
Abstract
Objectives
We evaluated the prognostic value of the neurotrophic tyrosine receptor kinase (NTRK) gene fusions by comparing the survival of patients with NTRK+ tumours with patients without NTRK+ tumours.
Methods
We used genomic and clinical registry data from the Center for Personalized Cancer Treatment (CPCT-02) study containing a cohort of cancer patients who were treated in Dutch clinical practice between 2012 and 2020. We performed a propensity score matching analysis, where NTRK+ patients were matched to NTRK− patients in a 1:4 ratio. We subsequently analysed the survival of the matched sample of NTRK+ and NTRK− patients using the Kaplan–Meier method and Cox regression, and performed an analysis of credibility to evaluate the plausibility of our result.
Results
Among 3556 patients from the CPCT-02 study with known tumour location, 24 NTRK+ patients were identified. NTRK+ patients were distributed across nine different tumour types: bone/soft tissue, breast, colorectal, head and neck, lung, pancreas, prostate, skin and urinary tract. NTRK fusions involving the NTRK3 gene (46%) and NTRK1 gene (33%) were most common. The survival analysis rendered a hazard ratio (HR) of 1.44 (95% CI 0.81–2.55) for NTRK+ patients. Using the point estimates of three prior studies on the prognostic value of NTRK fusions, our finding that the HR is > 1 was deemed plausible.
Conclusions
NTRK+ patients may have an increased risk of death compared with NTRK− patients. When using historic control data to assess the comparative effectiveness of TRK inhibitors, the prognostic value of the NTRK fusion biomarker should therefore be accounted for.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
We estimated the prognostic value of neurotrophic tyrosine receptor kinase (NTRK) gene fusions to support the estimation of the comparative effectiveness of TRK inhibitors. |
The hazard ratio (HR) for the survival of patients with tumours harbouring NTRK gene fusions compared with patients without NTRK fusions was found to be 1.44 (95% CI 0.81–2.55). While the result is not statistically significant, it is consistent with HR estimates from prior studies. |
Although the TRK inhibitors are marketed as ‘tumour agnostic’, we argue that there is likely to be heterogeneity in (comparative) treatment effectiveness and cost-effectiveness across tumour histologies and tissues of origin. We therefore encourage further research into methods that might be used to perform subgroup analyses on small patient samples. |
Furthermore, we encourage more widespread collection of clinic-genomic data and better linking of existing databases to facilitate improved estimates of treatment effectiveness for targeted therapies. |
1 Introduction
In line with an increased focus on genetic markers to better target cancer care, larotrectinib and entrectinib came to the market as the first two molecularly targeted therapies with a histology-independent (also called tissue or tumour agnostic) label. Larotrectinib received marketing authorisation from the US Food and Drug Administration (FDA) in November 2018 and was conditionally approved by the European Medicines Agency (EMA) in September 2019. Entrectinib followed in August 2019 and July 2020, respectively. Both pharmaceuticals are indicated for patients who have locally advanced or metastatic solid tumours with neurotrophic tyrosine receptor kinase (NTRK) gene fusions. They can be prescribed regardless of the tissue of origin of the tumour, and therefore are classed as histology-independent therapies [1].
While the prevalence of NTRK gene fusions varies significantly across different cancers, NTRK gene fusions are generally rare. A retrospective study conducted in the Netherlands found that NTRK gene fusions were identified in only 0.93% of patients referred for NTRK testing [2]. Indeed, the estimated prevalence across all cancer patients is only 0.30% [3]. NTRK fusions result from chromosomal rearrangements that cause the 3′ region of the NTRK gene to join the 5′ region of a fusion partner gene. Such fusions may result in TRK fusion proteins with constitutively active tyrosine kinases, which can lead to tumour growth [4]. In this paper, we will refer to patients who have cancer tumours with (without) NTRK gene fusions as NTRK+ (NTRK−) patients.
Larotrectinib and entrectinib are inhibitors of the three most common types of TRK protein: TRKA, TRKB and TRKC (encoded by the NTRK1, NTRK2 and NTRK3 genes, respectively). Trial results for both larotrectinib and entrectinib appear promising. A pooled analysis for three phase I/II clinical trials for larotrectinib, which included 244 patients, found a 67% objective response rate (ORR) [95% confidence interval (CI) 63–75] and a median duration of response (DoR) of 32.9 months (95% CI 27.3–41.7) [5]. A pooled analysis for three phase I/II trials for entrectinib, including 121 patients with a median follow-up of 25.8 months, found an ORR of 63% (95% CI 31–89) and median DoR of 22.1 months (95% CI 7.4 to not estimable) [6]. However, larotrectinib and entrectinib were evaluated on a subset of tumour types, therefore there is uncertainty on the efficacy of these treatments for other tumour types. Additionally, the trials for larotrectinib and entrectinib were single-arm trials. Due to the lack of a control arm, the comparative effectiveness of the two TRK inhibitors cannot be established from the trial data alone.
Additional data on the effectiveness of standard of care (SoC) for NTRK+ patients are needed. Briggs et al. outlined three methods for estimating the counterfactual in the absence of direct comparative data, using larotrectinib as a case study [7]. Two of these methods require access to the patient-level trial data. The first method uses the progression-free survival (PFS) that trial patients experienced during the previous line of therapy (i.e. before receiving the TRK inhibitor) as a proxy for the comparator arm and assumes the relationship between PFS and overall survival (OS) to be the same for both the TRK inhibitor and the comparator. The second method uses the PFS and OS for non-responders in the trial (i.e. those with stable or progressive disease after receiving the TRK inhibitor) as a proxy for the comparator arm. When patient-level data are not available, a third method can be used, which involves the use of a historical cohort to estimate survival in the control arm. In their study, Briggs et al. performed a systematic literature review for SoC treatment outcomes for each tumour type included in the larotrectinib trial and subsequently weighted the obtained data according to the distribution of tumour types in the trial.
However, estimates of the effectiveness of SoC are generally not available for NTRK+ patients specifically, given that cancer treatments have mostly been prescribed based on the tissue of origin (e.g. breast, pancreas), without identifying patients’ NTRK status. If patients with NTRK fusions have a different disease prognosis from patients without NTRK fusions, historical data combining NTRK+ and NTRK− patients may provide biased estimates of SoC effectiveness for NTRK+ patients. To establish whether historical data are appropriate, it is important to identify the prognostic value of NTRK fusions. If needed, the estimated prognostic value can subsequently also be used to adjust historical estimates of SoC effectiveness to better reflect the NTRK+ population. That is, when (extrapolated) survival data are available for a sample of NTRK− patients, the estimated hazard ratio can be applied to the survival times to get an estimate of the survival had the population been NTRK+.
In this study, we estimated a hazard ratio (HR) for the survival of NTRK+ patients relative to NTRK− patients. We performed a retrospective matching analysis on the Hartwig Medical Foundation (HMF) database, which comprises genomic and clinical data for metastatic cancer patients. We also used a Bayesian framework alongside the frequentist method to evaluate how plausible it is that there is indeed a difference in survival prognosis between NTRK+ and NTRK− patients (i.e. the effect of carrying an NTRK fusion on survival is non-zero, or HR ≠ 1) through an analysis of credibility [8, 9].
2 Methods
2.1 Data
The HMF database encompasses de-identified genomic and clinical registry data for cancer patients who were treated in Dutch clinical practice. We used data from the Center for Personalized Cancer Treatment (CPCT-02) study (NCT01855477), which is a subcohort in the HMF database. The study was approved by medical ethical committees of the University Medical Center Utrecht and the Netherlands Cancer Institute and was conducted in concordance with the Declaration of Helsinki, Dutch law and Good Clinical Practice. In the CPCT-02 study, whole-genome sequencing of tumour DNA was performed for thousands of patients from 44 academic, teaching and general hospitals in the Netherlands, over the period from 2012 until 2020. Patients were eligible for enrolment in the CPCT-02 study if (1) their age was ≥ 18 years, (2) they had a locally advanced or metastatic solid tumour, (3) they had an indication for a new line of systemic treatment with registered anti-cancer agents, (4) performing a biopsy on tumour tissue was safe according to the treating physician and (5) frozen blood and tissue samples were available and sufficient for whole-genome sequencing (WGS). All included patients gave explicit consent for the use of their genomic and clinical data for research purposes.
From the HMF database we obtained various genetic markers that were identified in patients’ tumour DNA, including NTRK gene fusions and other markers that are known as actionable targets for treatment. Detailed information on sample collection and the WGS procedure can be found elsewhere [10,11,12]. We also extracted data on several clinical variables, including the age and sex of the patient, the tumour type (i.e. tissue of origin), the year(s) in which tumour biopsies were performed, the starting date of the first post-biopsy treatment, the number of previous lines of therapy, a binary variable indicating whether the patient had died during the period of the study and, for patients remaining alive, the last known date at which they were still alive.
2.2 Matching Analysis
Patients were classified into two cohorts: NTRK+ patients and NTRK− patients. Given that the CPCT-02 study provides sequencing data of the tumour DNA, it cannot be known with certainty whether identified NTRK gene fusions are functional, i.e. whether they lead to the expression of fusion TRK proteins that have constitutively tyrosine kinase activity. Nonetheless, two necessary conditions for an NTRK gene fusion to be functional can be determined from the tumour DNA, namely an NTRK1, NTRK2 or NTRK3 gene with a complete tyrosine kinase domain is present on the 3′ end of the (postulated) transcript and the fusion gene (likely) encodes for an in-frame protein. Only patients who had NTRK gene fusions meeting the conditions were included in the NTKR+ cohort, while patients with NTRK gene fusions that did not meet the conditions were included in the NTRK− cohort.
To increase comparability between the NTRK+ and NTRK− patient cohorts, we only included NTRK− patients who had one of the tumour types appearing in the NTRK+ cohort. In both cohorts, patients who had received experimental treatments were excluded, given that our aim was to estimate the effectiveness of standard care. Patients for whom survival time could not be estimated because of missing dates on their appointment logs were also excluded.
We subsequently performed a propensity score matching analysis to identify a subgroup of NTRK− patients similar to the group of NTRK+ patients. Within each tumour type, patients were matched on the available demographic and clinical variables in the HMF database, i.e. age, sex, year of biopsy and number of previous lines of therapy. Age and sex are well-reported factors influencing expected disease outcomes, hence were included. The ‘year of biopsy’ variable was included to address possible changes in treatment patterns and treatment effectiveness over the included time period (2012–2020). The number of previous lines of treatment was included as a binary variable (≤ 2 or > 2 previous lines) and served as a proxy reflecting patients’ severity of disease, given that patients who have had many treatments already may be in a more advanced stage of disease. We used the optimal matching method [12] (see Online resource S1 for more details) without replacement, with a ratio of 1:4 (NTRK+: NTRK−) and a caliper width of 0.5 times the pooled estimate of the common standard deviation of the logits of the propensity scores. With smaller calipers, it was not possible to find a feasible optimal fixed ratio matching. Given the small sample size, no interaction terms or higher orders of the covariates were used. To assess whether the NTRK+ cohort and the matched NTRK− cohort were sufficiently similar to enable reliable estimation of the prognostic value of NTRK gene fusions, we used the three conditions outlined by Rubin [13]. First, the difference in the means of the propensity scores in the NTRK+ and NTRK− groups must be small, with the standardised measure Rubin’s B < 0.25. Also, the ratio of the variances of the propensity scores in the two groups (Rubin’s R), as well as the ratio of the variances of the residuals of the covariates after adjusting for the propensity score, must be between 0.5 and 2.
Although NTRK gene fusions are generally seen to be driver gene alterations (i.e. the alteration causing the onset and progression of tumour growth), they might in some cases not be the (only) oncogenic driver. We therefore performed a sensitivity analysis where we excluded NTRK+ patients whose tumour DNA contained other (known) oncogenic biomarkers. The remaining NTRK+ patients were matched to NTRK− patients using the same method as in the main analysis. Based on current knowledge about actionable biomarkers, we included mutations in the ALK, BRAF, EGFR, ERBB2, KRAS or ROS1 genes, as well as high tumour mutational burden (TMB) and microsatellite instability (MSI). The HMF database includes an estimate of driver likelihood between 0 and 1. The principle behind the likelihood assessment is that the likelihood of a passenger variant occurring in a particular sample should be approximately proportional to the tumor mutational burden and hence variants in samples with lower mutational burden are more likely to be drivers [14].
2.3 Survival Analysis
We analysed the survival of patients with and without NTRK gene fusions using the Kaplan–Meier method and Cox regression. To calculate patients’ overall survival (OS), we estimated the period between the start of the first post-biopsy treatment and the time of death or censor. Patients who were not recorded as dead were censored at their last known date of being alive, which was the date of their last appointment to assess response to treatment.
The survival analysis was also performed on the sensitivity analysis dataset described in Section 2.2.
2.4 Analysis of Credibility
Because of their small sample sizes, studies on the prognostic value of rare mutations such as NTRK gene fusions suffer from a lack of power in frequentist inference. This may lead to statistically insignificant study results. Also, p-values have been argued to be poor indicators of whether an effect is truly present (or absent) [8]. Instead, we used the analysis of credibility (AnCred) method, which originates from Bayesian methods, and is seen as a more nuanced alternative for evaluating the plausibility of study findings than the ‘pass/fail’ dichotomy posed by the p-value threshold of 0.05 [15]. In AnCred, the study finding (expressed as a point estimate and 95% CI) is used to calculate a critical prior interval (CPI) (see Online Resource S2 for more details) [9, 15]. The CPI indicates the level of support needed from prior studies to have credible evidence for a non-zero effect. For example, when the study finding of interest is non-significant, previous studies will make the finding plausible of a non-zero effect size if their point estimates fall within the CPI. This process is an inversion of the Bayes Theorem, as the study finding is used to deduce the range of prior effect sizes—the CPI—leading to a posterior interval that excludes no effect.
3 Results
3.1 Patient Characteristics
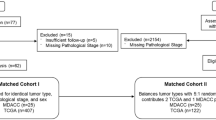
Among 3556 patients from the CPCT-02 study with known tumour location, 24 had tumours harbouring a likely functional NTRK gene fusion (Fig. 1). NTRK+ patients were spread across nine different tumour types: bone/soft tissue, breast, colorectal, head and neck, lung, pancreas, prostate, skin and urinary tract. The distribution of the different NTRK genes (NTRK1/NTRK2/NTRK3) across the tumour types is shown in Fig. 2. Among the remaining 2719 patients without an NTRK gene fusion, 2069 had one of the tumour types occurring in the NTRK+ cohort hence were included in the NTRK− cohort (Fig. 1).

Study schema. CPCT-02, Center for Personalized Cancer Treatment, NTRK, neurotrophic tyrosine receptor kinase

Distribution of tumour types in the NTRK+ cohort
In the NTRK+ cohort, the median age was 59 years (range 55–67 years), and 13 patients (54%) were female (Table 1). A minority of patients (33%) had received more than two lines of prior therapy. Most NTRK fusions involved the NTRK3 gene (11 patients, 46%) or NTRK1 gene (8 patients, 33%) (Fig. 2). Of the 24 different fusion partners identified, 20 were novel fusions that have not yet been reported in the Quiver database, a curated database of known oncogenic gene fusions. Other biomarkers found among NTRK+ patients were mutations in the BRAF, EGFR, ERBB2 and KRAS genes, as well as high TMB and MSI (Table 2) [16].
In the (non-matched) NTRK− cohort, the median age was higher (63 years, range 55–70 years), as was the percentage of patients with more than two lines of prior therapy (47%) (Table 1). The tumour distributions also differed between the non-matched NTRK− cohort and the NTRK+ cohort.
3.2 Matching and Survival Analysis
In the propensity score matching analysis, the 24 patients in the NTRK+ cohort were matched with 96 NTRK− patients. Standardized mean difference between groups were reduced for all covariates after propensity score matching (Table 1). Rubin’s B was 0.02 after matching, well below the recommended upper limit of 0.25. The variance ratios (Rubin’s R) of the propensity score and the covariates were also within the recommended range of 0.5–2 (Online Resource S3). Moreover, the box plot of the distribution of the logit of the propensity score shows an optimal overlap for the matched observations (Online Resource S4). Similarly, balance was obtained in the propensity score matching sensitivity analysis (Online Resources S5 and S6). Median OS of 12.7 months (95% CI 6.3–17.4) and 11.6 months (95% CI 7.8–17.9) were observed in the NTRK+ cohort and the matched NTRK− cohort, respectively. Despite the longer median OS for NTRK+ patients, the survival analysis rendered an HR of 1.44 (95% CI 0.81–2.55) (Fig. 3), meaning that NTRK+ patients are at higher risk of dying than NTRK− patients. The adjusted Cox regression provided an HR very close to the unadjusted, i.e. HR of 1.41 (95% CI 0.79–2.52). This result is in line with the reduction in the standardized mean difference between the covariates used for the propensity score (PS), which is below 0.10, for which performing a double adjustment is not necessary [17].

Kaplan-Meier plot for OS analysis
Additionally, a restricted mean survival time (RMST) analysis was conducted up to 40 months, representing the minimum of the largest observed event time within the NTRK− cohort. A 16.3 month RMST (95% CI 13.0–19.7) was estimated for NTRK+ patients, compared with 12.5 months RMST (95% CI 9.3–16.3) for NTRK− patients, supporting the results of the survival analysis.
In the sensitivity analysis, where NTRK+ patients with concurrent oncogenic biomarkers were excluded, we found a lower HR than in the main analysis (1.20, 95% CI 0.61–2.36) (Fig. 4).

Kaplan-Meier plot for OS sensitivity analysis
3.3 Analysis of Credibility
The point estimate (1.44) and 95% CI (0.81–2.55) in our main analysis show that the central effect is in the direction of a positive effect (i.e. HR > 1). However, HR values smaller than 1 are included in the 95% CI, and so the estimated point value is statistically non-significant.
The CPI associated with our results was calculated to be 1.0–11.2 (see Online Resource S2 for details), meaning that prior studies with estimates falling within this range make it more plausible that the HR for the survival of patients with NTRK+ tumours is larger than 1. To our knowledge, only three other studies have estimated the prognostic value of NTRK fusions. Two used the Flatiron Health-Foundation Medicine clinic-genomic database and one used the Genomic England database. Hibar et al. found an HR of 1.6 (95% CI 1.0–2.5) on survival analysis of 28 NTRK+ patients and 280 matched NTRK− patients. Bazhenova et al. found an HR of 1.44 (95% CI 0.61–3.37) in an analysis of 27 NTRK+ and 107 matched NTRK− patients [18]. Bridgewater et al. analysed 18 NTRK+ and 72 matched NTRK− patients and found a similar HR value of 1.47 (95% CI 0.39–5.57) [19]. Given that the point estimates of all three studies fall within the CPI, the studies support the plausibility of our finding that the survival HR for NTRK+ patients is > 1. That is, it is plausible that NTRK+ patients have a worse prognosis than NTRK− patients.
4 Discussion
Our study describes the clinical characteristics and survival of NTRK+ patients with advanced or metastatic disease who have previously been treated in Dutch clinical practice with SoC therapies other than targeted TRK inhibitors. NTRK+ patients appeared to have worse survival compared with NTRK− patients.
As the focus on better targeted, or ‘personalised’, cancer care continues, NTRK+ patients may be the first of many small patient groups with a specific genetic marker for whom treatment effectiveness must be evaluated. It has been argued that randomised controlled trials (RCTs), the preferred option to reliably estimate effectiveness [20, 21], may be difficult and time consuming to conduct for such small patient groups [22]. Although adapted, more flexible versions of the RCT design have been suggested [23, 24], pharmaceutical companies have so far mostly resorted to single-arm trials [23, 24]. Single-arm trials are poorly equipped to provide estimates of relative treatment effectiveness, due to the absence of a control arm reflecting the effectiveness of standard care. Briggs et al. [7] outlined possible ways to construct a control arm when faced with single-arm trial data for tumour-agnostic (i.e. genetic marker-focussed) treatments, including the use of historical data. We have expanded their work by arguing that, when evaluating the effectiveness of a treatment targeting a specific genetic marker, historical data may have to be adjusted for the prognostic value of said genetic marker. In this study we focussed on estimating the prognostic value of NTRK gene fusions. How the results can subsequently be used in an economic model evaluating treatment effectiveness can be found elsewhere [25]. While the results indicate that NTRK+ is a prognostic factor for earlier death relative to NTRK−, when using the HR on extrapolated survival estimated on NTRK− patients, the proportional hazards assumption is adopted for the entire forecasted period. Looking at the Kaplan–Meier survival plots, it is uncertain whether this assumption holds true.
As mentioned in the “Results” section, the prognostic value of NTRK gene fusions has been estimated in three prior studies focussing on the UK and the USA, using the Genomic England and FlatIron Health-Foundation Medicine databases. All studies to date, including ours, have been retrospective. A number of key differences among the studies can be noted. Firstly, in our study the median age of the patients was around 60 years while Bridgewater et al. included paediatric patients. Our study excluded patients treated with either TRK inhibitor or unlabelled therapy. Even though Bazhenova et al. conducted their study prior to the approval of larotrectinib and entrectinib in the USA, one patient with NTRK+ disease had received an unknown investigational agent in a clinical trial. Our cohort included some patients with tumour types not found in other studies (e.g. prostate and urothelial cancer). Also, the subtype of tumour was missing for some patients in the HMF database. This may be the reason why our study includes head and neck cancer as a broad tumour type, which potentially includes tumour in the salivary gland. Other differences in methodology need further consideration. The index date from which OS was measured varied between studies; Bazhenova et al. used the date of gene sequencing report in their primary analysis, Hibar et al. and Bridgewater et al. used the date of diagnosis. Hibar et al. used the start of last available treatment line before the NGS report in a sensitivity analysis. In our study, we used the date of first post-biopsy treatment to avoid potential immortal bias between the date of biopsy and the start of the treatment [26].
Despite these differences, all studies reported the same direction of effect, i.e. NTRK+ status increases the risk of mortality, with varying degrees of uncertainty.
The AnCred methodology has typically been used to interpret study results in the light of prior studies that have demonstrated an effect. Our application of AnCred is slightly different as there was no previous conclusive evidence, but rather previous uncertain evidence due to the sample size restrictions. Hence, we interpret our results in light of these previous studies to reflect a credible direction of effect. As EMA increasingly approves drugs based on evidence from single-arm studies, the challenge of dealing with uncertainty in HTA and reimbursement decision making is increasing. Against this background, it is important to use different means of managing uncertainty, one of which is the comparison of the previous results with the critical prior interval of AnCred.
We add to the literature by presenting findings obtained in a different country setting and using a different clinic-genomic database. Our sample distribution over age and primary tumour type broadly aligns with figures on solid cancer incidence in Western Europe, suggesting our Netherlands-focused research results may be applicable to Western Europe more broadly [27, 28].
4.1 Limitations
The aim of the CPCT-02 study was to identify patients eligible for clinical trials of targeted therapies (NCT01855477). That is, most patients enrolled in CPCT-02 had little to no SoC alternatives remaining. This is in line with the therapeutic indications for TRK inhibitors entrectinib and larotrectinib, both of which are for patients ‘who have no satisfactory treatment options’ according to the EMA [29, 30]. Nonetheless, there appear to be differences in patient characteristics between our study and a recent study focussing on NTRK testing in Dutch routine care [2], suggesting that the population included in the CPCT-02 study may not be fully representative of the population subject to NTRK testing (and treatment) in clinical practice. It is unknown to what extent such differences might affect our estimated HR for overall survival.
Because of limited availability of clinical data in the HMF database, we may not have included all relevant covariates in the matching process. Residual confounding can therefore not be ruled out. For example, known predictors of mortality [31] such as disease stage, severity of disease [e.g. measured by Eastern Cooperative Oncology Group (ECOG) performance status], serum albumin and platelet count, were not available in the dataset. For lack of explicit data on patients’ severity of disease, we used ‘the number of previous lines of therapy’ as a proxy. We theorise that patients who have had many treatments already are likely to be in a more advanced stage of disease, but this might not always be true as severely ill patients may be too weak to receive many lines of treatment.
In this study, we estimated a single HR value for all NTRK+ patients. However, evidence suggests heterogeneity in the prognostic value of NTRK gene fusions across tumour types [32]. We deemed our sample of 24 patients with NTRK+ tumours too small to obtain meaningful results from a subgroup analysis. Nonetheless, we encourage further research into methods that might be used to perform subgroup analyses on small patient samples [32].
When excluding NTRK+ patients with concurrent oncogenic biomarkers, we found a lower HR (1.20, 95% CI 0.61–2.36). When concurrent biomarkers are oncogenic drivers, there may be an interplay between said oncogenic drivers and the NTRK fusion gene, whereby collaborating oncogenic pathways are activated and tumour growth may be increased [33]. Thus, including patients with concurrent biomarkers in the NTRK+ cohort, as we did in the main analysis, may lead to an overestimation of the prognostic value of NTRK gene fusions per se. Nonetheless, the HR value estimated in the sensitivity analysis is larger than 1, suggesting that even if the HR value was overestimated in the main analysis, NTRK+ patients are still faced with worse survival than NTRK− patients.
4.2 Research and Policy Considerations
The advent of tumour-agnostic cancer care expands treatment opportunities and possibly enables better targeting of care. However, pooling patients in a tumour-agnostic manner when estimating treatment effectiveness may be inappropriate. There is likely heterogeneity in treatment effectiveness across tumours with different histologies and tissues of origin, for example because of differences in survival between tumour types and differences in the prognostic value of oncogenic drivers (e.g. it has been found that the tumour-promoting activity of oncogenic drivers may depend on the tissue of origin) [33, 34]. We therefore recommend that treatment effectiveness is estimated not only for the whole patient population with a specific genetic marker but for relevant subgroups as well. We acknowledge that doing so would reduce the sample sizes per disease indication even further. Solutions may be found during the running of the trial (e.g. stopping rules in an adaptive trial design framework) and in applying statistical methods that do not assume identical treatment effect between tumour types (e.g. exchangeability assumption in the Bayesian approach), as well as in more extensive collection of (real-world) data [35,36,37].
Beyond heterogeneity in treatment effect, there may also be heterogeneity in comparative effectiveness and cost-effectiveness, due to differences in the effectiveness and costs of comparative therapies across tumour types, as well as differences in existing testing protocols [e.g. broad genetic testing is already commonplace for non-small cell lung cancer (NSCLC) in the Netherlands, making the additional cost of testing for NTRK gene fusions negligible]. Reimbursement decisions for tumour-agnostic treatments may therefore also have to be specified for relevant subgroups instead of the whole population with the genetic marker.
Our research on the prognostic value of NTRK fusions and, relatedly, the treatment effectiveness of larotrectinib and entrectinib [25], was hampered by limited data. With a larger database and data on more clinical variables, we might have been able to provide further insights. Given that genetic marker-based pharmaceuticals (and single-arm trials) are likely to become more frequent, we encourage policymakers to consider more widespread collection of clinic-genomic data, and better linking of existing databases. As pharmaceutical trials have been notoriously Caucasian- and male focused [38, 39], we would like to stress the importance of ensuring that the populations included in clinic-genomic databases reflect real-life populations.
In conclusion, our findings suggest that patients with tumours harbouring an NTRK fusion gene may have an increased, or at least similar, risk of death compared with matched patients with tumours harbouring NTRK wild-type genes. This emphasises the relevance of NTRK gene fusions as actionable drug targets and provides support for the potential clinical benefit of TRK inhibitor therapy. By showing that survival may differ between NTRK+ and NTRK− patients, our study underscores the need to correct historic control data for the prognostic value of biomarkers when assessing comparative effectiveness.
References
Dunn DB. Larotrectinib and entrectinib: TRK inhibitors for the treatment of pediatric and adult patients with NTRK gene fusion. J Adv Pract Oncol. 2020. https://doi.org/10.6004/jadpro.2020.11.4.9.
Koopman B, et al. Detection of NTRK fusions and TRK expression and performance of pan-trk immunohistochemistry in routine diagnostics: results from a nationwide community-based cohort. Diagnostics. 2022;12(3):668. https://doi.org/10.3390/diagnostics12030668.
Westphalen CB, et al. Genomic context of NTRK1/2/3 fusion-positive tumours from a large real-world population. NPJ Precis Oncol. 2021;5(1):69. https://doi.org/10.1038/s41698-021-00206-y.
Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30:viii4–15. https://doi.org/10.1093/annonc/mdz383.
Drilon AE, et al. Long-term efficacy and safety of larotrectinib in a pooled analysis of patients with tropomyosin receptor kinase (TRK) fusion cancer. J Clin Oncol. 2022;40(16_suppl):3100. https://doi.org/10.1200/JCO.2022.40.16_suppl.3100.
Demetri GD, et al. Updated integrated analysis of the efficacy and safety of entrectinib in patients with NTRK fusion-positive solid tumors. Clin Cancer Res. 2022;28(7):1302–12. https://doi.org/10.1158/1078-0432.CCR-21-3597.
Briggs A, et al. Comparison of alternative methods to assess the cost-effectiveness of tumor-agnostic therapies: a triangulation approach using larotrectinib as a case study. Value Health J Int Soc Pharmacoecon Outcomes Res. 2022;25(6):1002–9. https://doi.org/10.1016/j.jval.2021.11.1354.
Matthews RAJ. Moving towards the post p < 0.05 era via the analysis of credibility. Am Stat. 2019;73(sup1):202–12. https://doi.org/10.1080/00031305.2018.1543136.
Matthews RAJ. Beyond “significance”: principles and practice of the analysis of credibility. R Soc Open Sci. 2018;5(1): 171047. https://doi.org/10.1098/rsos.171047.
Priestley P, et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature. 2019;575(7781):210–6. https://doi.org/10.1038/s41586-019-1689-y.
Samsom KG, et al. Study protocol: whole genome sequencing implementation in standard diagnostics for every cancer patient (WIDE). BMC Med Genomics. 2020;13(1):169. https://doi.org/10.1186/s12920-020-00814-w.
Hartwig Medical Foundation—Pipeline5. [Online]. https://github.com/hartwigmedical/pipeline5. Accessed Aug 2022.
Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Methodol. 2001;2(3/4):169–88. https://doi.org/10.1023/A:1020363010465.
‘Driver Catalog’. [Online]. https://github.com/hartwigmedical/hmftools/blob/master/purple/DriverCatalog.md. Accessed Aug 2022.
Held L, Matthews R, Ott M, Pawel S. Reverse-Bayes methods for evidence assessment and research synthesis. Res Synth Methods. 2022;13(3):295–314. https://doi.org/10.1002/jrsm.1538.
Archer DX Inc. Quiver database, 2021 [Internet]. [cited March 12, 2021]. http://quiver.archerdx.com. Accessed Aug 2022.
Nguyen T-L, et al. Double-adjustment in propensity score matching analysis: choosing a threshold for considering residual imbalance. BMC Med Res Methodol. 2017;17(1):78. https://doi.org/10.1186/s12874-017-0338-0.
Bazhenova L, et al. TRK fusion cancer: patient characteristics and survival analysis in the real-world setting. Target Oncol. 2021;16(3):389–99. https://doi.org/10.1007/s11523-021-00815-4.
Bridgewater J, et al. Prognosis and oncogenomic profiling of patients with tropomyosin receptor kinase fusion cancer in the 100,000 genomes project. Cancer Treat Res Commun. 2022;33: 100623. https://doi.org/10.1016/j.ctarc.2022.100623.
Levi M, McGovern DPB, Summerskill WSM, Valori RM. Key topics evidence-based medicine. Boca Raton: CRC Press; 2001.
Evans D. Hierarchy of evidence: a framework for ranking evidence evaluating healthcare interventions. J Clin Nurs. 2003;12(1):77–84. https://doi.org/10.1046/j.1365-2702.2003.00662.x.
Parmar MKB, Sydes MR, Morris TP. How do you design randomised trials for smaller populations? A framework. BMC Med. 2016;14(1):183. https://doi.org/10.1186/s12916-016-0722-3.
Burnett T, Mozgunov P, Pallmann P, Villar SS, Wheeler GM, Jaki T. Adding flexibility to clinical trial designs: an example-based guide to the practical use of adaptive designs. BMC Med. 2020;18(1):352. https://doi.org/10.1186/s12916-020-01808-2.
Superchi C, et al. Study designs for clinical trials applied to personalised medicine: a scoping review. BMJ Open. 2022;12(5): e052926. https://doi.org/10.1136/bmjopen-2021-052926.
Huygens S, et al. Cost-effectiveness analysis of treating patients with NTRK-positive cancer with the histology-independent therapy entrectinib. Value Health J Int Soc Pharmacoecon Outcomes Res. 2023;26(2):193–203. https://doi.org/10.1016/j.jval.2022.08.006.
Yadav K, Lewis RJ. Immortal time bias in observational studies. JAMA. 2021;325(7):686–7. https://doi.org/10.1001/jama.2020.9151.
Sung H, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Ferlay J, et al. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021. https://doi.org/10.1002/ijc.33588.
European Medicines Agency (EMA). Assessment report VITRAKVI International non-proprietary name: larotrectinib. July 25, 2019. Available https://www.ema.europa.eu/en/documents/assessment-report/vitrakvi-epar-public-assessment-report_en.pdf.
European Medicines Agency (EMA). CHMP assessment report Rozlytrek International non-proprietary name: entrectinib. May 28, 2020. Available https://www.ema.europa.eu/en/documents/assessment-report/rozlytrek-epar-public-assessment-report_en.pdf.
Owusuaa C, Dijkland SA, Nieboer D, van der Heide A, van der Rijt CCD. Predictors of mortality in patients with advanced cancer-a systematic review and meta-analysis. Cancers. 2022;14(2):328. https://doi.org/10.3390/cancers14020328.
Murphy P, et al. Modelling approaches for histology-independent cancer drugs to inform NICE appraisals: a systematic review and decision-framework. Health Technol Assess Winch Engl. 2021;25(76):1–228. https://doi.org/10.3310/hta25760.
El Tekle G, et al. Co-occurrence and mutual exclusivity: what cross-cancer mutation patterns can tell us. Trends Cancer. 2021;7(9):823–36. https://doi.org/10.1016/j.trecan.2021.04.009.
Hoadley KA, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173(2):291-304.e6. https://doi.org/10.1016/j.cell.2018.03.022.
Liu R, Liu Z, Ghadessi M, Vonk R. Increasing the efficiency of oncology basket trials using a Bayesian approach. Contemp Clin Trials. 2017;63:67–72. https://doi.org/10.1016/j.cct.2017.06.009.
Ventz S, Barry WT, Parmigiani G, Trippa L. Bayesian response-adaptive designs for basket trials. Biometrics. 2017;73(3):905–15. https://doi.org/10.1111/biom.12668.
Bokemeyer C, et al. Overall survival (OS) of patients with TRK fusion–positive cancer receiving larotrectinib versus standard of care (SoC): a matching-adjusted indirect comparison (MAIC) using real-world data (RWD). J Clin Oncol. 2022;40(16_suppl):6579–6579. https://doi.org/10.1200/JCO.2022.40.16_suppl.6579.
Camidge DR, et al. Race and ethnicity representation in clinical trials: findings from a literature review of phase I oncology trials. Future Oncol Lond Engl. 2021;17(24):3271–80. https://doi.org/10.2217/fon-2020-1262.
Woods-Burnham L, Johnson JR, Hooker SE, Bedell FW, Dorff TB, Kittles RA. The role of diverse populations in US clinical trials. Med N Y N. 2021;2(1):21–4. https://doi.org/10.1016/j.medj.2020.12.009.
Acknowledgements
We thank the Hartwig Medical Foundation for providing the genetic and clinical data. The authors express their gratitude to the oncologists and clinical geneticists who provided insights into clinical practice for oncology patients and explained various details about the diagnostic methods for identifying NTRK gene fusions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The study took place within the context of an EU-funded project ‘Health economics for personalised medicine’ abbreviated to HEcoPerMed. The HEcoPerMed project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement number 824997.
Conflicts of Interest
Irene Santi (IS), Heleen Vellekoop (HV), Matthijs Versteegh (MV), Simone Huygens (SH), Winand N.M. Dinjens (WD), Maureen Rutten-van Mölken (MRvM) have no conflicts of interest that are directly or indirectly related to the content of this article.
Availability of Data and Materials
Data sharing is not applicable to this article as no datasets were generated during the current study.
Ethics Approval
Not applicable.
Informed Consent
Not applicable.
Author Contributions
IS contributed to the concept of the study, acquired, analysed and interpreted the data and took the lead in writing the manuscript. MV and MRvM obtained the funds, contributed to the concept of the study, interpretation of the results and to the writing of the manuscript. HV, SH and WD contributed to the concept of the study, interpretation of the results and to the writing of the manuscript.
Code Availability
Not applicable.
Role of the Funder/Sponsor
The funder had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review and approval of the manuscript; nor the decision to submit the manuscript for publication.
Consent to participate
The participants have already given informed consent when included in the CPCT-02 study.
Consent for publication
All authors provide this consent.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Santi, I., Vellekoop, H., M Versteegh, M. et al. Estimating the Prognostic Value of the NTRK Fusion Biomarker for Comparative Effectiveness Research in The Netherlands. Mol Diagn Ther 28, 319–328 (2024). https://doi.org/10.1007/s40291-024-00704-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-024-00704-2