialysis within the first week after transplantation [23]. DGF is reported in nearly 50% of kidney transplant recipients and is associated with greater health care costs and reduced long-term graft survival [73]. Ischemia reperfusion (IR) injury is involved in the pathogenesis of DGF. Tissue hypoxia and ATP depletion that occur at the time of organ procurement and preservation and the subsequent generation of free oxygen radicals upon reperfusion lead to local cytokine release and complement activation. The inflammation that follows, results in tubular injury and renal allograft dysfunction [22]. Zhou et al. studied IR injury in C3, C4, C5, and C6 deficient mice [74]. While C3, C5, and C6 deficient mice were protected from IR injury, neither C4 deficient mice nor mice treated with an antibody to block C5a were protected from IR injury. They concluded that the C5b-9 membrane attack complex is vital to IR injury. Several lines of animal model data provide evidence of local activation of the alternative complement pathway and ensuing inflammation leading to IR injury [22]. In human studies, de Vries et al. noted transient release of soluble C5b-9 from reperfused deceased donor kidneys and not living donor kidneys [75]. Given all this evidence solidifying the role of complement as a vital element in the pathophysiology of IR injury and DGF, two ongoing clinical trials are examining the role of eculizumab in the prevention of DGF in deceased donor renal transplant recipients (NCT02145182, NCT01919346) [76, 77].
Conclusions
Several ongoing clinical trials hope to ascertain the value of available complement inhibitors both in the prevention and treatment of a host of diseases that afflict transplanted organs. Novel agents that target various elements in the complement cascade are in development. These include naturally occurring complement regulatory proteins like soluble MCP and soluble CD59 (sCD59) as well as antibodies and synthetic peptides that target specific proteins like C5a and C5a receptor (C5aR) [78•]. Since the complement system plays an important role in defense against infections and clearance of immune complexes, the real benefits of pharmacologic complement blockade will only be realized by mitigating emergent infections with appropriate prophylactic and/or therapeutic interventions. Escalating financial costs of currently available complement modulatory agents and those in development pose a challenge that needs to be addressed. Nevertheless, pharmacologic complement inhibition promises to be a significant addition to the growing armamentarium of therapeutic tools available to the transplant physician.
Change history
25 November 2017
The article Pharmacologic Complement Inhibition in Clinical Transplantation, written by Vasishta S. Tatapudi and Robert A. Montgomery, was originally published Online First without open access.
References
Papers of particular interest have been highlighted as: • Of importance •• Of major importance
Nankivell BJ, Alexander SI. Rejection of the kidney allograft. N Engl J Med. 2010;363(15):1451–62.
Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med. 1999;341(23):1725–30.
Evans RW, Manninen DL, Garrison Jr LP, Hart LG, Blagg CR, Gutman RA, et al. The quality of life of patients with end-stage renal disease. N Engl J Med. 1985;312:553–9.
United States Renal Data System, 2015 Annual Data Report (Accessed on September 25, 2016 at https://www.usrds.org/2015/download/vol2_11_MedicareExpenditures_15.pdf).
•• Patel R, Terasaki PI. Significance of the positive crossmatch test in kidney transplantation. N Engl J Med. 1969;280(14):735–9. This paper described the importance of detection of preformed donor-specific antibodies in identifying patients at risk of immediate graft failure and established favorable cross-match testing as a pre-requisite to transplantation.
Stegall MD, Chedid MF, Cornell LD. The role of complement in antibody-mediated rejection in kidney transplantation. Nat Rev Nephrol. 2012;8(11):670–8.
• Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, MJ MI, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Engl J Med. 2000;342(9):605–12. This paper depicted the improved outcomes in renal transplantation that resulted from introduction of CNI in the 1980s.
•• Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant. 2004;4(3):378–83. This paper emphasized that improved acute rejection rates due to the advent of CNI did not translate in to improved long-term renal allograft survival and recognized the importance of investigating the etiologies that underlie late graft loss.
Lodhi SA, Lamb KE, Meier-Kriesche HU. Solid organ allograft survival improvement in the United States: the long-term does not mirror the dramatic short-term success. Am J Transplant. 2011;11(6):1226–35.
Gaston RS, Cecka JM, Kasiske BL, Fieberg AM, Leduc R, Cosio FC, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation. 2010;90(1):68–74.
Halloran PF. Call for revolution: a new approach to describing allograft deterioration. Am J Transplant. 2002;2(3):195–200.
Solez K, Colvin RB, Racusen LC, Sis B, Halloran PF, Birk PE, et al. Banff ‘05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN’). Am J Transplant. 2007;7(3):518–26.
El-Zoghby ZM, Stegall MD, Lager DJ, Kremers WK, Amer H, Gloor JM, et al. Identifying specific causes of kidney allograft loss. Am J Transplant. 2009;9(3):527–35.
Briganti EM, Russ GR, McNeil JJ, Atkins RC, Chadban SJ. Risk of renal allograft loss from recurrent glomerulonephritis. N Engl J Med. 2002;347(2):103–9.
Racusen LC, Colvin RB, Solez K, Mihatsch MJ, Halloran PF, Campbell PM, et al. Antibody-mediated rejection criteria–an addition to the Banff ‘97 classification of renal allograft rejection. Am J Transplant. 2003;3(6):708–14.
Karpinski M, Rush D, Jeffery J, Exner M, Regele H, Dancea S, et al. Flow cytometric crossmatching in primary renal transplant recipients with a negative anti-human globulin enhanced cytotoxicity crossmatch. J Am Soc Nephrol. 2001;12(12):2807–14.
Tait BD, Hudson F, Cantwell L, Brewin G, Holdsworth R, Bennett G, et al. Review article: Luminex technology for HLA antibody detection in organ transplantation. Nephrology (Carlton). 2009;14(2):247–54.
•• Feucht HE, Felber E, Gokel MJ, Hillebrand G, Nattermann U, Brockmeyer C, et al. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin Exp Immunol. 1991;86(3):464–70. This paper was the first to report the deposition of C4d in renal allografts undergoing rejection and recognized that it signifies activation of complement by DSA.
Haas M, Sis B, Racusen LC, Solez K, Glotz D, Colvin RB, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant. 2014;14(2):272–83.
Loupy A, Lefaucheur C, Vernerey D, Prugger C, Duong van Huyen JP, Mooney N, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med. 2013;369(13):1215–26.
Yell M, Muth BL, Kaufman DB, Djamali A, Ellis TM. C1q binding activity of de novo donor-specific HLA antibodies in renal transplant recipients with and without antibody-mediated rejection. Transplantation. 2015;99(6):1151–5.
Cravedi P, Heeger PS. Complement as a multifaceted modulator of kidney transplant injury. J Clin Invest. 2014;124(6):2348–54.
Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant. 2011;11(11):2279–96.
Sellarés J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, Hidalgo LG, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12(2):388–99.
Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344(14):1058–66.
Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis-a new look at an old entity. N Engl J Med. 2012;366(12):1119–31.
•• Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–43. This paper describes the clinical trial that established the safety and efficacy of eculizumab in the treatment of PNH and led to the approval of this drug by the FDA for this indication.
Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. French study group for aHUS/C3G. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643–57.
•• Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–81. This paper describes the clinical trial that established the safety and efficacy of eculizumab in the treatment of aHUS and led to the approval of this drug by the FDA for this indication.
• Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363(6):513–22. This paper describes the clinical trial that established the safety and efficacy of C1 inhibitor in the treatment of HAE and led to the approval of this drug by the FDA for this indication.
Jordan SC, Choi J, Kahwaji J, Vo A. Complement inhibition for prevention and treatment of antibody-mediated rejection in renal allograft recipients. Transplant Proc. 2016;48(3):806–8.
Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012;8(11):634–42.
Bresin E, Daina E, Noris M, Castelletti F, Stefanov R, Hill P, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1(1):88–99.
Nester C, Stewart Z, Myers D, Jetton J, Nair R, Reed A, et al. Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2011;6(6):1488–94.
Remuzzi G, Ruggenenti P, Codazzi D, Noris M, Caprioli J, Locatelli G, et al. Combined kidney and liver transplantation for familial haemolytic uraemic syndrome. Lancet. 2002;359(9318):1671–2.
Zimmerhackl LB, Hofer J, Cortina G, Mark W, Würzner R, Jungraithmayr TC, Khursigara G, et al. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med. 2010;362(18):1746–8.
Weitz M, Amon O, Bassler D, Koenigsrainer A, Nadalin S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr Nephrol. 2011;26(8):1325–9.
Krid S, Roumenina LT, Beury D, Charbit M, Boyer O, Frémeaux-Bacchi V, et al. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am J Transplant. 2012;12(7):1938–44.
• Matar D, Naqvi F, Racusen LC, Carter-Monroe N, Montgomery RA, Alachkar N. Atypical hemolytic uremic syndrome recurrence after kidney transplantation. Transplantation. 2014;98(11):1205–12. This paper reports one of the largest series of renal transplant recipients with aHUS and details a single center’s experience with eculizumab in patients with post-transplant recurrence of aHUS.
Bomback AS, Smith RJ, Barile GR, Zhang Y, Heher EC, Herlitz L, et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol. 2012;7(5):748–56.
McCaughan JA, O'Rourke DM, Courtney AE. Recurrent dense deposit disease after renal transplantation: an emerging role for complementary therapies. Am J Transplant. 2012;12(4):1046–51.
•• Montgomery RA, Lonze BE, King KE, Kraus ES, Kucirka LM, Locke JE, et al. Desensitization in HLA-incompatible kidney recipients and survival. N Engl J Med. 2011;365(4):318–26. First published single-center data demonstrating the superiority of HLA-incompatible transplantation to remaining on the wait-list in highly sensitized ESRD patients.
•• Orandi BJ, Luo X, Massie AB, Garonzik-Wang JM, Lonze BE, Ahmed R, et al. Survival benefit with kidney transplants from HLA-incompatible live donors. N Engl J Med. 2016;374(10):940–50. Multicenter data demonstrating the superiority of HLA-incompatible transplantation to remaining on the waiting-list in highly sensitized ESRD patients. This study corroborated findings of the aforementioned single-center study (reference 44).
Vo AA, Lukovsky M, Toyoda M, Wang J, Reinsmoen NL, Lai CH, et al. Rituximab and intravenous immune globulin for desensitization during renal transplantation. N Engl J Med. 2008;359(3):242–51.
Burns JM, Cornell LD, Perry DK, Pollinger HS, Gloor JM, Kremers WK, et al. Alloantibody levels and acute humoral rejection early after positive crossmatch kidney transplantation. Am J Transplant. 2008;8(12):2684–94.
•• Stegall MD, Diwan T, Raghavaiah S, Cornell LD, Burns J, Dean PG, et al. Terminal complement inhibition decreases antibody-mediated rejection in sensitized renal transplant recipients. Am J Transplant. 2011;11(11):2405–13. This is the first study to suggest that eculizumab could lower incidence of acute AMR in recipients of HLA-incompatible kidney transplants.
•• Cornell LD, Schinstock CA, Gandhi MJ, Kremers WK, Stegall MD. Positive crossmatch kidney transplant recipients treated with eculizumab: outcomes beyond 1 year. Am J Transplant. 2015;15(5):1293–302. This study, involving long-term follow-up of HLA-incompatible kidney transplant recipients who received prophylactic eculizumab (reference 47), noted that despite lowering rates of early acute AMR, terminal complement blockade did not decrease rates of chronic AMR in patients with persistent DSA.
• Alexion provides update on phase 2 clinical trial with eculizumab in antibody-mediated rejection (amr) in living-donor kidney transplant recipients alexion provides update on phase 2 clinical trial with eculizumab in antibody-mediated rejection (amr) in living-donor kidney transplant recipients Alexion Pharmaceuticals. http://news.alexionpharma.com/press-release/company-news/alexion-provides-update-phase-2-clinical-trial-eculizumab-antibody-mediat. This is the first randomized-controlled trial to examine the role of eculizumab in prevention of AMR in HLA-incompatible kidney transplant recipients. However, this study was unable to demonstrate such benefit from eculizumab.
• Vo AA, Zeevi A, Choi J, Cisneros K, Toyoda M, Kahwaji J, et al. A phase I/II placebo-controlled trial of C1-inhibitor for prevention of antibody-mediated rejection in HLA sensitized patients. Transplantation. 2015;99(2):299–308. This is the first randomized-controlled trial to examine the role of C1-inhibitor in prevention of AMR in HLA-incompatible kidney transplant recipients.
Archdeacon P, Chan M, Neuland C, Velidedeoglu E, Meyer J, Tracy L, et al. Summary of FDA antibody-mediated rejection workshop. Am J Transplant. 2011;11(5):896–906.
Locke JE, Zachary AA, Haas M, Melancon JK, Warren DS, Simpkins CE, et al. The utility of splenectomy as rescue treatment for severe acute antibody mediated rejection. Am J Transplant. 2007;7(4):842–6.
• Montgomery RA, Orandi BJ, Racusen L, Jackson AM, Garonzik-Wang JM, Shah T, et. al. Plasma-derived C1 esterase inhibitor for acute antibody-mediated rejection following kidney transplantation: results of a randomized double-blind placebo-controlled pilot study. Am J Transplant. 2016 This is the first randomized clinical trial to test the safety and efficacy of C1 inhibitor to treat AMR.
• Locke JE, Magro CM, Singer AL, Segev DL, Haas M, Hillel AT, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant. 2009;9(1):231–5. This is the first report of use of eculizumab to salvage a renal allograft undergoing severe AMR that was refractory to SOC.
Chandran S, Baxter-Lowe L, Olson JL, Tomlanovich SJ, Webber A. Eculizumab for the treatment of de novo thrombotic microangiopathy post simultaneous pancreas-kidney transplantation—a case report. Transplant Proc. 2011;43(5):2097–101.
Jackson AM, Kuperman MB, Montgomery RA. Multiple hyperacute rejections in the absence of detectable complement activation in a patient with endothelial cell reactive antibody. Am J Transplant. 2012;12(6):1643–9.
Noone D, Al-Matrafi J, Tinckam K, Zipfel PF, Herzenberg AM, Thorner PS, et al. Antibody mediated rejection associated with complement factor h-related protein 3/1 deficiency successfully treated with eculizumab. Am J Transplant. 2012;12(9):2546–53.
Stewart ZA, Collins TE, Schlueter AJ, Raife TI, Holanda DG, Nair R, et al. Case report: eculizumab rescue of severe accelerated antibody-mediated rejection after ABO-incompatible kidney transplant. Transplant Proc. 2012;44(10):3033–6.
Kocak B, Arpali E, Demiralp E, Yelken B, Karatas C, Gorcin S, et al. Eculizumab for salvage treatment of refractory antibody-mediated rejection in kidney transplant patients: case reports. Transplant Proc. 2013;45(3):1022–5.
Burbach M, Suberbielle C, Brochériou I, Ridel C, Mesnard L, Dahan K, et al. Report of the inefficacy of eculizumab in two cases of severe antibody-mediated rejection of renal grafts. Transplantation. 2014;98(10):1056–9.
Orandi BJ, Zachary AA, Dagher NN, Bagnasco SM, Garonzik-Wang JM, Van Arendonk KJ, et al. Eculizumab and splenectomy as salvage therapy for severe antibody-mediated rejection after HLA-incompatible kidney transplantation. Transplantation. 2014;98(8):857–63.
• https://clinicaltrials.gov/ct2/show/record/NCT01895127?term=eculizumab+antibody+mediated+rejection&rank=3. This is the first randomized-controlled trial examining the safety and efficacy of eculizumab in the treatment of AMR. The study was terminated due to lack of demonstrable efficacy of the study drug.
• Viglietti D, Gosset C, Loupy A, Deville L, Verine J, Zeevi A, et al. C1 inhibitor in acute antibody-mediated rejection nonresponsive to conventional therapy in kidney transplant recipients: a pilot study. Am J Transplant. 2016;16(5):1596–603. This is one of the first studies to investigate the role of C1 inhibitor in the treatment of AMR.
Wavamunno MD, O'Connell PJ, Vitalone M, Fung CL, Allen RD, Chapman JR, et al. Transplant glomerulopathy: ultrastructural abnormalities occur early in longitudinal analysis of protocol biopsies. Am J Transplant. 2007;7(12):2757–68.
https://clinicaltrials.gov/ct2/show/NCT02936479?term=berinert+lonze&rank=1
Cervera R, Piette JC, Font J, Khamashta MA, Shoenfeld Y, Camps MT, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum. 2002;46(4):1019–27.
Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–44.
Canaud G, Bienaimé F, Tabarin F, Bataillon G, Seilhean D, Noël LH, et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N Engl J Med. 2014;371(4):303–12.
Salmela B, Hartman J, Peltonen S, Albäck A, Lassila R. Thrombophilia and arteriovenous fistula survival in ESRD. Clin J Am Soc Nephrol. 2013;8(6):962–8.
• Lonze BE, Zachary AA, Magro CM, Desai NM, Orandi BJ, Dagher NN, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14(2):459–65. This paper is the first to describe the successful use of eculizumab in the prevention of recurrent APS and/or CAPS in a small series of patients.
Cervera R, Bucciarelli S, Plasín MA, Gómez-Puerta JA, Plaza J, Pons-Estel G, et al. Catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the “CAPS Registry”. J Autoimmun. 2009;32(3–4):240–5.
Fischetti F, Durigutto P, Pellis V, Debeus A, Macor P, Bulla R, et al. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood. 2005;106(7):2340–6.
Lonze BE, Singer AL, Montgomery RA. Eculizumab and renal transplantation in a patient with CAPS. N Engl J Med. 2010;362(18):1744–5.
Niemann CU, Feiner J, Swain S, Bunting S, Friedman M, Crutchfield M, et al. Therapeutic hypothermia in deceased organ donors and kidney-graft function. N Engl J Med. 2015;373(5):405–14.
Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, et al. Predominant role for C5b-9 in renal ischemia/reperfusion injury. J Clin Invest. 2000;105(10):1363–71.
de Vries DK, van der Pol P, van Anken GE, van Gijlswijk DJ, Damman J, Lindeman JH, et al. Acute but transient release of terminal complement complex after reperfusion in clinical kidney transplantation. Transplantation. 2013;95(6):816–20.
https://clinicaltrials.gov/ct2/results?term=NCT02145182&Search=Search
https://clinicaltrials.gov/ct2/show/NCT01919346?term=NCT01919346&rank=1
• Wagner E, Frank MM. Therapeutic potential of complement modulation. Nat Rev Drug Discov. 2010;9(1):43–56. This review highlights various complement targeted therapies that are in development.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Robert Montgomery received research grants from Alexion (manufacturer of Soliris–Eculizumab) and Shire ViroPharma; served as a paid consultant for Alexion, Shire ViroPharma and CSL, Behring; and received travel honoraria from Alexion and Shire VioPharma during the conduct of this study. He served on advisory boards for Genentech Scientific/ROCHE, True North/iPerian, Novartis, and Hansa Medical; received consulting feels from OrbidMed, GuidePoint Global, Sucampo, Astellas, and Shire; and received research grants from Immune Tolerance Network, ViroPharma, Hansa and Alexion. Vasishta Tatapudi declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Kidney Transplantation
A correction to this article is available online at https://doi.org/10.1007/s40472-017-0174-5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tatapudi, V.S., Montgomery, R.A. Pharmacologic Complement Inhibition in Clinical Transplantation. Curr Transpl Rep 4, 91–100 (2017). https://doi.org/10.1007/s40472-017-0148-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40472-017-0148-7