
Abstract
Alcohol-use disorders (AUDs) are a major public health concern in the United States. To better understand the etiology of alcohol dependence and to identify physiological and behavioral markers that predict alcohol use progression, research has focused on linking diagnostic phenotypes with genetic variation. In recent years, neurobiological endophenotypes have largely surpassed clinical symptoms as the major phenotypes of interest, because they are typically more proximal to underlying genetic mechanisms, and can help to fill the gaps between genetic variation and clinical diagnosis. To date, numerous useful neurobiological endophenotypes for alcohol dependence have been uncovered, including those related to reward dysregulation, impulsivity, and subjective response to alcohol, In general, further work is needed to demonstrate direct associations between AUD endophenotypes and specific genetic variation. Future research would also benefit from applying a theoretical framework emphasizing the shifting imbalance between reward and control networks that occurs during the typical progression from recreational drinking to alcohol dependence. Identifying endophenotypes characteristic of different stages of addiction could have important diagnostic and treatment implications.
Similar content being viewed by others


Introduction
Alcohol-use disorders (AUDs) are an economically burdensome and widespread problem, with lifetime prevalence an estimated 18 % in the United States [37]. AUDs cost approximately $235 billion per year, accounting for approximately 13 % of annual health-care costs, or 2.7 % of the total gross domestic product [86]. In addition to the financial impact, numerous deleterious health outcomes are associated with alcohol misuse, including cardiovascular disease, cancer, liver disease, and damage to brain structure and function [76,86,102]. Given the substantial heterogeneity among AUDs in terms of clinical symptoms and response to treatment [43], research in this field is tending towards development of a more nuanced understanding of their etiology and maintenance.
Numerous studies over the last 30 years have suggested that risk for AUDs is at least partially determined by genetics—specifically, AUD risk seems to be 50–60 % heritable [38,53,81,91]. Although genetic variation is a crucial differentiating factor [40], current research has yet to characterize genetic variations that can be used to effectively identify at-risk individuals or prescribe effective personalized treatments. Because of the difficulty of linking genetic variation to complex diagnostic phenotypes, the field has shifted its focus to delineating “intermediate” phenotypes, in the hope they might simplify the connection between genes, neurobiological mechanisms, and the development and maintenance of AUDs [66,84].
The term “intermediate phenotype” is generally indicative of a mechanism that mediates the effect of genetic variation on a more distal clinical phenotype. Intermediate phenotypes can relate to a broad range of observable behavior, including behavior patterns (i.e., repeated failed efforts to control drinking) that are not necessarily a form of diagnosed psychopathology. The “endophenotype” concept was first used over 30 years ago [36], to describe particular intermediate phenotypes ranging from neuroanatomical features to metabolic processes to psychological characteristics to electrophysiological or hemodynamic brain responses. These physiological markers are more proximal to genes than are diagnostic phenotypes, and can help form the link between upstream genes and downstream phenotypes. Use of endophenotypes in psychiatric genetics research may aid diagnosis, classification, treatment, and the development of animal models [35]. Moreover, better understanding of these components could enable the development of more customized and efficacious treatment options, including improved pharmacotherapy for AUDs [43,66].
It should be noted that the commonly used term “biomarker” refers to any “endogenous, measurable characteristic that indicates either risk for or manifestation of a psychiatric illness” [5], whereas endophenotypes—an important subtype of biomarkers—are subject to a more specific set of defining criteria. Cannon & Keller [17] have outlined the characteristics that must be present for a behavior or response to be considered a genetically informative endophenotype:
-
1.
it must be heritable;
-
2.
it must be associated with the causes rather than the effects of a disorder (i.e., the level of the endophenotype should enable prediction of the individual level of genetic risk, and the endophenotype should be expressed in the “deviant range” before and after an individual manifests a particular disorder);
-
3.
it must be less complex than its associated disorder (and numerous endophenotypes may be related to a complex disorder);
-
4.
it must vary continuously in the general population;
-
5.
it should be measureable across more than one level of analysis; and
-
6.
endophenotypes that affect multiple disorders should be found for genetically related disorders [17].
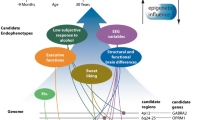
It may also be useful to add an additional criterion, namely, that the endophenotype should fit into a conceptual model for which prior data already provide strong theoretical support. For instance, several commonly accepted models of addiction suggest that substance-use disorders are maintained, in part, through an imbalance between an individual’s incentive reward network [48], which creates the urge to use a substance, and the control network [6], which affects whether these impulses are acted upon [43,44]. Briefly, the incentive network refers to structures involved in reward and/or reinforcement, including the ventral tegmental area (VTA), the nucleus accumbens, the thalamus, the insula, and the amygdala [29,52,57,69], and the control network refers to structures involved in executive functioning, including the inferior frontal gyrus (IFG), the orbitofrontal cortex (OFC), and the dorsolateral prefrontal cortex (dlPFC) [12,20,52]. Thus, useful endophenotypes might examine the balance between reward and control networks.
Relatedly, the three-stage model [56] suggests that addiction develops in three stages:
-
binge/intoxication;
-
withdrawal/negative effect; and
-
preoccupation/anticipation
and that this progression is accompanied by neurobiological adaptations that promote the addicted state. Recently, Karoly and colleagues [52] have extended the three-stage theoretical model to demonstrate the shifting imbalance between reward and control networks across each stage of the addiction cycle, as an individual progresses from recreational use to an addicted (dependent) neural state. Briefly, the extended model states that during stage 1 (the “binge/intoxication” stage), the reward network seems to be dysregulated with repeated drug use, such that responses to drug-related rewards increase whereas the incentive value of natural reinforcers decreases [4,27,74,88]. Repeated drug use is associated with an increase in connectivity between regions involved in negative effect and withdrawal [13,68,75,99]. These changes lead to a downstream strengthening of reward areas in response to drug-related cues, and concurrent weakening of control regions [45,108], which serves to perpetuate compulsive substance use, characteristic of the “preoccupation/anticipation” stage. An example of an endophenotype derived from the three-stage model might involve targeting neurobiological adaptations characteristic of the “withdrawal/negative effect” stage, and might measure brain activation in response to visual alcohol cues among heavy drinkers undergoing acute or protracted withdrawal (additional examples are given by [52].
One notable concern regarding the development of endophenotypes for complex psychological disorders is the issue of disorder specificity. Although criterion 6, above, notes that an endophenotype can affect several disorders if those disorders are genetically related, an endophenotype associated with numerous forms of psychopathology (i.e., executive functioning deficits) may have less clinical utility. For AUDs, one possible solution to this problem is testing the “gene X alcohol exposure” interaction for a given endophenotype. If a particular gene alters the effect of alcohol exposure on some endophenotype (i.e., executive control) this interaction would suggest a more specific genetic link between the endophenotype and the disorder. Further, longitudinal studies examining whether a given endophenotype affects future alcohol use or treatment outcome could indicate the presence of a causal relationship between the endophenotype and AUDs.
Endophenotypes for AUDs
Given the complex biopsychosocial etiology of AUDs [70,109] and the significant individual variability in subjective responses to alcohol consumption [23,82], much research effort over the past 10–15 years has attempted to uncover endophenotypes for AUDs [8]. Early work focused on hypotheses related to electrophysiology and alcohol metabolism. Electrophysiological endophenotypes include cortical synchronization [107], the P300 component of the event-related potential [18,80] and event-related brain oscillations during cognitively demanding tasks [49]. Individual differences in alcohol metabolism—the rate at which alcohol is converted to acetaldehyde—is also predictive of disordered alcohol use, given that accumulation of acetaldehyde affects alcohol sensitivity and subjective drinking experience [41]. Much other evidence has linked variation in alcohol-metabolism genes to AUD risk [63,106]. However, although these early endophenotypes are genetically informative and clinically relevant, they are less applicable in the context of more recent conceptual models of loss of control, reward dysfunction and alcohol dependence. Therefore, electrophysiology and alcohol metabolism research will not be reviewed comprehensively here.
In this review, we will discuss three major categories of AUD endophenotypes, noting advantages and limitations of each, and, when appropriate, identifying the stage(s) within our theoretical model of addiction in which each endophenotype is likely to confer AUD risk. Broadly, we will focus on endophenotypes related to:
-
1.
craving and reward dysregulation;
-
2.
impulsivity; and
-
3.
subjective response to alcohol (Table 1).
Table 1 Proposed endophenotypes related to three AUD-relevant hypotheses
We will then further discuss the development of theoretically derived endophenotypes for AUDs, and comment on the limitations of current endophenotypes in the context of such models. Finally, we will suggest theoretically derived endophenotypes that simultaneously probe reward and control networks to overcome some of these limitations.
Craving and Reward Dysregulation
Much evidence suggests that neural reward processing that promotes craving in response to drug-relevant cues is altered in substance abusers [64,99,108]. PET imaging studies have revealed that among addicted individuals, impaired dopamine signaling is associated with compulsive drug use, as drug rewards increase in value while natural rewards become less reinforcing [105]. Dysfunctional adaptations in neural reward networks tend to worsen as addiction progresses [58], probably involving numerous brain areas and neurotransmitter systems. For example, while dopaminergic dysfunction seems to promote the loss of control observed in the initial stages of addiction, impaired glutamatergic projections to the nucleus accumbens have been implicated in the reward dysfunction characteristic of later stages of addiction [48]. Further, disturbances in different aspects of reward processing may be a pre-morbid risk factor predisposing individuals to the development of AUDs [79,97•]. Thus, dysfunction within neural reward pathways has been a crucial target for investigation as a possible AUD endophenotype.
Accordingly, neuroimaging research by our group has attempted to identify activation related to craving and AUD severity [19••]. This study demonstrated that brain activation in reward regions (including the nucleus accumbens, amygdala, precuneus, insula, and dorsal striatum) elicited by exposure to the taste of a favorite alcohol beverage was predictive of AUD severity. This study also found activation in brain areas associated with craving, including the prefrontal cortex, striatum, and ventral tegmental area, and this activation was positively correlated with measures of alcohol problems. Increased neural responses to alcohol taste cues within the mesocorticolimbic pathway have been linked to variation in two genes, DRD4 and OPRM1, which regulate receptors within the same pathway [28]. Similar activation was found in a study using olfactory alcohol cues among high-risk drinkers [50], and activation in reward and craving areas (i.e., the prefrontal cortex, ventral striatum, thalamus, and insula) after presentation of alcohol images was greater for alcohol-dependent subjects than for healthy control subjects [33,39,46•,73].
Taken together, this work suggests that brain activation in response to alcohol cues is likely to be a useful biomarker for AUDs, and dysfunctional craving and/or reward-responsivity is likely to confer significant risk in the binge/intoxication and preoccupation/anticipation stages of addiction. Additional work should implement cue-elicited craving paradigms among individuals experiencing acute or protracted alcohol withdrawal, to examine the role of craving and reward-responsivity during the withdrawal/negative effect stage. In addition, to determine whether this biomarker is a true endophenotype, studies should examine neural activation in response to alcohol cues among the unaffected siblings of individuals with AUDs. As far as we are aware, no studies have yet tried to directly answer this question. However, one study of individuals who are family history-positive for alcoholism demonstrated altered neural reward processing among unaffected family members [2•]. This finding is promising initial support for craving and/or reward-responsivity as a true endophenotype for AUDs.
Impulsivity
In response-inhibition tasks impulsive responses and poor performance tend to be characteristics of heavy drinkers [11,59,71]. The term “impulsivity” involves both the ability to inhibit pre-potent responses (a measure of “stopping impulsivity”), when necessary, and the ability to determine the subjective value of delayed rewards (a measure of “waiting impulsivity”) [21]. Although AUDs are likely to be related to several deficits in executive functioning, impulsivity is a particularly useful endophenotype because it is a heritable [9], pre-morbid risk factor [22] present in AUD sufferers’ unaffected siblings [26], and it appears to worsen with increased AUD severity [59]. Neuroimaging research has leveraged several effective impulsivity tasks in an attempt to characterize the altered neural activation patterns observed among heavy drinkers and those at risk for AUDs
First, neural and behavioral responses to stop–signal task (SST; [62]) paradigms (i.e., “stopping impulsivity”) may be a particularly informative endophenotype for AUDs. Altered processing during stop–signal inhibition is observed more often among alcohol-dependent individuals than among healthy controls, and higher levels of in-task craving is predictive of impaired frontal activation [61], which suggests that response to this task may be particularly informative during the preoccupation/anticipation stage. In addition, intravenous infusion of alcohol during an SST significantly reduced stop-trial minus go-trial activation in the right PFC among family history-negative but not family history-positive subjects [51••], which suggests that stopping impulsivity may also confer a significant risk at the binge/intoxication stage. Further support for this hypothesis is provided by studies that demonstrate effects of acute intoxication on stop–signal inhibition [30,72], but not other aspects of task performance, for example reaction time. The Kareken study also indicates that genetic variation may affect individual differences in SST performance and neural activation. As far as we are aware, particular genes related to stop–signal inhibition have not yet been identified. Determining how specific genes affect SST response would lend additional support to its utility as an AUD endophenotype.
In addition, behavioral response to delayed reward discounting (DRD) tasks (i.e., “waiting impulsivity”) has received substantial support as a behavioral AUD endophenotype [65], and more recent fMRI research has largely corroborated this view. In a study of heavy drinkers, greater discounting of delayed monetary rewards during an fMRI task and greater activation in the supplementary motor area, insula, inferior frontal gyrus, and precuneus were observed for subjects with more severe clinical symptoms [20]. Importantly, abnormally enhanced temporal discounting seems to persist even among abstinent alcohol-dependent subjects [71], and seems to be at least partially genetically mediated. In particular, DRD has been linked to genetic variation in the COMT gene [100], and DRD may confer AUD risk from family history [78]. Studies examining the effects of acute alcohol administration on DRD have furnished inconsistent results [24,77,87], so further research is needed to determine which stage(s) of the cycle may be affected by DRD.
Finally, recent neuroimaging work targeting resting-state connectivity in AUDs has uncovered another potential endophenotype related to inhibitory control. Resting state connectivity is a measure of the brain’s functional organization based on communication between brain regions [31]. Alcohol-dependent individuals in early abstinence who later relapsed had significantly less connectivity in executive control networks than those who remained abstinent, and this decreased connectivity was associated with deficits in a response-inhibition task [14]. In contrast, individuals in long-term abstinence had reduced connectivity in limbic reward areas and increased connectivity in executive control regions [15]. Evidently, as individuals progress from short to long-term abstinence, resting state connectivity progressively decreases within the reward network and increases within the control network [16] which is consistent with our theoretical model, which posits that these changes occur in the opposite direction during the progression from recreational use to addiction. Further, a recent study using an alcohol-cue reactivity (rather than a resting state study) paradigm demonstrated altered functional connectivity in abstinent individuals who later relapsed compared with those who remained abstinent [7]. Alcohol intoxication also seems to affect functional connectivity acutely [54]. Taken together, this evidence suggests that both resting-state and task functional connectivity data may be useful for predicting treatment outcome and assessing AUD progression and severity.
Subjective Response to Alcohol
Subjective responses to alcohol depend on how alcohol-relevant neural pathways are organized within an individual [84], and subjective response to the acute effects of alcohol is known to be a major determinant of individual risk of developing an AUD [93,96]. More specifically, it seems that alcohol causes greater stimulating (rewarding) effects and lower sedative (unpleasant) effects in heavy compared with light drinkers, and these subjective effects predict future alcohol consumption [55]. Higher alcohol-induced stimulation is, thus, a useful AUD vulnerability marker, given its association with greater reinforcement and increased consumption [60].
Subjective responses to alcohol are heritable [95,104], and differences in subjective response to alcohol have been linked to variation in particular genes [85,94]. Ray & Hutchison [83] found that individuals with at least one copy of the A118G allele had greater response to subjective intoxication, sedation, stimulation, and changes in mood states. Level of response to alcohol has also been significantly associated with two SNPs—rs1051730 and rs8034191—within the 15q25.1 region [47], and Hendershot et al. [40] showed that alcohol sensitivity mediated the effects of the ALDH2 genotype on drinking behavior.
One particularly well-studied construct is the “low level of response” to alcohol (LR) endophenotype, which has been consistently supported as an AUD risk marker. Briefly, LR refers to the genetically affected trait that leads some individuals (low responders) to require higher doses of alcohol to obtain the desired effect [90,92]. LR seems to be heritable [91], present in unaffected family members [25], and genetically mediated [42,47]. A recent fMRI study identified differential activation in the inferior frontal and cingulate regions associated with LR [103], and continued investigation of potential neural correlates of LR is warranted. Evidently, subjective responses to alcohol are affected by numerous interrelated factors, including alcohol pharmacodynamics and metabolism, and the obvious environmental, social, and/or psychological considerations discussed elsewhere [98], and are thus likely to confer risk differentially throughout the addiction cycle [84].
Future Directions in Theoretically-Derived Endophenotype Development
The extent to which the reward network overpowers the control network at a given time theoretically determines whether an individual will act upon the craving for, or urge to use, a substance. Accordingly, the strongest determinant of loss of control over substance use may be the different strength of these two networks [4,10,34]. We propose that an informative endophenotype for AUD should fit within a theoretical framework emphasizing the emergence of a network imbalance in which the increasingly dysregulated reward network (which can become overly responsive to alcohol-related rewards after repeated alcohol use) eventually overpowers the weakened control network, thus promoting alcohol misuse by some individuals. Such a “reward–control–endophenotype” could be refined even further in the context of the three-stage model, given that the balance between reward and control networks shifts as addiction progresses [52]. Although study of the endophenotypes proposed above have clarified several AUD vulnerability factors that confer different risk at different stages of addiction, further research in this area is needed. Neuroimaging of endophenotypes focusing on the relative strengths of the reward and control networks could have significant diagnostic utility in terms of characterizing neural markers of disorder progression by identifying the stage of addiction of an individual at a given time.
Improved AUD Endophenotypes: Reward and Control
In the context of this theoretical model, it should be noted that some of the existing endophenotypes for AUDs are limited in their ecological validity—this is particularly true of tasks that use alcohol cues to probe the reward network, but fail to simultaneously access the control system because the individual is not asked to inhibit behavior while the cue is being presented. In “real life” situations in which an individual attempts to control his or her drinking, cue-induced craving places high demands on both reward and control systems [1], as the individual decides whether or not to drink in response to alcohol cues. For this reason, fMRI tasks aimed at defining an endophenotype for AUDs should ideally involve simultaneous alcohol cue presentation and an inhibitory control task. Implementation of an fMRI task aimed at characterizing the neural basis for the reward vs. control network imbalance could help to classify individuals in terms of AUD severity, and may have implications for optimizing personalized treatments.
For this reason, our group is developing an fMRI task designed to identify neural activation patterns that characterize the extent to which dysregulated reward processing in response to acute alcohol cue exposure compounds the behavioral effects of impaired response inhibition. This task combines the standard SST paradigm with visual alcohol and control cues. We hypothesize that our task will elicit differential neural activation during stop–signal inhibition, depending upon which cue is presented, and that this difference will depend on severity of alcohol dependence. Similarly, Fryer et al. [32••] have implemented a visual–oddball paradigm using alcohol and control images, and have used this task to categorize altered neural activation patterns throughout the stages of addiction.
In general, existing reward-based decision-making tasks [67] have proved useful for examining the relationship between control processes and specific aspects of reward (i.e., reward prediction, anticipation, etc.). For instance, gambling paradigms have been useful for examining cognitive control and reward expectation [89] and delay discounting tasks have been used to study impulsivity in the context of reward anticipation [101]. However, a task that taxes control systems while simultaneously activating the reward network using craving-inducing cues (rather than the real or imaginary monetary rewards typically used in reward-based decision-making tasks) may have particular relevance for better understanding AUDs, given the clinical implications of altered neural responses to alcohol cues, and the relationship between cue-induced craving and relapse [99]. Ultimately, such a task could be further specified to examine control over drinking behavior itself, perhaps through an fMRI compatible adaptation of a laboratory drinking paradigm [3].
Conclusions
Numerous biologically based endophenotypes that mediate the effects of genetic variations on AUD severity have been identified. Well-supported AUD endophenotypes include alcohol metabolism, electrophysiology measures, and subjective responses to acute alcohol consumption. In recent years, neuroimaging techniques have been used to examine potential endophenotypes related to reward dysregulation and impulsivity. We propose that further research would benefit from expanding this work, and focusing on endophenotypes consistent with a theoretical framework emphasizing the imbalance between neural reward and control networks, and the shifting network balance that can occur as individuals progress from recreational drinking to dependence. Gaining a deeper understanding of the interplay between these two networks—and identifying genetic variation and neural adaptations that affect reward and control processes—are likely to have important implications for diagnosis and treatment of AUDs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance and •• Of major importance
Abroms BD, Fillmore MT, Marczinski CA. Alcohol-induced impairment of behavioral control: effects on the alteration and suppression of prepotent responses. J Stud Alcohol. 2003;64(5):687–95.
Andrews MM, Meda SA, Thomas AD, Potenza MN, Krystal JH, Worhunsky P, et al. Individuals family history positive for alcoholism show functional magnetic resonance imaging differences in reward sensitivity that are related to impulsivity factors. Biol Psychiatry. 2011;69(7):675–83. doi:10.1016/j.biopsych.2010.09.049. By examining both reward sensitivity and impulsivity, this study exemplifies theory-driven endophenotype research. The finding of altered neural reward sensitivity in a family history-positive population supports reward dysfunction as a genetically informative endophenotype through demonstrating that it is present in relatives of individuals with AUDs.
Anton RF, Drobes DJ, Voronin K, Durazo-Avizu R, Moak D. Naltrexone effects on alcohol consumption in a clinical laboratory paradigm: temporal effects of drinking. Psychopharmacology (Berl). 2004;173(1–2):32–40. doi:10.1007/s00213-003-1720-7.
Baler RD, Volkow ND. Drug addiction: the neurobiology of disrupted self-control. Trends Mol Med. 2006;12(12):559–66. doi:10.1016/j.molmed.2006.10.005.
Beauchaine TP. The Role of Biomarkers and Endophenotypes in Prevention and Treatment of Psychopathological Disorders. Biomark Med. 2009;3(1):1–3. doi:10.2217/17520363.3.1.1.
Bechara A. Decision making, impulse control and loss of willpower to resist drugs: a neurocognitive perspective. Nat Neurosci. 2005;8(11):1458–63. doi:10.1038/nn1584.
Beck A, Wustenberg T, Genauck A, Wrase J, Schlagenhauf F, Smolka MN, et al. Effect of brain structure, brain function, and brain connectivity on relapse in alcohol-dependent patients. Arch Gen Psychiatry. 2012;69(8):842–52. doi:10.1001/archgenpsychiatry.2011.2026.
Begleiter H, Porjesz B. What is inherited in the predisposition toward alcoholism? A proposed model. Alcohol Clin Exp Res. 1999;23(7):1125–35.
Bezdjian S, Baker LA, Tuvblad C. Genetic and environmental influences on impulsivity: a meta-analysis of twin, family and adoption studies. Clin Psychol Rev. 2011;31(7):1209–23. doi:10.1016/j.cpr.2011.07.005.
Bickel WK, Miller ML, Yi R, Kowal BP, Lindquist DM, Pitcock JA. Behavioral and neuroeconomics of drug addiction: competing neural systems and temporal discounting processes. Drug Alcohol Depend. 2007;90 Suppl 1:S85–91. doi:10.1016/j.drugalcdep.2006.09.016.
Bjork JM, Hommer DW, Grant SJ, Danube C. Impulsivity in abstinent alcohol-dependent patients: relation to control subjects and type 1-/type 2-like traits. Alcohol. 2004;34(2–3):133–50.
Boettiger CA, Kelley EA, Mitchell JM, D’Esposito M, Fields HL. Now or Later? An fMRI study of the effects of endogenous opioid blockade on a decision-making network. Pharmacol Biochem Behav. 2009;93(3):291–9. doi:10.1016/j.pbb.2009.02.008.
Breese GR, Sinha R, Heilig M. Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther. 2011;129(2):149–71. doi:10.1016/j.pharmthera.2010.09.007.
Camchong J, Stenger A, Fein G. Resting-state synchrony during early alcohol abstinence can predict subsequent relapse. Cereb Cortex. 2013;23(9):2086–99. doi:10.1093/cercor/bhs190.
Camchong J, Stenger A, Fein G. Resting-state synchrony in long-term abstinent alcoholics. Alcohol Clin Exp Res. 2013;37(1):75–85. doi:10.1111/j.1530-0277.2012.01859.x.
Camchong J, Stenger VA, Fein G. Resting-state synchrony in short-term versus long-term abstinent alcoholics. Alcohol Clin Exp Res. 2013;37(5):794–803. doi:10.1111/acer.12037.
Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–90. doi:10.1146/annurev.clinpsy.2.022305.095232.
Carlson SR, Iacono WG, McGue M. P300 amplitude in adolescent twins discordant and concordant for alcohol use disorders. Biol Psychol. 2002;61(1–2):203–27.
Claus ED, Ewing SW, Filbey FM, Sabbineni A, Hutchison KE. Identifying neurobiological phenotypes associated with alcohol use disorder severity. Neuropsychopharmacology. 2011;36(10):2086–96. doi:10.1038/npp.2011.99. This study demonstrates the effective implementation of a cue-elicited craving model in fMRI research. Further, the brain regions that were significantly activated in response to alcohol taste cues in this study are all important candidates for future investigation in the context of alcohol craving and reward responsivity.
Claus ED, Kiehl KA, Hutchison KE. Neural and behavioral mechanisms of impulsive choice in alcohol use disorder. Alcohol Clin Exp Res. 2011;35(7):1209–19. doi:10.1111/j.1530-0277.2011.01455.x.
Dalley JW, Everitt BJ, Robbins TW. Impulsivity, compulsivity, and top-down cognitive control. Neuron. 2011;69(4):680–94. doi:10.1016/j.neuron.2011.01.020.
de Wit H. Impulsivity as a determinant and consequence of drug use: a review of underlying processes. Addict Biol. 2009;14(1):22–31. doi:10.1111/j.1369-1600.2008.00129.x.
de Wit H, Uhlenhuth EH, Pierri J, Johanson CE. Individual differences in behavioral and subjective responses to alcohol. Alcohol Clin Exp Res. 1987;11(1):52–9.
Dougherty DM, Marsh-Richard DM, Hatzis ES, Nouvion SO, Mathias CW. A test of alcohol dose effects on multiple behavioral measures of impulsivity. Drug Alcohol Depend. 2008;96(1–2):111–20. doi:10.1016/j.drugalcdep.2008.02.002.
Eng MY, Schuckit MA, Smith TL. The level of response to alcohol in daughters of alcoholics and controls. Drug Alcohol Depend. 2005;79(1):83–93. doi:10.1016/j.drugalcdep.2005.01.002.
Ersche KD, Turton AJ, Pradhan S, Bullmore ET, Robbins TW. Drug addiction endophenotypes: impulsive versus sensation-seeking personality traits. Biol Psychiatry. 2010;68(8):770–3. doi:10.1016/j.biopsych.2010.06.015.
Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8(11):1481–9. doi:10.1038/nn1579.
Filbey FM, Ray L, Smolen A, Claus ED, Audette A, Hutchison KE. Differential neural response to alcohol priming and alcohol taste cues is associated with DRD4 VNTR and OPRM1 genotypes. Alcohol Clin Exp Res. 2008;32(7):1113–23. doi:10.1111/j.1530-0277.2008.00692.x.
Filbey FM, Schacht JP, Myers US, Chavez RS, Hutchison KE. Marijuana craving in the brain. Proc Natl Acad Sci U S A. 2009;106(31):13016–21. doi:10.1073/pnas.0903863106.
Fillmore MT, Vogel-Sprott M. Response inhibition under alcohol: effects of cognitive and motivational conflict. J Stud Alcohol. 2000;61(2):239–46.
Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8(9):700–11. doi:10.1038/nrn2201.
Fryer SL, Jorgensen KW, Yetter EJ, Daurignac EC, Watson TD, Shanbhag H, et al. Differential brain response to alcohol cue distractors across stages of alcohol dependence. Biol Psychol. 2013;92(2):282–91. doi:10.1016/j.biopsycho.2012.10.004. This innovative study uses a novel task to simultaneously examine neural reward and control networks among individuals with different levels of alcohol use, and demonstrates how these findings may be useful for characterizing and differentiating the different stages of alcohol dependence. As the authors of this review recommended, such an investigation of theory-driven AUD endophenotypes is likely to have useful implications for diagnosis and treatment.
George MS, Anton RF, Bloomer C, Teneback C, Drobes DJ, Lorberbaum JP, et al. Activation of prefrontal cortex and anterior thalamus in alcoholic subjects on exposure to alcohol-specific cues. Arch Gen Psychiatry. 2001;58(4):345–52.
Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159(10):1642–52.
Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160(4):636–45.
Gottesman II, Shields J. Genetic theorizing and schizophrenia. Br J Psychiatry. 1973;122(566):15–30.
Hasin DS, Stinson FS, Ogburn E, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2007;64(7):830–42. doi:10.1001/archpsyc.64.7.830.
Heath AC, Bucholz KK, Madden PA, Dinwiddie SH, Slutske WS, Bierut LJ, et al. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol Med. 1997;27(6):1381–96.
Heinz A, Siessmeier T, Wrase J, Hermann D, Klein S, Grusser SM, et al. Correlation between dopamine D(2) receptors in the ventral striatum and central processing of alcohol cues and craving. Am J Psychiatry. 2004;161(10):1783–9. doi:10.1176/appi.ajp.161.10.1783.
Hendershot CS, Witkiewitz K, George WH, Marlatt GA. Relapse prevention for addictive behaviors. Subst Abuse Treat Prev Policy. 2011;6:17. doi:10.1186/1747-597x-6-17.
Higuchi S, Matsushita S, Masaki T, Yokoyama A, Kimura M, Suzuki G, et al. Influence of genetic variations of ethanol-metabolizing enzymes on phenotypes of alcohol-related disorders. Ann N Y Acad Sci. 2004;1025:472–80. doi:10.1196/annals.1316.058.
Hu X, Oroszi G, Chun J, Smith TL, Goldman D, Schuckit MA. An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alcohol Clin Exp Res. 2005;29(1):8–16.
Hutchison KE. Substance use disorders: realizing the promise of pharmacogenomics and personalized medicine. Annu Rev Clin Psychol. 2010;6:577–89. doi:10.1146/annurev.clinpsy.121208.131441.
Hutchison KE, Haughey H, Niculescu M, Schacht J, Kaiser A, Stitzel J, et al. The incentive salience of alcohol: translating the effects of genetic variant in CNR1. Arch Gen Psychiatry. 2008;65(7):841–50. doi:10.1001/archpsyc.65.7.841.
Hyman SE. The neurobiology of addiction: implications for voluntary control of behavior. Am J Bioeth. 2007;7(1):8–11. doi:10.1080/15265160601063969.
Ihssen N, Cox WM, Wiggett A, Fadardi JS, Linden DE. Differentiating heavy from light drinkers by neural responses to visual alcohol cues and other motivational stimuli. Cereb Cortex. 2011;21(6):1408–15. doi:10.1093/cercor/bhq220. This study uses a neural reward–responsivity endophenotype to classify individuals on the basis of drinking behavior. The work is a good example of how neuroimaging phenotypes can be used for diagnostic purposes.
Joslyn G, Brush G, Robertson M, Smith TL, Kalmijn J, Schuckit M, et al. Chromosome 15q25.1 genetic markers associated with level of response to alcohol in humans. Proc Natl Acad Sci U S A. 2008;105(51):20368–73. doi:10.1073/pnas.0810970105.
Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162(8):1403–13. doi:10.1176/appi.ajp.162.8.1403.
Kamarajan C, Porjesz B, Jones KA, Choi K, Chorlian DB, Padmanabhapillai A, et al. The role of brain oscillations as functional correlates of cognitive systems: a study of frontal inhibitory control in alcoholism. Int J Psychophysiol. 2004;51(2):155–80.
Kareken DA, Claus ED, Sabri M, Dzemidzic M, Kosobud AE, Radnovich AJ, et al. Alcohol-related olfactory cues activate the nucleus accumbens and ventral tegmental area in high-risk drinkers: preliminary findings. Alcohol Clin Exp Res. 2004;28(4):550–7.
Kareken DA, Dzemidzic M, Wetherill L, Eiler 2nd W, Oberlin BG, Harezlak J, et al. Family history of alcoholism interacts with alcohol to affect brain regions involved in behavioral inhibition. Psychopharmacology (Berl). 2013;228(2):335–45. doi:10.1007/s00213-013-3038-4. This study, by examining the interaction of genetic factors and alcohol consumption in neural control regions, furnished compelling evidence that inhibitory control is meaningfully (and perhaps specifically) related to alcohol use.
Karoly HC, Harlaar N, Hutchison KE. Substance use disorders: a theory-driven approach to the integration of genetics and neuroimaging. Ann N Y Acad Sci. 2013;1282:71–91. doi:10.1111/nyas.12074.
Kendler KS, Heath AC, Neale MC, Kessler RC, Eaves LJ. A population-based twin study of alcoholism in women. JAMA. 1992;268(14):1877–82.
Khalili-Mahani N, Zoethout RM, Beckmann CF, Baerends E, de Kam ML, Soeter RP, et al. Effects of morphine and alcohol on functional brain connectivity during "resting state": a placebo-controlled crossover study in healthy young men. Hum Brain Mapp. 2012;33(5):1003–18. doi:10.1002/hbm.21265.
King AC, de Wit H, McNamara PJ, Cao D. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry. 2011;68(4):389–99. doi:10.1001/archgenpsychiatry.2011.26.
Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278(5335):52–8.
Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24(2):97–129. doi:10.1016/s0893-133x(00)00195-0.
Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2009;35(1):217–38.
Lawrence AJ, Luty J, Bogdan NA, Sahakian BJ, Clark L. Impulsivity and response inhibition in alcohol dependence and problem gambling. Psychopharmacology (Berl). 2009;207(1):163–72. doi:10.1007/s00213-009-1645-x.
Lewis MJ, June HL. Neurobehavioral studies of ethanol reward and activation. Alcohol. 1990;7(3):213–9.
Li CS, Luo X, Yan P, Bergquist K, Sinha R. Altered impulse control in alcohol dependence: neural measures of stop signal performance. Alcohol Clin Exp Res. 2009;33(4):740–50. doi:10.1111/j.1530-0277.2008.00891.x.
Logan GD. On the ability to inhibit thought and action: a user’s guide to the stop signal paradigm. In: Dagenbach D, Carr TH, editors. Inhibitory Processes in Attention, Memory, and Language. San Diego: Academic Press; 1994. p. 189–239.
Luczak SE, Glatt SJ, Wall TL. Meta-analyses of ALDH2 and ADH1B with alcohol dependence in Asians. Psychol Bull. 2006;132(4):607–21. doi:10.1037/0033-2909.132.4.607.
Maas LC, Lukas SE, Kaufman MJ, Weiss RD, Daniels SL, Rogers VW, et al. Functional magnetic resonance imaging of human brain activation during cue-induced cocaine craving. Am J Psychiatry. 1998;155(1):124–6.
MacKillop J. Integrating behavioral economics and behavioral genetics: delayed reward discounting as an endophenotype for addictive disorders. J Exp Anal Behav. 2013;99(1):14–31. doi:10.1002/jeab.4.
Mann K, Hermann D. Individualised treatment in alcohol-dependent patients. Eur Arch Psychiatry Clin Neurosci. 2010;260 Suppl 2:S116–20. doi:10.1007/s00406-010-0153-7.
Marschner A, Mell T, Wartenburger I, Villringer A, Reischies FM, Heekeren HR. Reward-based decision-making and aging. Brain Res Bull. 2005;67(5):382–90. doi:10.1016/j.brainresbull.2005.06.010.
McClernon FJ, Kozink RV, Lutz AM, Rose JE. 24-h smoking abstinence potentiates fMRI-BOLD activation to smoking cues in cerebral cortex and dorsal striatum. Psychopharmacology (Berl). 2009;204(1):25–35. doi:10.1007/s00213-008-1436-9.
McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21(21):8655–63.
Miller NS. Comorbidity of psychiatric and alcohol/drug disorders: interactions and independent status. J Addict Dis. 1993;12(3):5–16. doi:10.1300/J069v12n03_02.
Mitchell JM, Fields HL, D’Esposito M, Boettiger CA. Impulsive responding in alcoholics. Alcohol Clin Exp Res. 2005;29(12):2158–69.
Mulvihill LE, Skilling TA, Vogel-Sprott M. Alcohol and the ability to inhibit behavior in men and women. J Stud Alcohol. 1997;58(6):600–5.
Myrick H, Anton RF, Li X, Henderson S, Drobes D, Voronin K, et al. Differential brain activity in alcoholics and social drinkers to alcohol cues: relationship to craving. Neuropsychopharmacology. 2004;29(2):393–402. doi:10.1038/sj.npp.1300295.
Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2(2):119–28. doi:10.1038/35053570.
O’Daly OG, Trick L, Scaife J, Marshall J, Ball D, Phillips ML, et al. Withdrawal-associated increases and decreases in functional neural connectivity associated with altered emotional regulation in alcoholism. Neuropsychopharmacology. 2012;37(10):2267–76. doi:10.1038/npp.2012.77.
O’Keefe JH, Bybee KA, Lavie CJ. Alcohol and cardiovascular health: the razor-sharp double-edged sword. J Am Coll Cardiol. 2007;50(11):1009–14. doi:10.1016/j.jacc.2007.04.089.
Ortner CN, MacDonald TK, Olmstead MC. Alcohol intoxication reduces impulsivity in the delay-discounting paradigm. Alcohol Alcohol. 2003;38(2):151–6.
Petry NM, Kirby KN, Kranzler HR. Effects of gender and family history of alcohol dependence on a behavioral task of impulsivity in healthy subjects. J Stud Alcohol. 2002;63(1):83–90.
Piray P, Keramati MM, Dezfouli A, Lucas C, Mokri A. Individual differences in nucleus accumbens dopamine receptors predict development of addiction-like behavior: a computational approach. Neural Comput. 2010;22(9):2334–68. doi:10.1162/NECO_a_00009.
Prabhu VR, Porjesz B, Chorlian DB, Wang K, Stimus A, Begleiter H. Visual p3 in female alcoholics. Alcohol Clin Exp Res. 2001;25(4):531–9.
Prescott CA, Kendler KS. Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. Am J Psychiatry. 1999;156(1):34–40.
Quinn PD, Fromme K. Subjective response to alcohol challenge: a quantitative review. Alcohol Clin Exp Res. 2011;35(10):1759–70. doi:10.1111/j.1530-0277.2011.01521.x.
Ray LA, Hutchison KE. A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res. 2004;28(12):1789–95.
Ray LA, Mackillop J, Monti PM. Subjective responses to alcohol consumption as endophenotypes: advancing behavioral genetics in etiological and treatment models of alcoholism. Subst Use Misuse. 2010;45(11):1742–65. doi:10.3109/10826084.2010.482427.
Ray LA, Miranda Jr R, Tidey JW, McGeary JE, MacKillop J, Gwaltney CJ, et al. Polymorphisms of the μ-opioid receptor and dopamine D4 receptor genes and subjective responses to alcohol in the natural environment. Journal of abnormal psychology. 2010;119(1):115.
Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223–33. doi:10.1016/s0140-6736(09)60746-7.
Reynolds B, Richards JB, de Wit H. Acute-alcohol effects on the Experiential Discounting Task (EDT) and a question-based measure of delay discounting. Pharmacol Biochem Behav. 2006;83(2):194–202. doi:10.1016/j.pbb.2006.01.007.
Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18(3):247–91.
Satterthwaite TD, Green L, Myerson J, Parker J, Ramaratnam M, Buckner RL. Dissociable but inter-related systems of cognitive control and reward during decision making: evidence from pupillometry and event-related fMRI. Neuroimage. 2007;37(3):1017–31. doi:10.1016/j.neuroimage.2007.04.066.
Schuckit MA. Self-rating of alcohol intoxication by young men with and without family histories of alcoholism. J Stud Alcohol and Drugs. 1980;41(03):242.
Schuckit MA. Genetics of the risk for alcoholism. Am J Addict. 2000;9(2):103–12.
Schuckit MA. An overview of genetic influences in alcoholism. J Subst Abuse Treat. 2009;36(1):S5–14.
Schuckit MA, Smith TL. An 8-year follow-up of 450 sons of alcoholic and control subjects. Arch Gen Psychiatry. 1996;53(3):202–10.
Schuckit MA, Smith TL, Kalmijn J. The search for genes contributing to the low level of response to alcohol: patterns of findings across studies. Alcohol Clin Exp Res. 2004;28(10):1449–58.
Schuckit MA, Smith TL, Kalmijn J, Danko GP. A cross-generational comparison of alcohol challenges at about age 20 in 40 father-offspring pairs. Alcohol Clin Exp Res. 2005;29(11):1921–7.
Schuckit MA, Tsuang JW, Anthenelli RM, Tipp JE, Nurnberger Jr JI. Alcohol challenges in young men from alcoholic pedigrees and control families: a report from the COGA project. J Stud Alcohol. 1996;57(4):368–77.
Setiawan E, Pihl RO, Dagher A, Schlagintweit H, Casey KF, Benkelfat C, et al. Differential Striatal Dopamine Responses Following Oral Alcohol in Individuals at Varying Risk for Dependence. Alcohol Clin Exp Res. 2013. doi:10.1111/acer.12218. This work supports striatal dopamine as a probable AUD endophenotype, through demonstrating that extracellular dopamine level after alcohol ingestion is related to subjective response to alcohol, alcohol use problems, and family history.
Sher KJ. Subjective effects of alcohol: the influence of setting and individual differences in alcohol expectancies. J Stud Alcohol. 1985;46(2):137–46.
Sinha R, Li CS. Imaging stress- and cue-induced drug and alcohol craving: association with relapse and clinical implications. Drug Alcohol Rev. 2007;26(1):25–31. doi:10.1080/09595230601036960.
Smith CT, Boettiger CA. Age modulates the effect of COMT genotype on delay discounting behavior. Psychopharmacology (Berl). 2012;222(4):609–17. doi:10.1007/s00213-012-2653-9.
Stoeckel LE, Murdaugh DL, Cox JE, Cook 3rd EW, Weller RE. Greater impulsivity is associated with decreased brain activation in obese women during a delay discounting task. Brain Imaging Behav. 2013;7(2):116–28. doi:10.1007/s11682-012-9201-4.
Sullivan EV, Harris RA, Pfefferbaum A. Alcohol’s Effects on Brain and Behavior. Alcohol Res Health. 2010;33(1):127–43.
Trim RS, Simmons AN, Tolentino NJ, Hall SA, Matthews SC, Robinson SK, et al. Acute ethanol effects on brain activation in low- and high-level responders to alcohol. Alcohol Clin Exp Res. 2010;34(7):1162–70. doi:10.1111/j.1530-0277.2010.01193.x.
Viken RJ, Rose RJ, Morzorati SL, Christian JC, Li TK. Subjective intoxication in response to alcohol challenge: heritability and covariation with personality, breath alcohol level, and drinking history. Alcohol Clin Exp Res. 2003;27(5):795–803. doi:10.1097/01.alc.0000067974.41160.95.
Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56:3–8.
Wall TL, Shea SH, Luczak SE, Cook TA, Carr LG. Genetic associations of alcohol dehydrogenase with alcohol use disorders and endophenotypes in white college students. J Abnorm Psychol. 2005;114(3):456–65. doi:10.1037/0021-843x.114.3.456.
Winterer G, Enoch MA, White KV, Saylan M, Coppola R, Goldman D. EEG phenotype in alcoholism: increased coherence in the depressive subtype. Acta Psychiatr Scand. 2003;108(1):51–60.
Wrase J, Schlagenhauf F, Kienast T, Wustenberg T, Bermpohl F, Kahnt T, et al. Dysfunction of reward processing correlates with alcohol craving in detoxified alcoholics. Neuroimage. 2007;35(2):787–94. doi:10.1016/j.neuroimage.2006.11.043.
Zucker RA, Gomberg ES. Etiology of alcoholism reconsidered. The case for a biopsychosocial process. Am Psychol. 1986;41(7):783–93.
Compliance with Ethics Guidelines
Conflict of Interest
Hollis C. Karoly, Sarah L. Hagerty, and Kent E. Hutchison declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Karoly, H.C., Hagerty, S.L. & Hutchison, K.E. Developing Neurobiological Endophenotypes that Reflect Failure to Control Alcohol Consumption and Dependence. Curr Addict Rep 1, 10–18 (2014). https://doi.org/10.1007/s40429-013-0007-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40429-013-0007-2